
Brefeldin A-Inhibited Guanine Nucleotide-Exchange Factor 1 (BIG1) Governs the Recruitment of Tumor Necrosis Factor Receptor-Associated Factor 2 (TRAF2) to Tumor Necrosis Factor Receptor 1 (TNFR1) Signaling Complexes

 ,
,
Abstract
:
1. Introduction
2. Results
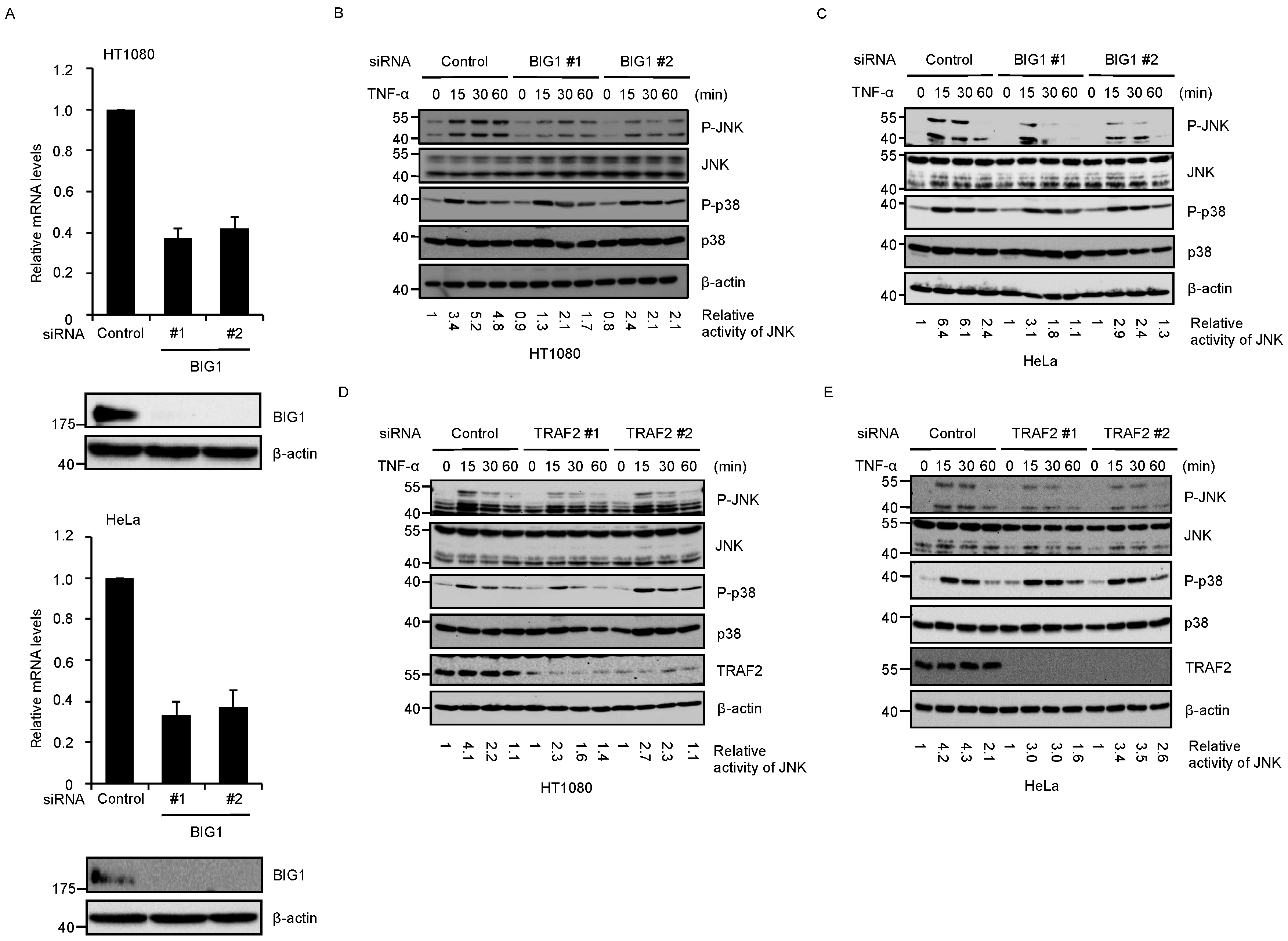
2.1. BIG1 Is Involved in TNF-α-Induced JNK Activation
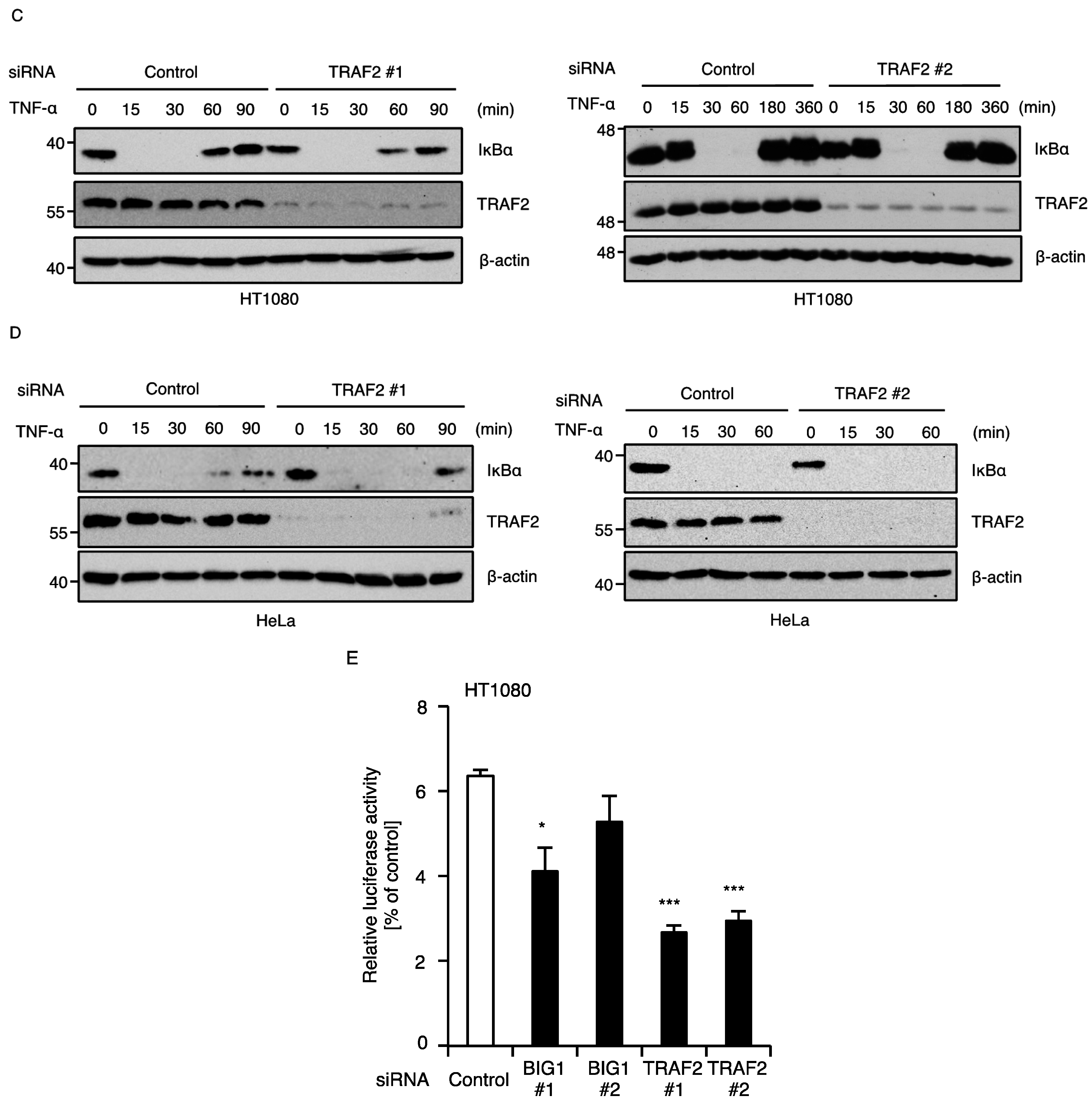
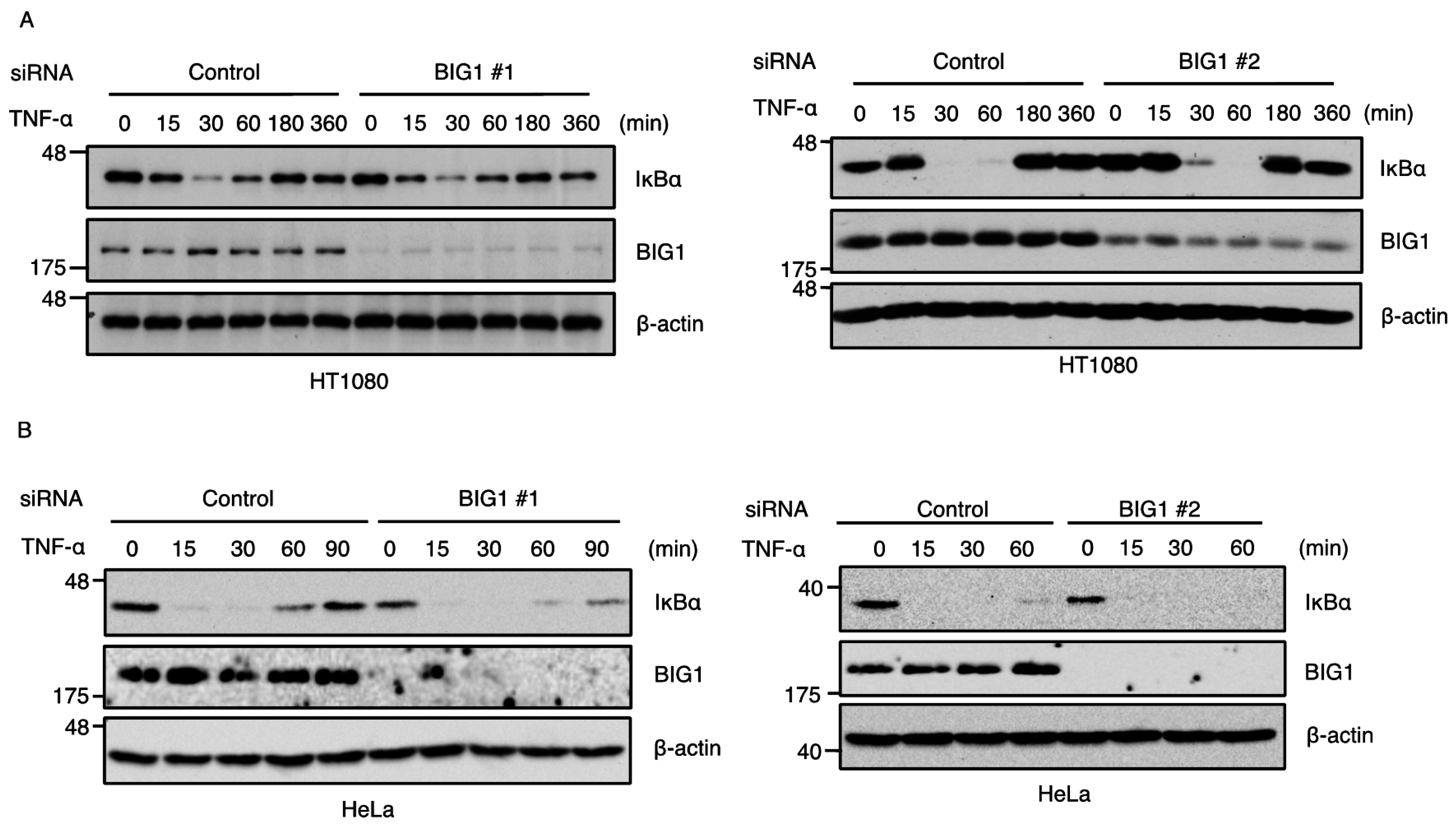
2.2. BIG1 Is Dispensable for TNF-α-Induced Degradation of IκBα
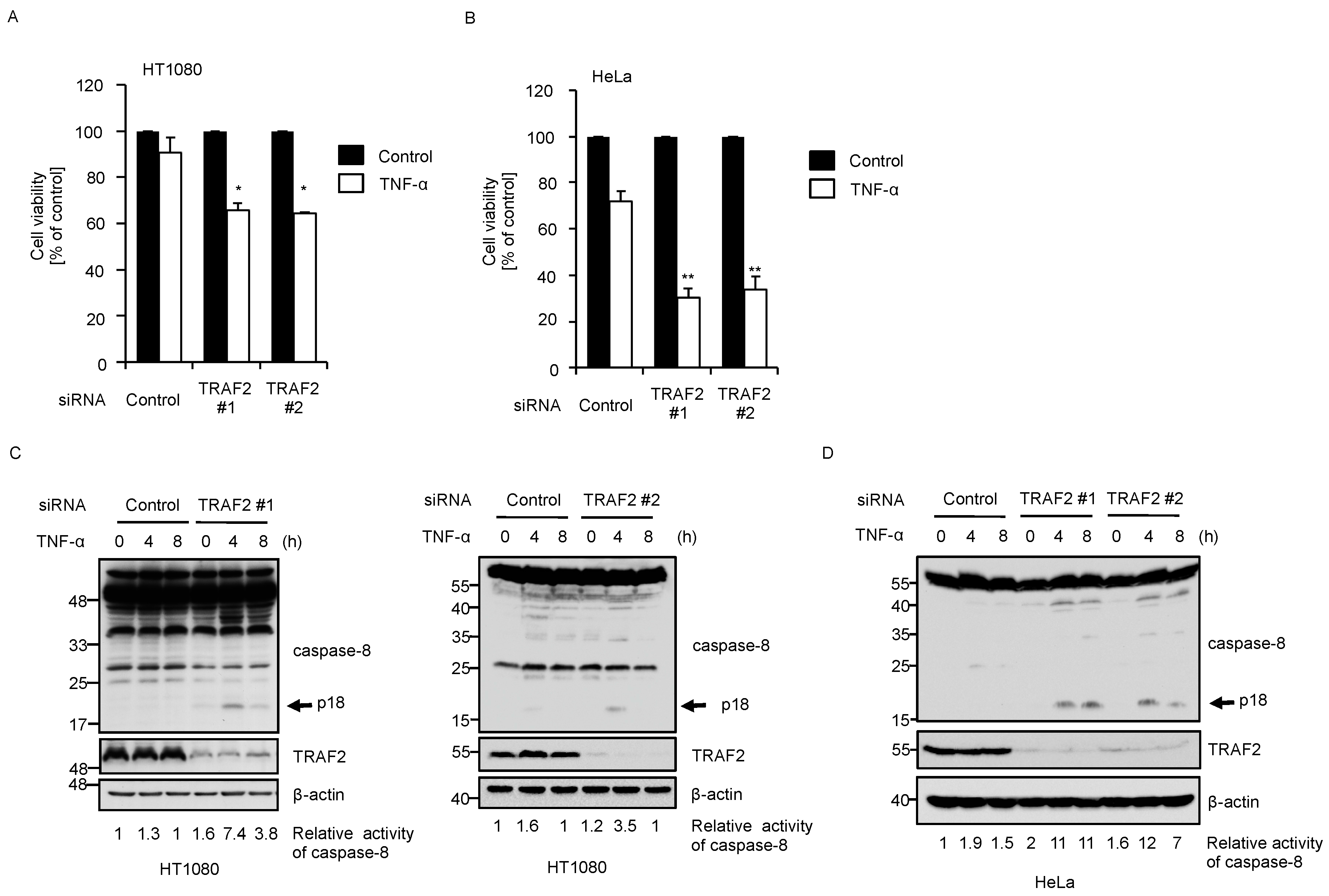
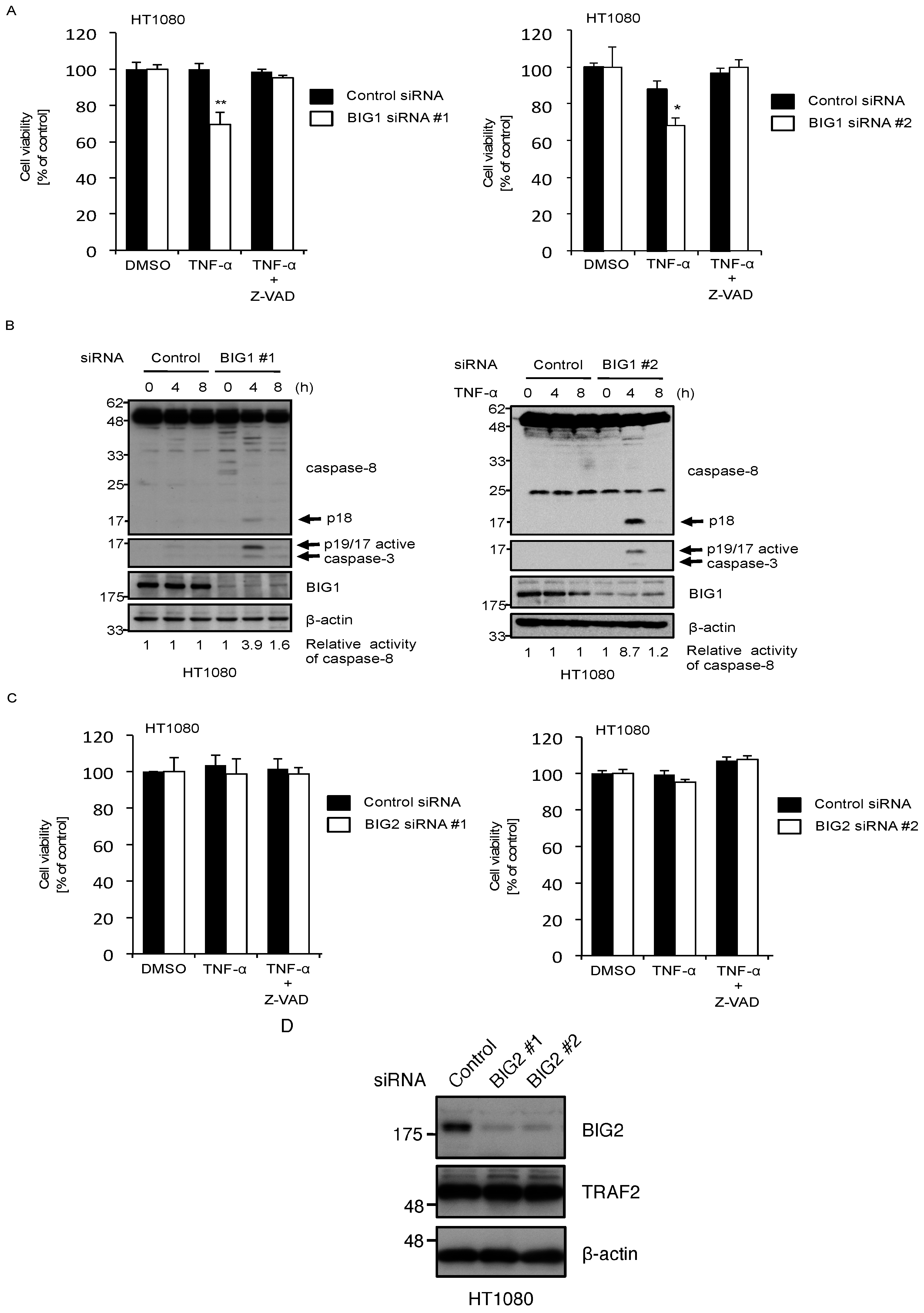
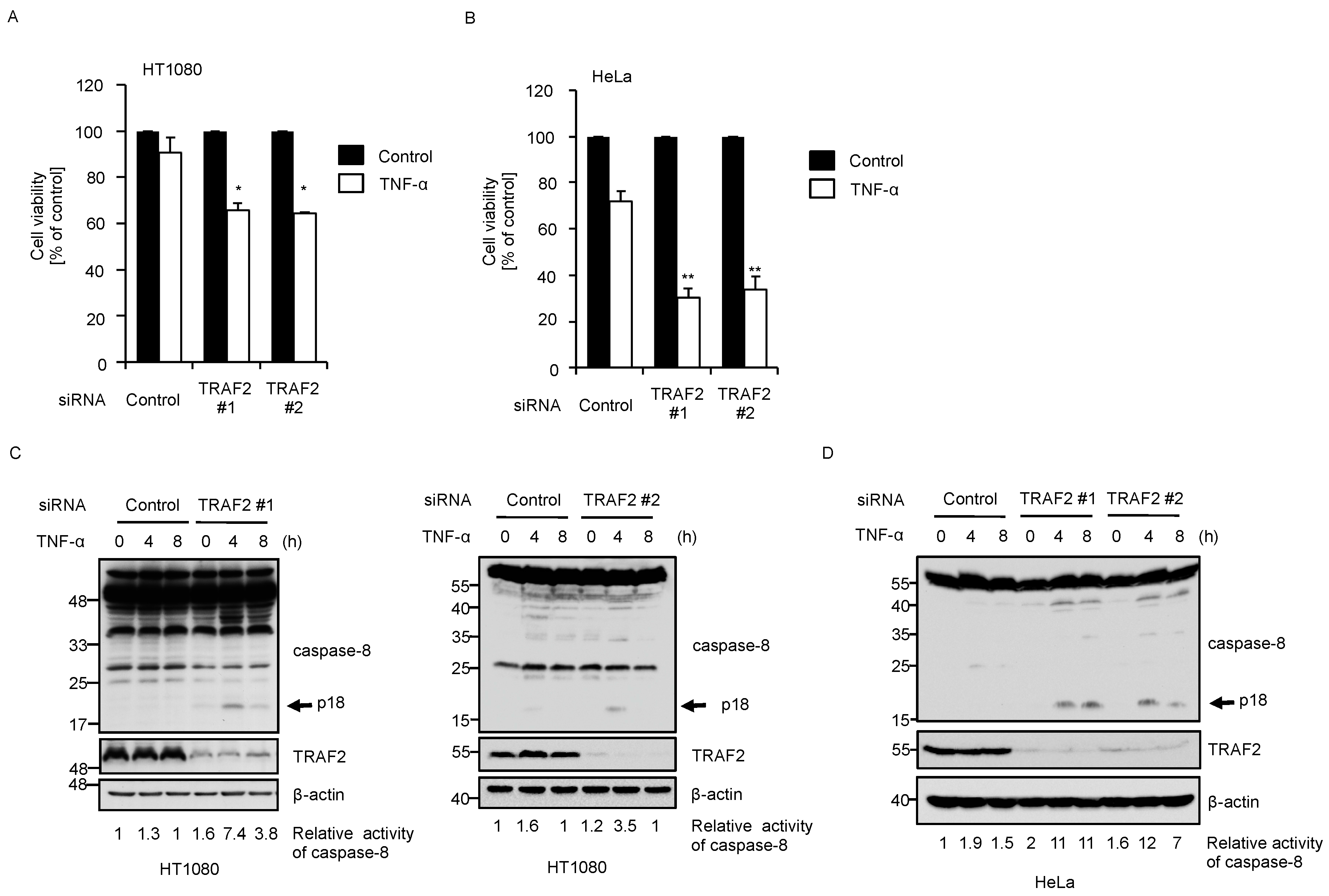
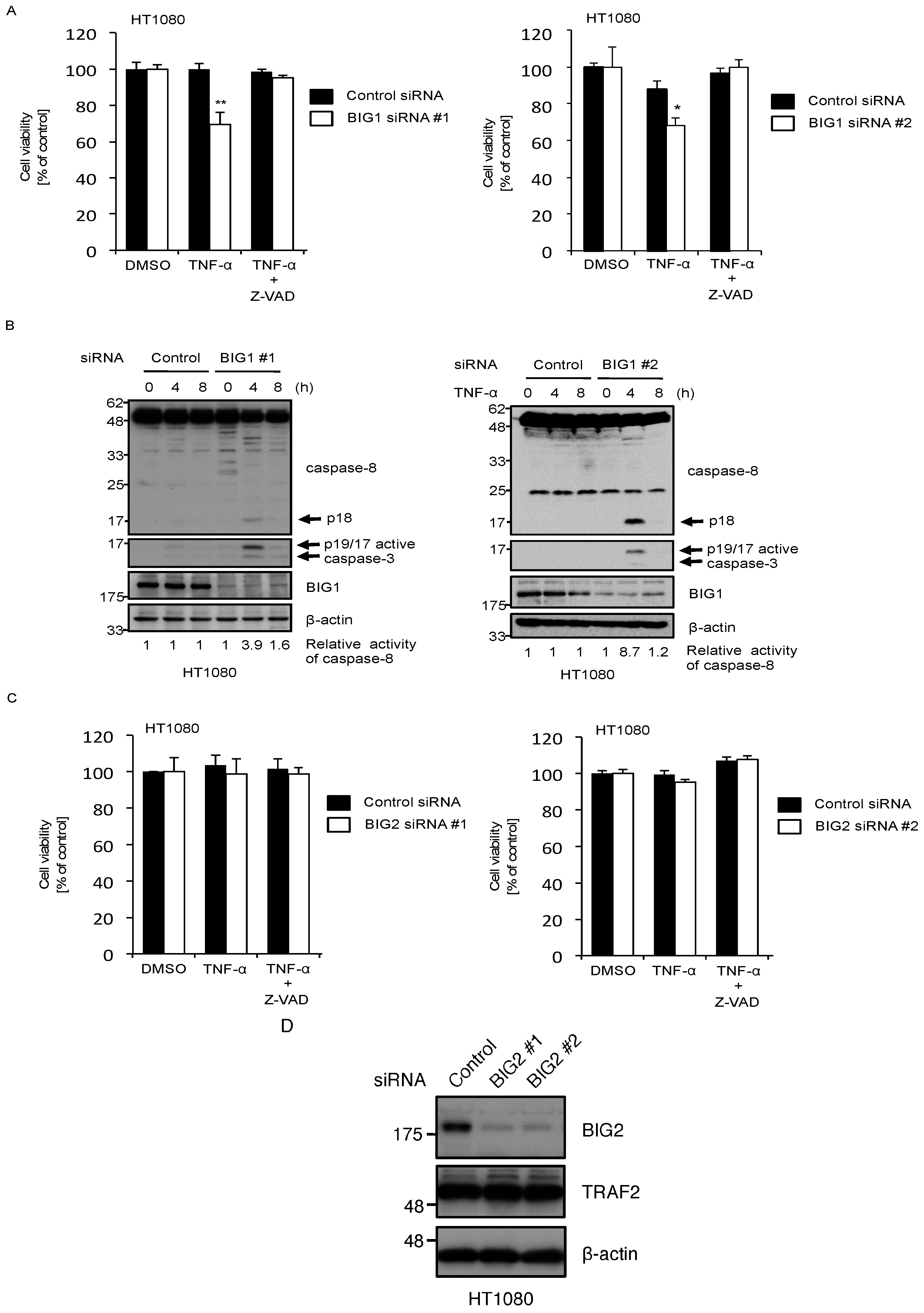
2.3. Knockdown of BIG1 Increases Sensitivity to TNF-α-Induced Apoptosis
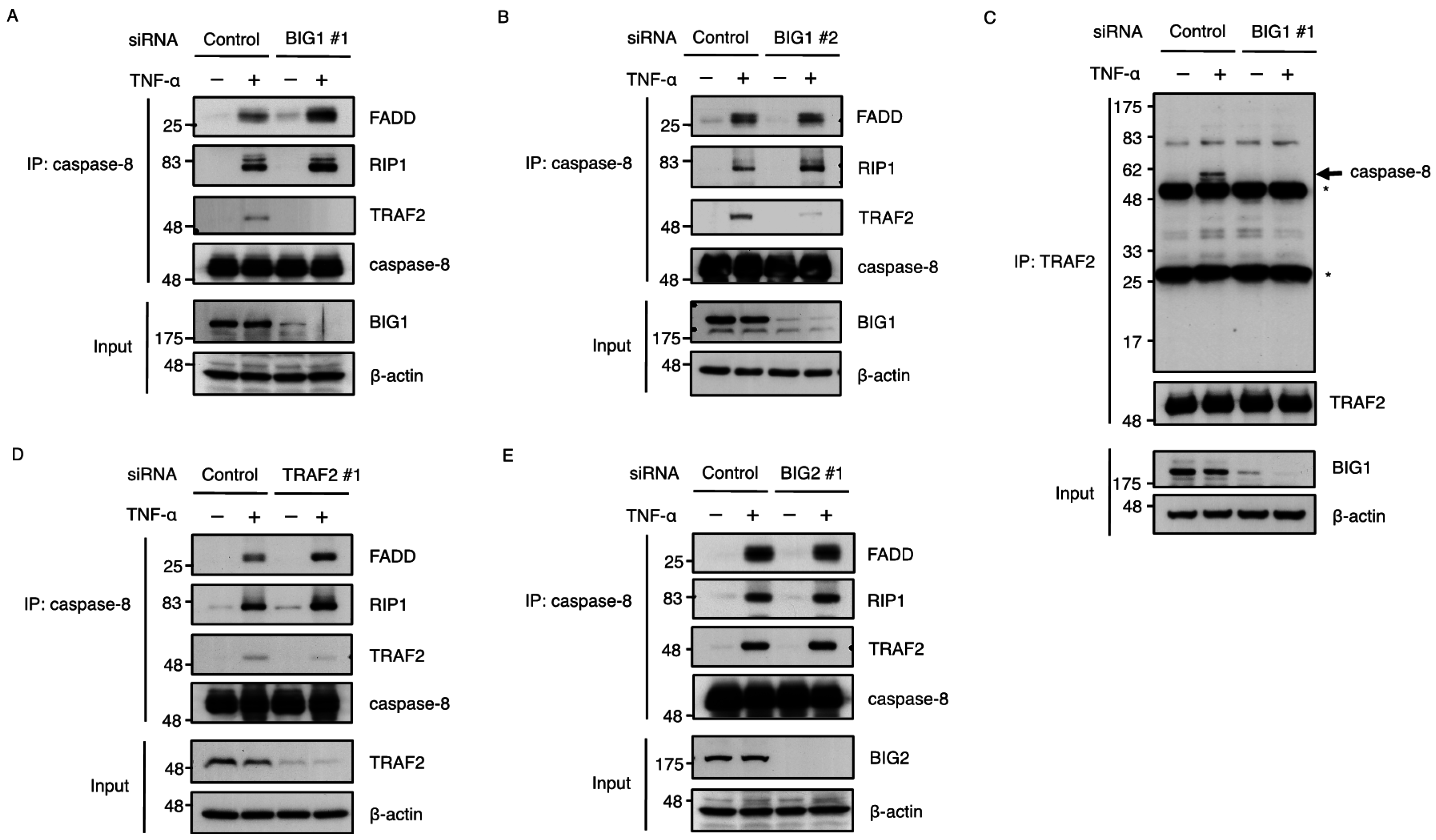
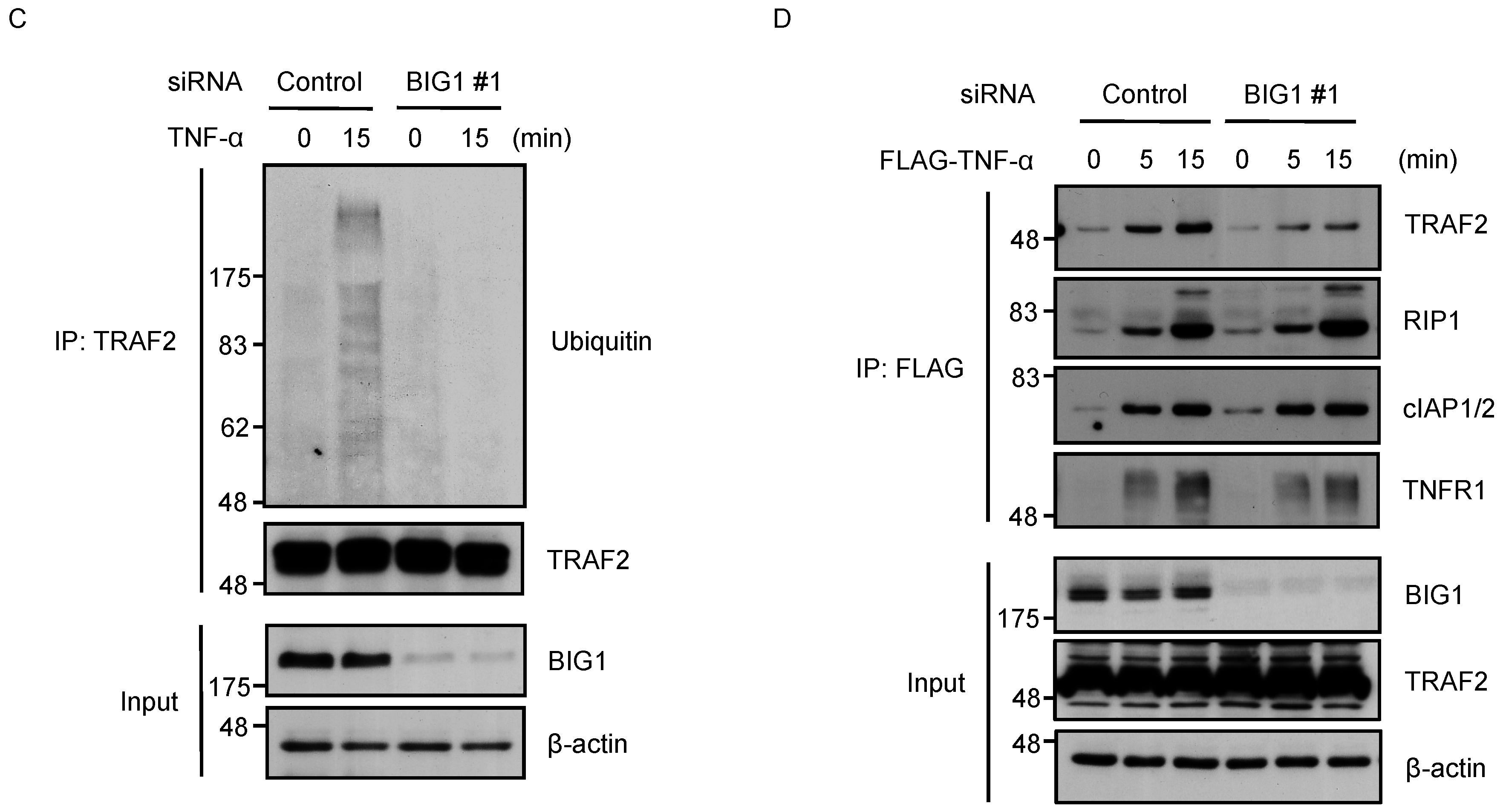
2.4. BIG1 Is Involved in the Recruitment of TRAF2 to the TNFR1 Signaling Complex
2.5. BIG1 Is Involved in the Recruitment of TRAF2 to the Death-Inducing Complex
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Immunoblot
4.3. NF-κB Reporter Assay
4.4. Colorimetric Cell Viability Assay
4.5. TNFR1 Complex Analysis by Immunoprecipitation
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Species, Name (Source) |
|---|---|
| β-actin | rabbit pAb, ab8337 (Abcam or Santa Cruz) |
| BIG1 | rabbit pAb, H-200 (Santa Cruz) |
| BIG2 | rabbit pAb (Bethyl Laboratories) |
| caspase-3 cleaved | rabbit pAb (Cell Signaling) |
| caspase-8 for IP | goat pAb, p20 C-20 (Santa Cruz) |
| caspase-8 for IB | mouse mAb, Clone 12F5 (Enzo Life Sciences) |
