
Transcriptome Sequencing and De Novo Assembly of Golden Cuttlefish Sepia esculenta Hoyle

Abstract
:1. Introduction
2. Results
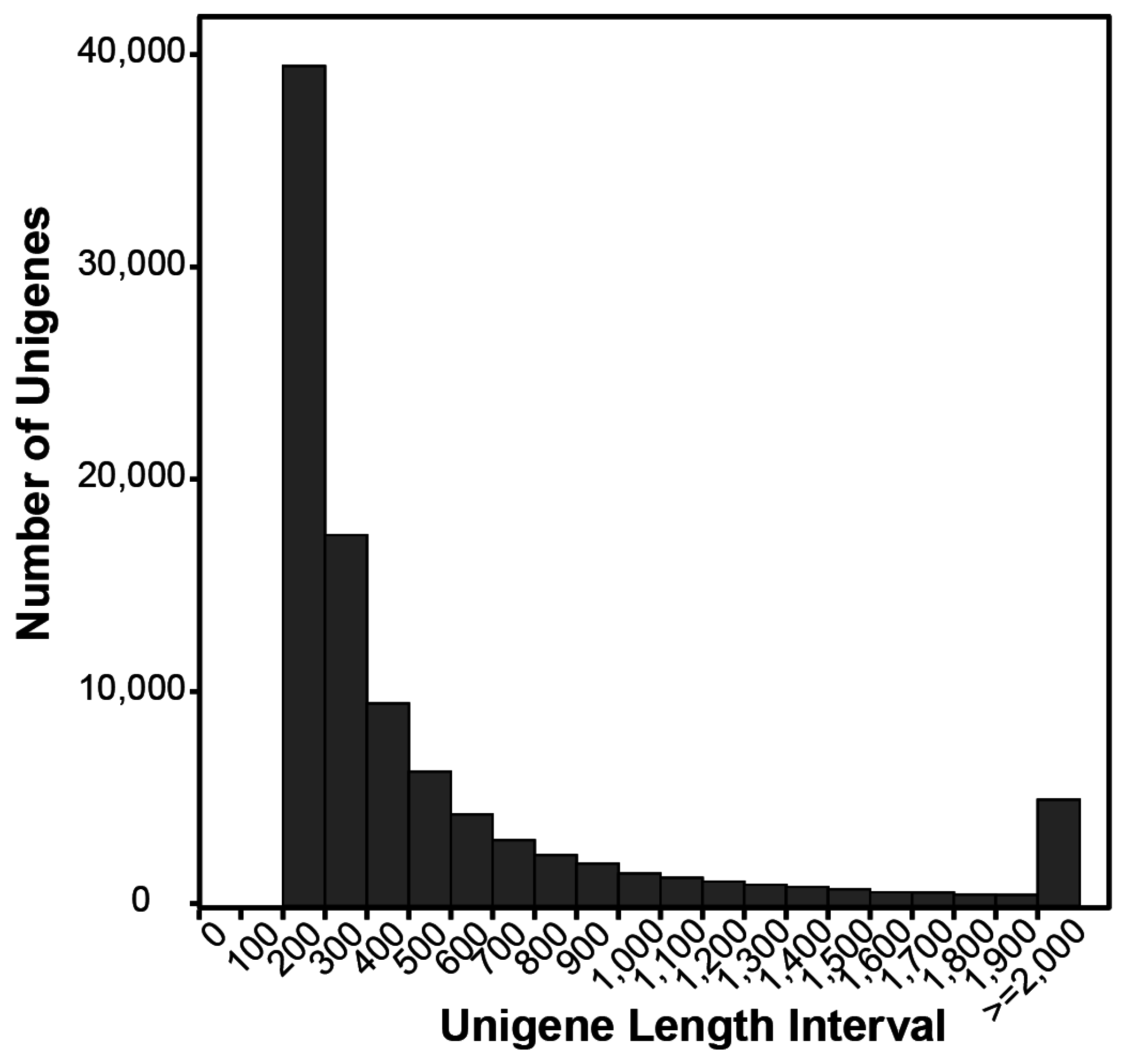
2.1. Illumina Sequencing and Assembly
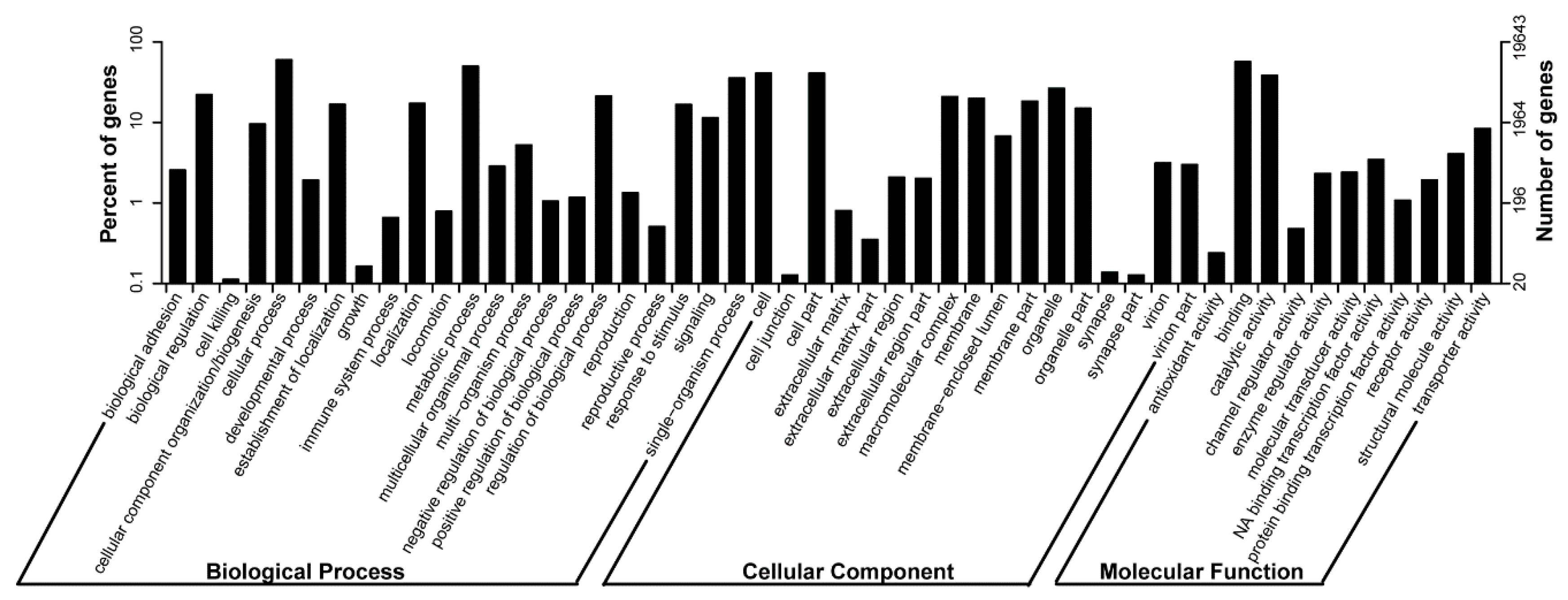
2.2. Functional Categorization
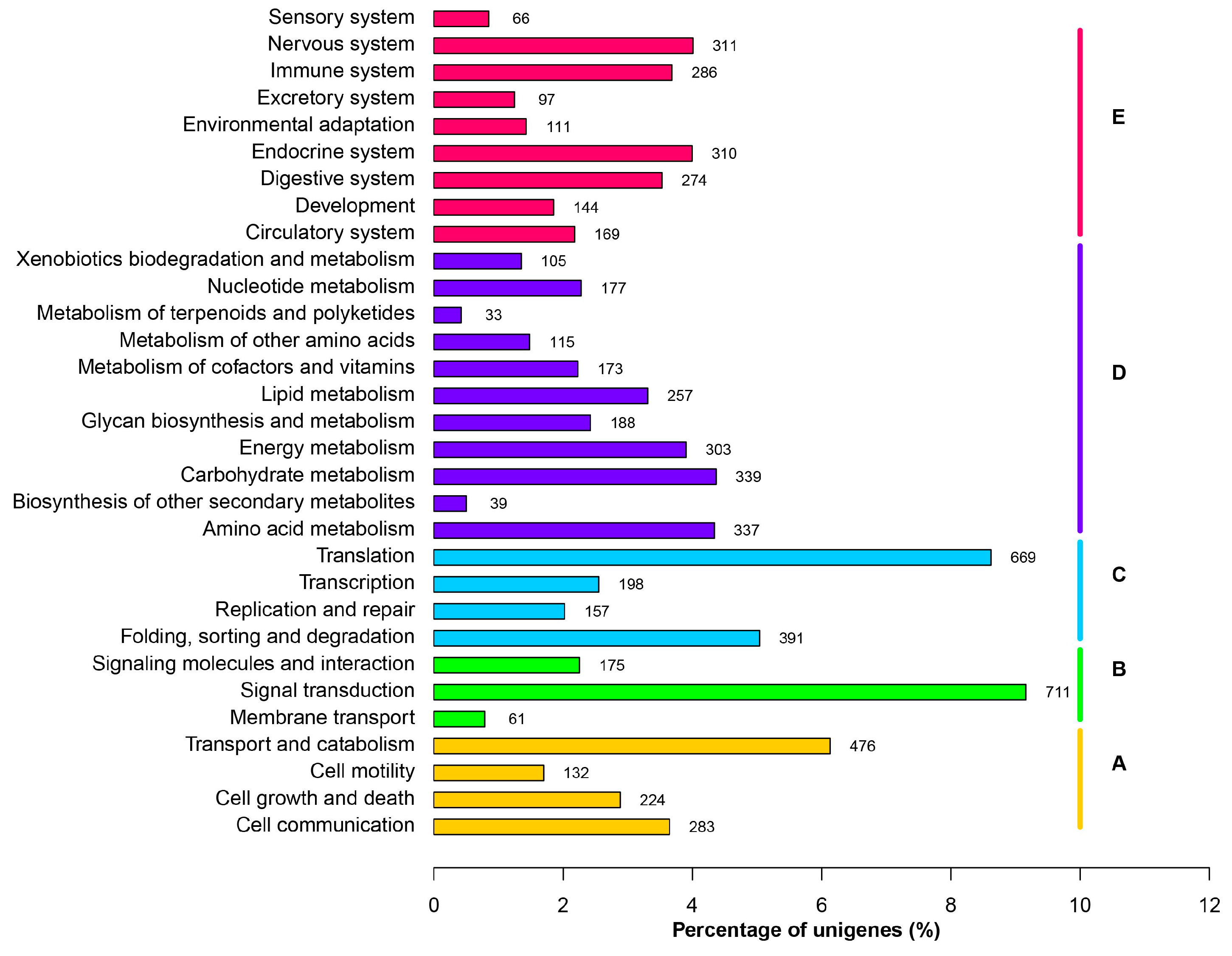
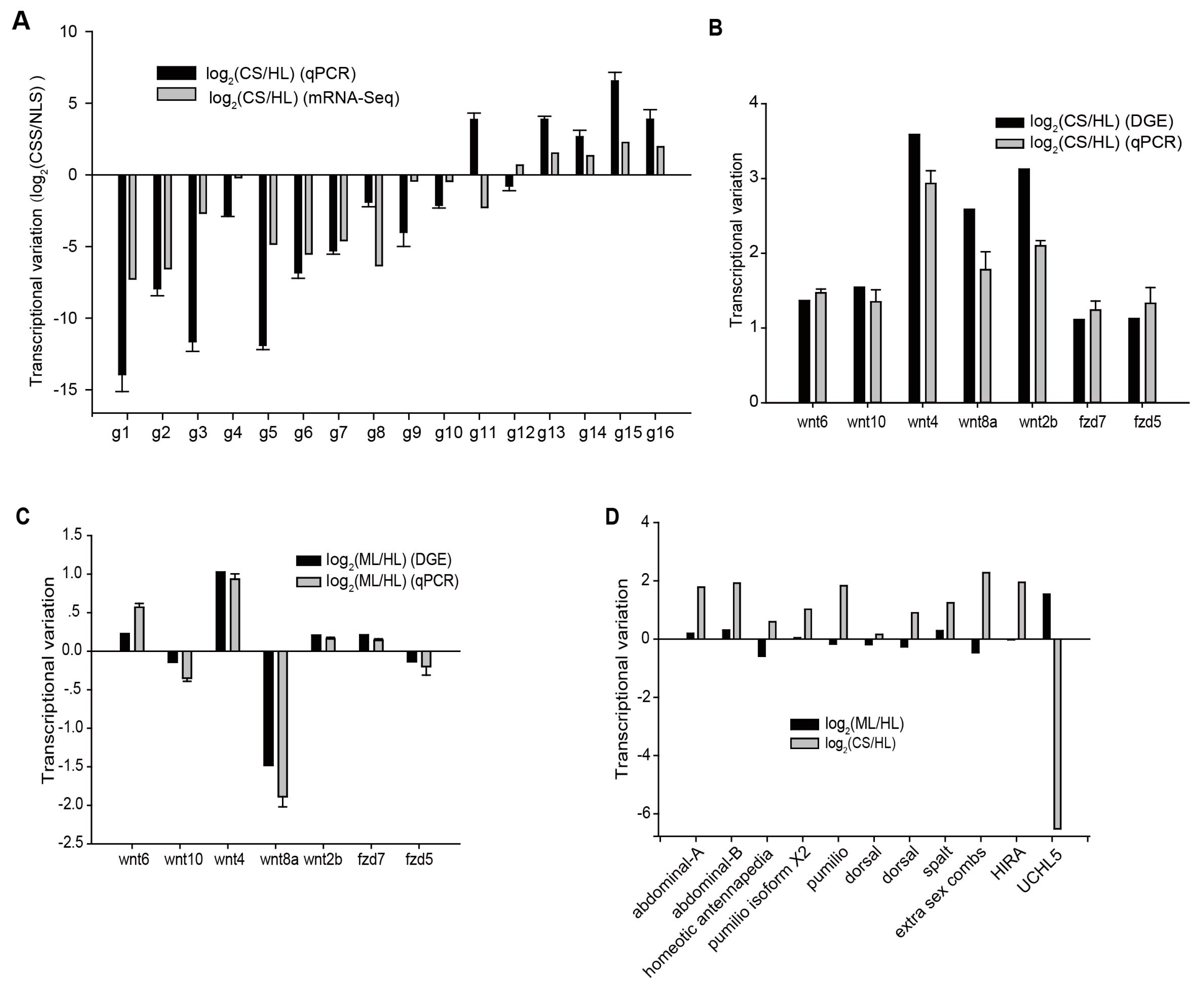
2.3. Differentially Transcribed Genes among Cleavage Stage Embryos and Healthy Larvae
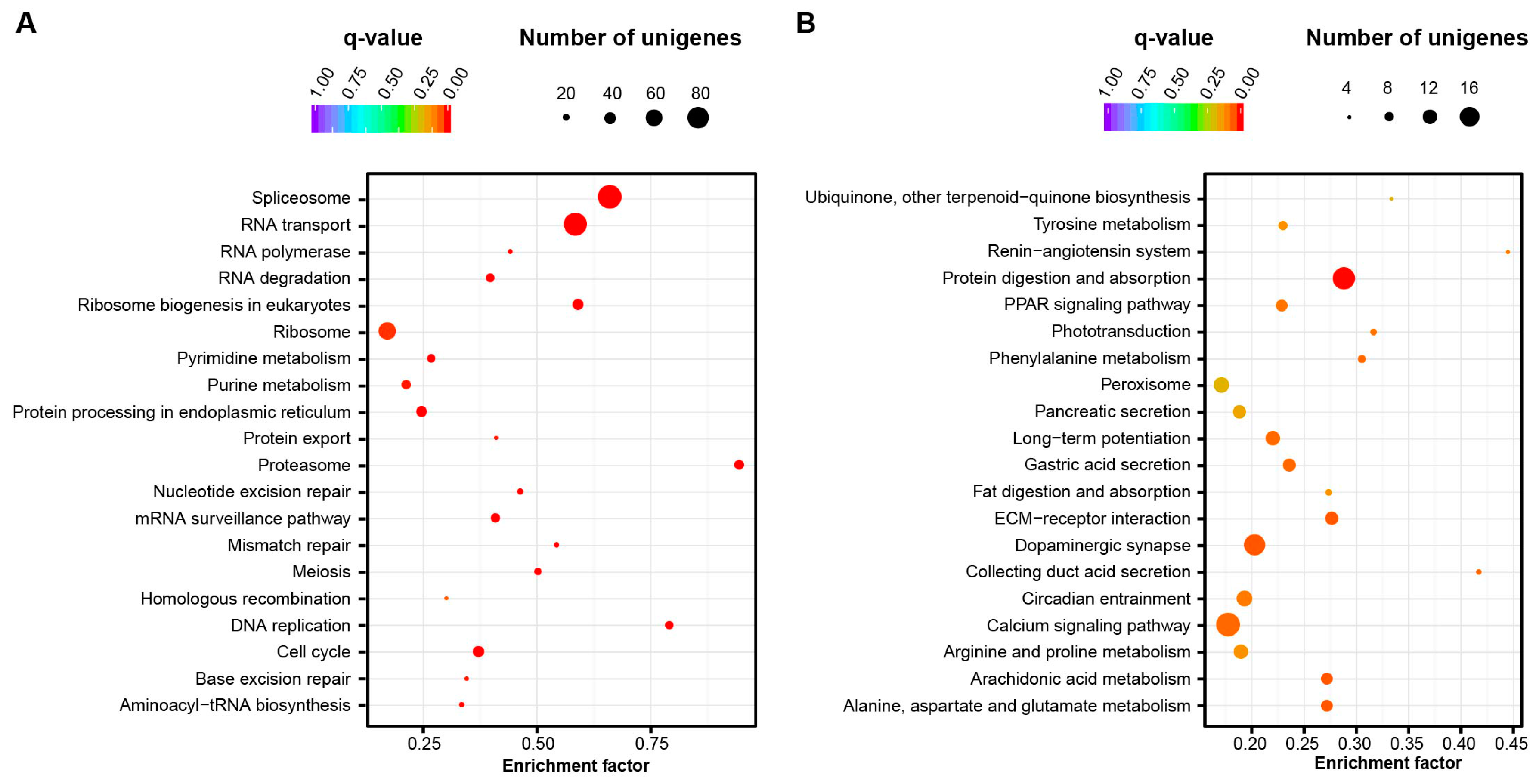
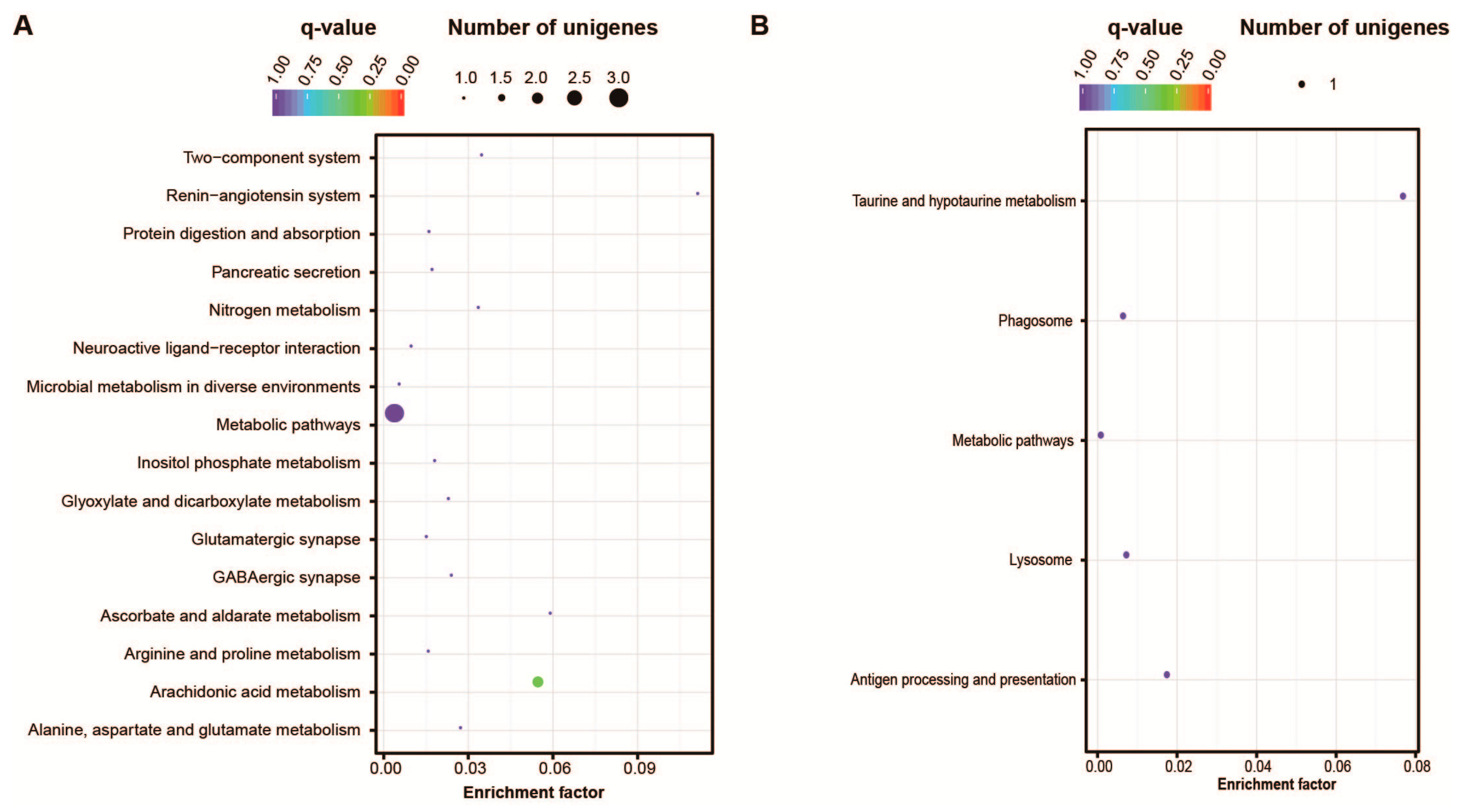
2.4. Signaling Pathways Related to Embryo Development
3. Discussion
4. Experimental Section
4.1. Sample Collection
4.2. RNA Isolation and Illumina Sequencing
4.3. Transcriptome Assembly
4.4. Gene Annotation
4.5. Differential Gene Expression Analysis
4.6. Quantitative Real Time PCR
