
Dominant Suppression of β1 Integrin by Ectopic CD98-ICD Inhibits Hepatocellular Carcinoma Progression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Result
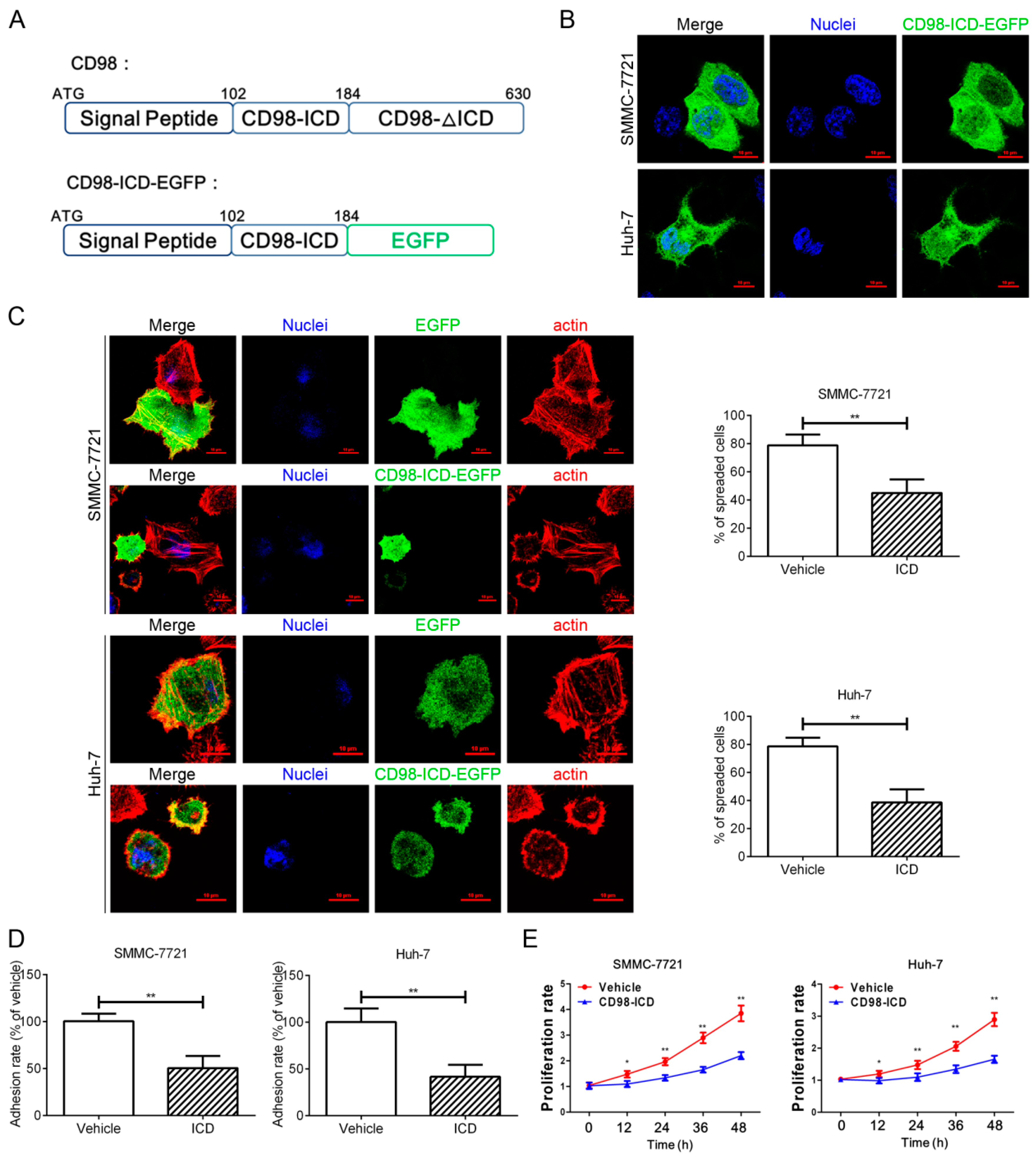