| cIAP1 | rat mAb, 1E1-1-10 (Enzo Life Sciences) |
| FADD | mouse mAb (Becton and Dickinson) |
| FLAG | mouse mAb, M2 (Sigma) |
| IκBα | rabbit pAb (Cell signaling) |
| p38, phospho | rabbit pAb (Cell signaling) |
| JNK, phospho | rabbit pAb (Biosource or Cell signaling) |
| RIP1 | mouse mAb (Becton and Dickinson) |
| TRAF2 | mouse mAb (Becton and Dickinson or Santa Cruz) |
| Ubiquitin | mouse mAb, P4D1 (Santa Cruz) |
References
- Jackson, C.L.; Bouvet, S. ARFS at a glance. J. Cell Sci. 2014, 127, 4103–4109. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.R.; Casanova, J.E. Regulation of actin cytoskeleton dynamics by Arf-family GTPases. Trends Cell Biol. 2008, 18, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Gillingham, A.K.; Munro, S. The small G proteins of the Arf family and their regulators. Annu. Rev. Cell Dev. Biol. 2007, 23, 579–611. [Google Scholar] [CrossRef] [PubMed]
- Casanova, J.E. Regulation of Arf activation: The Sec7 family of guanine nucleotide exchange factors. Traffic 2007, 8, 1476–1485. [Google Scholar] [CrossRef] [PubMed]
- Morinaga, N.; Tsai, S.C.; Moss, J.; Vaughan, M. Isolation of a brefeldin A-inhibited guanine nucleotide-exchange protein for ADP ribosylation factor (Arf) 1 and ARF3 that contains a Sec7-like domain. Proc. Natl. Acad. Sci. USA 1996, 93, 12856–12860. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.D.; Moss, J.; Vaughan, M. BIG1 and BIG2, brefeldin A-inhibited guanine nucleotide-exchange factors for ADP-ribosylation factors. Methods Enzymol. 2005, 404, 174–184. [Google Scholar] [PubMed]