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Domingues, P.; Sykes, A.; Sommerfield, A.; Andrade, J.P. Effects of feeding live or frozen prey on growth, survival and the life cycle of the cuttlefish, Sepia officinalis (Linnaeus, 1758). Aquac. Int. 2003, 11, 397–410. [Google Scholar] [CrossRef]
- Domingues, P.M.; Dimarco, F.P.; Andrade, J.P.; Lee, P.G. Effect of artificial diets on growth, survival and condition of adult cuttlefish, Sepia officinalis Linnaeus, 1758. Aquac. Int. 2005, 13, 423–440. [Google Scholar] [CrossRef]
- Domingues, P.M.; Kingston, T.; Sykes, A.; Andrade, J.P. Growth of young cuttlefish, Sepia officinalis (Linnaeus 1758) at the upper end of the biological distribution temperature range. Aquac. Res. 2001, 32, 923–930. [Google Scholar] [CrossRef]
- Lee, P.G.; Turk, P.E.; Forsythe, J.W.; DiMarco, F.P. Cephalopod Culture: Physiological, Behavioral and Environmental Requirements. Aquac. Sci. 1998, 46, 417–422. [Google Scholar]
- Sykes, A.V.; Domingues, P.M.; Andrade, J.P. Effects of using live grass shrimp (Palaemonetes varians) as the only source of food for the culture of cuttlefish, Sepia officinalis (Linnaeus, 1758). Aquac. Int. 2006, 14, 551–568. [Google Scholar] [CrossRef]
- Okutani, T. Cuttlefish and Squids of the World in Color; National Cooperative Association of Squid Processors: Tokyo, Japan, 1995; pp. 1–185. [Google Scholar]
- Hao, Z.; Zhang, X.; Zhang, P. Biological characteristics and multiplication techniques of Sepia esculenta. Chin. J. Ecol. 2007, 26, 601–606, (In Chinese with English Abstract). [Google Scholar]
- Bloor, I.S.M.; Attrill, M.J.; Jackson, E.L. A Review of the Factors Influencing Spawning, Early Life Stage Survival and Recruitment Variability in the Common Cuttlefish (Sepia officinalis). Adv. Mar. Biol. 2013, 65, 1–65. [Google Scholar] [PubMed]
- Yasuda, J. Some ecological notes on the cuttlefish, Sepia esculenta Hoyle. Bull. Jpn. Soc. Sci. Fish. 1951, 16, 350–356, (In Japanese with English Abstract). [Google Scholar] [CrossRef]
- Chen, S.; Liu, C.; Zhunag, Z.; Jia, W.; Sun, J.; Yu, G.; Liu, K.; Wang, X. Observations on the embryonic development of Sepia esculenta Hoyle. Mar. Fish. Res. 2010, 31, 1–7, (In Chinese with English Abstract). [Google Scholar]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Yamada, T.; Kanehisa, M.; Bork, P. iPath: Interactive exploration of biochemical pathways and networks. Trends Biochem. Sci. 2008, 33, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Bondos, S. Variations on a theme: Hox and Wnt combinatorial regulation during animal development. Sci. Signal. 2006, 2006, pe38. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Mlodzik, M. Wnt-Frizzled/planar cell polarity signaling: Cellular orientation by facing the wind (Wnt). Annu. Rev. Cell Dev. Biol. 2015, 31, 623–646. [Google Scholar] [CrossRef] [PubMed]
- Barile, N.B.; Cappabianca, S.; Antonetti, L.; Scopa, M.; Nerone, E.; Mascilongo, G.; Recchi, S.; D’Aloise, A. New protocols to improve the deposition and hatching of Sepia officinalis’ eggs. Vet. Ital. 2013, 49, 367–374. [Google Scholar] [PubMed]
- Kelman, E.J.; Osorio, D.; Baddeley, R.J. A review of cuttlefish camouflage and object recognition and evidence for depth perception. J. Exp. Biol. 2008, 211, 1757–1763. [Google Scholar] [CrossRef] [PubMed]
- Strobel, A.; Hu, M.Y.A.; Gutowska, M.A.; Lieb, B.; Lucassen, M.; Melzner, F.; Pörtner, H.O.; Mark, F.C. Influence of Temperature, Hypercapnia, and Development on the Relative Expression of Different Hemocyanin Isoforms in the Common Cuttlefish Sepia officinalis. J. Exp. Zool. Part A 2012, 317, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.H.; Sun, L.L.; Chi, C.F.; Liu, H.H.; Zhou, L.Q.; Lv, Z.M.; Wu, C.W. Molecular