- Yamaji, R.; Adamik, R.; Takeda, K.; Togawa, A.; Pacheco-Rodriguez, G.; Ferrans, V.J.; Moss, J.; Vaughan, M. Identification and localization of two brefeldin A-inhibited guanine nucleotide-exchange proteins for ADP-ribosylation factors in a macromolecular complex. Proc. Natl. Acad. Sci. USA 2000, 97, 2567–2572. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, R.; Shin, H.W.; Mitsuhashi, H.; Nakayama, K. Redundant roles of BIG2 and BIG1, guanine-nucleotide exchange factors for ADP-ribosylation factors in membrane traffic between the trans-golgi network and endosomes. Mol. Biol. Cell 2008, 19, 2650–2660. [Google Scholar] [CrossRef] [PubMed]
- Boal, F.; Stephens, D.J. Specific functions of BIG1 and BIG2 in endomembrane organization. PLoS ONE 2010, 5, e9898. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Hong, M.S.; Moss, J.; Vaughan, M. BIG1, a brefeldin A-inhibited guanine nucleotide-exchange protein, is required for correct glycosylation and function of integrin β1. Proc. Natl. Acad. Sci. USA 2007, 104, 1230–1235. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Li, C.; Li, D.; Wang, Y.; Shao, W.; You, Y.; Peng, J.; Zhang, X.; Lu, L.; Shen, X. BIG1, a brefeldin A-inhibited guanine nucleotide-exchange protein regulates neurite development via PI3K-AKT and ERK signaling pathways. Neuroscience 2013, 254, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Li, C.C.; Kuo, J.C.; Waterman, C.M.; Kiyama, R.; Moss, J.; Vaughan, M. Effects of brefeldin A-inhibited guanine nucleotide-exchange (BIG) 1 and KANK1 proteins on cell polarity and directed migration during wound healing. Proc. Natl. Acad. Sci. USA 2011, 108, 19228–19233. [Google Scholar] [CrossRef] [PubMed]