cloning, expression analysis and cellular localization of an LFRFamide gene in the cuttlefish Sepiella japonica. Peptides 2016, 80, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Albertin, C.B.; Bonnaud, L.; Brown, C.T.; Crookes-Goodson, W.J.; da Fonseca, R.R.; di Cristo, C.; Dilkes, B.P.; Edsinger-Gonzales, E.; Freeman, R.M., Jr.; Hanlon, R.T.; et al. Cephalopod genomics: A plan of strategies and organization. Stand. Genom. Sci. 2012, 7, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Mao, Y.; Huang, Z.X.; Qu, M.; Chen, J.; Ding, S.; Hong, J.; Sun, T. Transcriptome Analysis of the Octopus vulgaris Central Nervous System. PLoS ONE 2012, 7, e40320. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.K.; Feng, Z.X.; Wang, X.; Wang, X.W.; Zhang, X.G. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.Z.; Huang, J.J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistics of Sequencing Data | Numbers |
|---|---|
| Raw reads of mRNA-Seq | 32,597,241 |
| Clean reads of mRNA-Seq | 31,840,631 (97.7%) |
| Unigenes assembled | 98,615 |
| Total length of unigenes (bp) | 61,246,386 |
| Ave. length of unigenes (bp) | 621 |
| N50 length of unigenes (bp) | 911 |
| Max. length of unigenes (bp) | 19,292 |
| Min. length of unigenes (bp) | 201 |
| Raw reads of the three DGE samples | 7,671,799 a/9,439,043 b/8,512,259 c |
| Clean reads of the three DGE samples | 7,562,335 a/9,305,149 b/8,376,805 c |
| Databases | Number of Unigenes | Percentage (%) |
|---|---|---|
| Annotated in NR 1 | 18,921 | 19.2 |
| Annotated in NT 2 | 2720 | 2.8 |
| Annotated in KEGG 3 | 7761 | 7.9 |
| Annotated in SwissProt | 15,078 | 15.3 |
| Annotated in PFAM 4 | 19,150 | 19.4 |
| Annotated in GO | 19,643 | 19.9 |
| Annotated in KOG 5 | 10,547 | 10.7 |
| Annotated in all databases | 1516 | 1.5 |
| Annotated in least one database | 25,462 | 25.8 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Zhao, F.; Yan, J.; Liu, C.; Liu, S.; Chen, S. Transcriptome Sequencing and De Novo Assembly of Golden Cuttlefish Sepia esculenta Hoyle. Int. J. Mol. Sci. 2016, 17, 1749. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101749
Liu C, Zhao F, Yan J, Liu C, Liu S, Chen S. Transcriptome Sequencing and De Novo Assembly of Golden Cuttlefish Sepia esculenta Hoyle. International Journal of Molecular Sciences. 2016; 17(10):1749. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101749
Chicago/Turabian StyleLiu, Changlin, Fazhen Zhao, Jingping Yan, Chunsheng Liu, Siwei Liu, and Siqing Chen. 2016. "Transcriptome Sequencing and De Novo Assembly of Golden Cuttlefish Sepia esculenta Hoyle" International Journal of Molecular Sciences 17, no. 10: 1749. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17101749