- Citterio, C.; Jones, H.D.; Pacheco-Rodriguez, G.; Islam, A.; Moss, J.; Vaughan, M. Effect of protein kinase a on accumulation of brefeldin A-inhibited guanine nucleotide-exchange protein 1 (BIG1) in HEPG2 cell nuclei. Proc. Natl. Acad. Sci. USA 2006, 103, 2683–2688. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.W.; Morinaga, N.; Noda, M.; Nakayama, K. BIG2, a guanine nucleotide exchange factor for ADP-ribosylation factors: Its localization to recycling endosomes and implication in the endosome integrity. Mol. Biol. Cell 2004, 15, 5283–5294. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Xu, K.F.; Fan, Q.; Pacheco-Rodriguez, G.; Moss, J.; Vaughan, M. Association of brefeldin A-inhibited guanine nucleotide-exchange protein 2 (BIG2) with recycling endosomes during transferrin uptake. Proc. Natl. Acad. Sci. USA 2006, 103, 2635–2640. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.M. Tumor necrosis factor. Cancer Lett. 2013, 328, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Han, J.; Zhong, C.Q.; Zhang, D.W. Programmed necrosis: Backup to and competitor with apoptosis in the immune system. Nat. Immunol. 2011, 12, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Borghi, A.; Verstrepen, L.; Beyaert, R. TRAF2 multitasking in TNF receptor-induced signaling to NF-κB, MAP kinases and cell death. Biochem. Pharmacol. 2016, 116, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xie, P. TRAF molecules in cell signaling and in human diseases. J. Mol. Signal. 2013, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Zelova, H.; Hosek, J. TNF-α signalling and inflammation: Interactions between old acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Tada, K.; Okazaki, T.; Sakon, S.; Kobarai, T.; Kurosawa, K.; Yamaoka, S.; Hashimoto, H.; Mak, T.W.; Yagita, H.; Okumura, K.; et al. Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF-κB activation and protection from cell death. J. Biol. Chem. 2001, 276, 36530–36534. [Google Scholar] [CrossRef] [PubMed]
- Yeh, W.C.; Shahinian, A.; Speiser, D.; Kraunus, J.; Billia, F.; Wakeham, A.; de la Pompa, J.L.; Ferrick, D.; Hum, B.; Iscove, N.; et al. Early lethality, functional NF-κB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity 1997, 7, 715–725. [Google Scholar] [CrossRef]
- Petersen, S.L.; Chen, T.T.; Lawrence, D.A.; Marsters, S.A.; Gonzalvez, F.; Ashkenazi, A. TRAF2 is a biologically important necroptosis suppressor. Cell Death Differ. 2015, 22, 1846–1857. [Google Scholar] [CrossRef] [PubMed]
- Karl, I.; Jossberger-Werner, M.; Schmidt, N.; Horn, S.; Goebeler, M.; Leverkus, M.; Wajant, H.; Giner, T. TRAF2 inhibits trail- and CD95L-induced apoptosis and necroptosis. Cell Death Dis. 2014, 5, e1444. [Google Scholar] [CrossRef] [PubMed]
- Gonzalvez, F.; Lawrence, D.; Yang, B.; Yee, S.; Pith, R.; Marsters, S.; Pham, V.C.; Stephan, J.P.; Lill, J.; Ashkenazi, A. TRAF2 sets a threshold for extrinsic apoptosis by tagging caspase-8 with a ubiquitin shutoff timer. Mol. Cell 2012, 48, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Brachert, W.; Heigl, U.; Ehrenschwender, M. Membrane trafficking of death receptors: Implications on signalling. Int. J. Mol. Sci. 2013, 14, 14475–14503. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Brachert, W.; Tchikov, V.; Neumeyer, J.; Jakob, M.; Winoto-Morbach, S.; Held-Feindt, J.; Heinrich, M.; Merkel, O.; Ehrenschwender, M.; Adam, D.; et al. Compartmentalization of TNF receptor 1 signaling: Internalized TNF receptosomes as death signaling vesicles. Immunity 2004, 21, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, B.; Bertsch, U.; Tchikov, V.; Winoto-Morbach, S.; Perrotta, C.; Jakob, M.; Adam-Klages, S.; Kabelitz, D.; Schutze, S. Caspase-8 and caspase-7 sequentially mediate proteolytic activation of acid sphingomyelinase in TNF-R1 receptosomes. EMBO J. 2011, 30, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Islam, A.; Shen, X.; Hiroi, T.; Moss, J.; Vaughan, M.; Levine, S.J. The brefeldin A-inhibited guanine nucleotide-exchange protein, BIG2, regulates the constitutive release of TNFR1 exosome-like vesicles. J. Biol. Chem. 2007, 282, 9591–9599. [Google Scholar] [CrossRef] [PubMed]
- Lang, I.; Fullsack, S.; Wyzgol, A.; Fick, A.; Trebing, J.; Arana, J.A.; Schafer, V.; Weisenberger, D.; Wajant, H. Binding studies of TNF receptor superfamily (TNFRSF) receptors on intact cells. J. Biol. Chem. 2016, 291, 5022–5037. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Nakabayashi, O.; Nakano, H. Flip the switch: Regulation of apoptosis and necroptosis by cFLIP. Int. J. Mol. Sci. 2015, 16, 30321–30341. [Google Scholar] [CrossRef] [PubMed]
- Napetschnig, J.; Wu, H. Molecular basis of NF-κB signaling. Annu. Rev. Biophys. 2013, 42, 443–468. [Google Scholar] [CrossRef] [PubMed]
- Habelhah, H.; Takahashi, S.; Cho, S.G.; Kadoya, T.; Watanabe, T.; Ronai, Z. Ubiquitination and translocation of TRAF2 is required for activation of JNK but not of p38 or NF-κB. EMBO J. 2004, 23, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Geserick, P.; Hupe, M.; Moulin, M.; Wong, W.W.; Feoktistova, M.; Kellert, B.; Gollnick, H.; Silke, J.; Leverkus, M. Cellular iaps inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J. Cell Biol. 2009, 187, 1037–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Gaeta, M.L.; Madge, L.A.; Yang, J.H.; Bradley, J.R.; Pober, J.S. Caveolin-1 associates with TRAF2 to form a complex that is recruited to tumor necrosis factor receptors. J. Biol. Chem. 2001, 276, 8341–8349. [Google Scholar] [CrossRef] [PubMed]
- Legler, D.F.; Micheau, O.; Doucey, M.A.; Tschopp, J.; Bron, C. Recruitment of tnf receptor 1 to lipid rafts is essential for TNFα-mediated NF-κB activation. Immunity 2003, 18, 655–664. [Google Scholar] [CrossRef]
- Holler, N.; Tardivel, A.; Kovacsovics-Bankowski, M.; Hertig, S.; Gaide, O.; Martinon, F.; Tinel, A.; Deperthes, D.; Calderara, S.; Schulthess, T.; et al. Two adjacent trimeric fas ligands are required for Fas signaling and formation of a death-inducing signaling complex. Mol. Cell. Biol. 2003, 23, 1428–1440. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P. Production of recombinant trail and trail receptor: Fc chimeric proteins. Methods Enzymol. 2000, 322, 325–345. [Google Scholar] [PubMed]
- Meylan, E.; Martinon, F.; Thome, M.; Gschwendt, M.; Tschopp, J. RIP4 (DIK/PKK), a novel member of the RIP kinase family, activates NF-κB and is processed during apoptosis. EMBO Rep. 2002, 3, 1201–1208. [Google Scholar] [CrossRef] [PubMed]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noguchi, T.; Tsuchida, M.; Kogue, Y.; Spadini, C.; Hirata, Y.; Matsuzawa, A. Brefeldin A-Inhibited Guanine Nucleotide-Exchange Factor 1 (BIG1) Governs the Recruitment of Tumor Necrosis Factor Receptor-Associated Factor 2 (TRAF2) to Tumor Necrosis Factor Receptor 1 (TNFR1) Signaling Complexes. Int. J. Mol. Sci. 2016, 17, 1869. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17111869
Noguchi T, Tsuchida M, Kogue Y, Spadini C, Hirata Y, Matsuzawa A. Brefeldin A-Inhibited Guanine Nucleotide-Exchange Factor 1 (BIG1) Governs the Recruitment of Tumor Necrosis Factor Receptor-Associated Factor 2 (TRAF2) to Tumor Necrosis Factor Receptor 1 (TNFR1) Signaling Complexes. International Journal of Molecular Sciences. 2016; 17(11):1869. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17111869
Chicago/Turabian StyleNoguchi, Takuya, Mei Tsuchida, Yosuke Kogue, Christian Spadini, Yusuke Hirata, and Atsushi Matsuzawa. 2016. "Brefeldin A-Inhibited Guanine Nucleotide-Exchange Factor 1 (BIG1) Governs the Recruitment of Tumor Necrosis Factor Receptor-Associated Factor 2 (TRAF2) to Tumor Necrosis Factor Receptor 1 (TNFR1) Signaling Complexes" International Journal of Molecular Sciences 17, no. 11: 1869. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17111869