2.1. CD98-ICD Inhibits HCC Cell Adhesion, Spreading and Proliferation
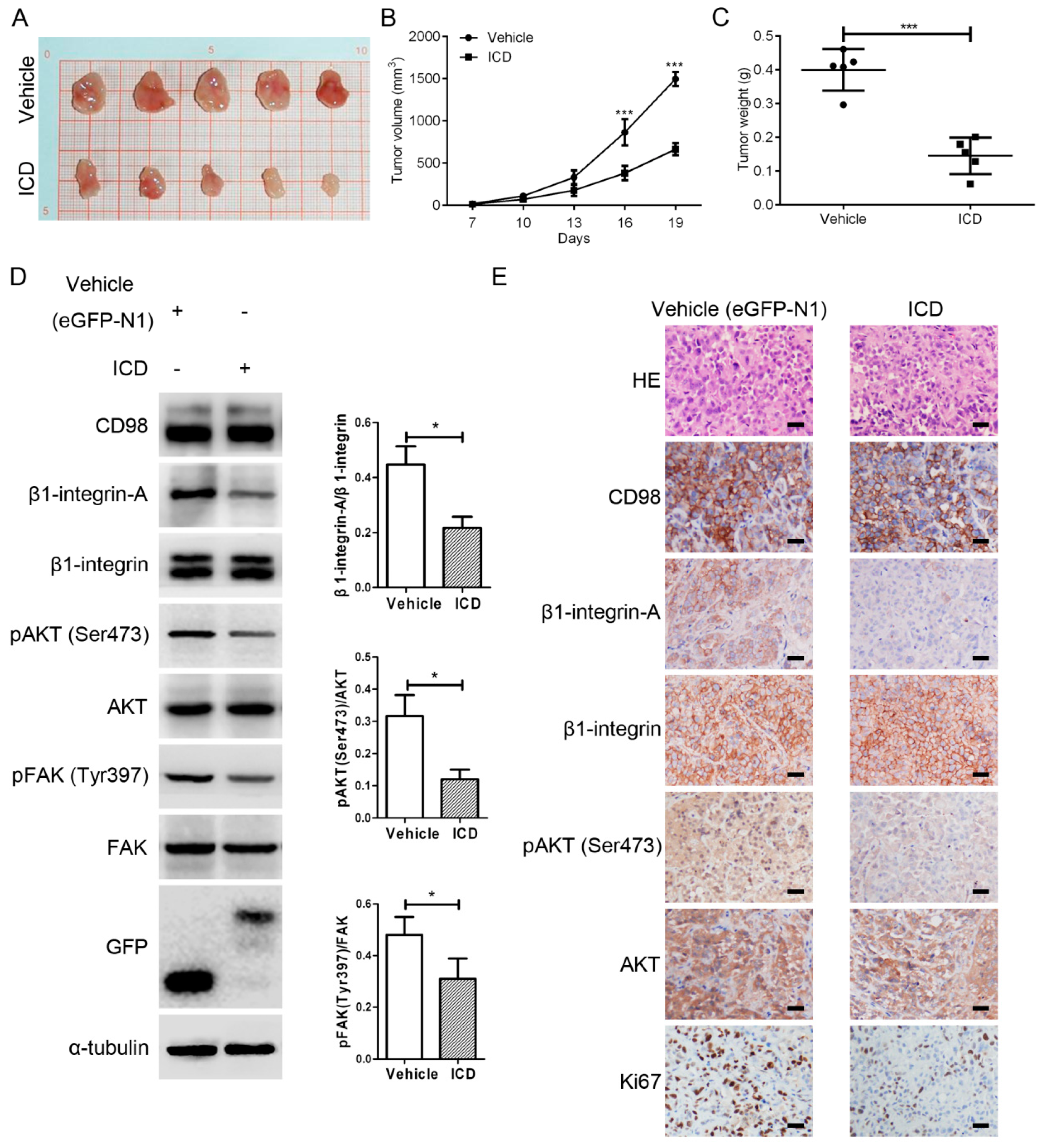
2.2. CD98-ICD Inhibits Tumorigenicity of SMMC-7721 Cells
2.3. Membrane CD98 Was Not Influenced by Exogenous CD98-ICD
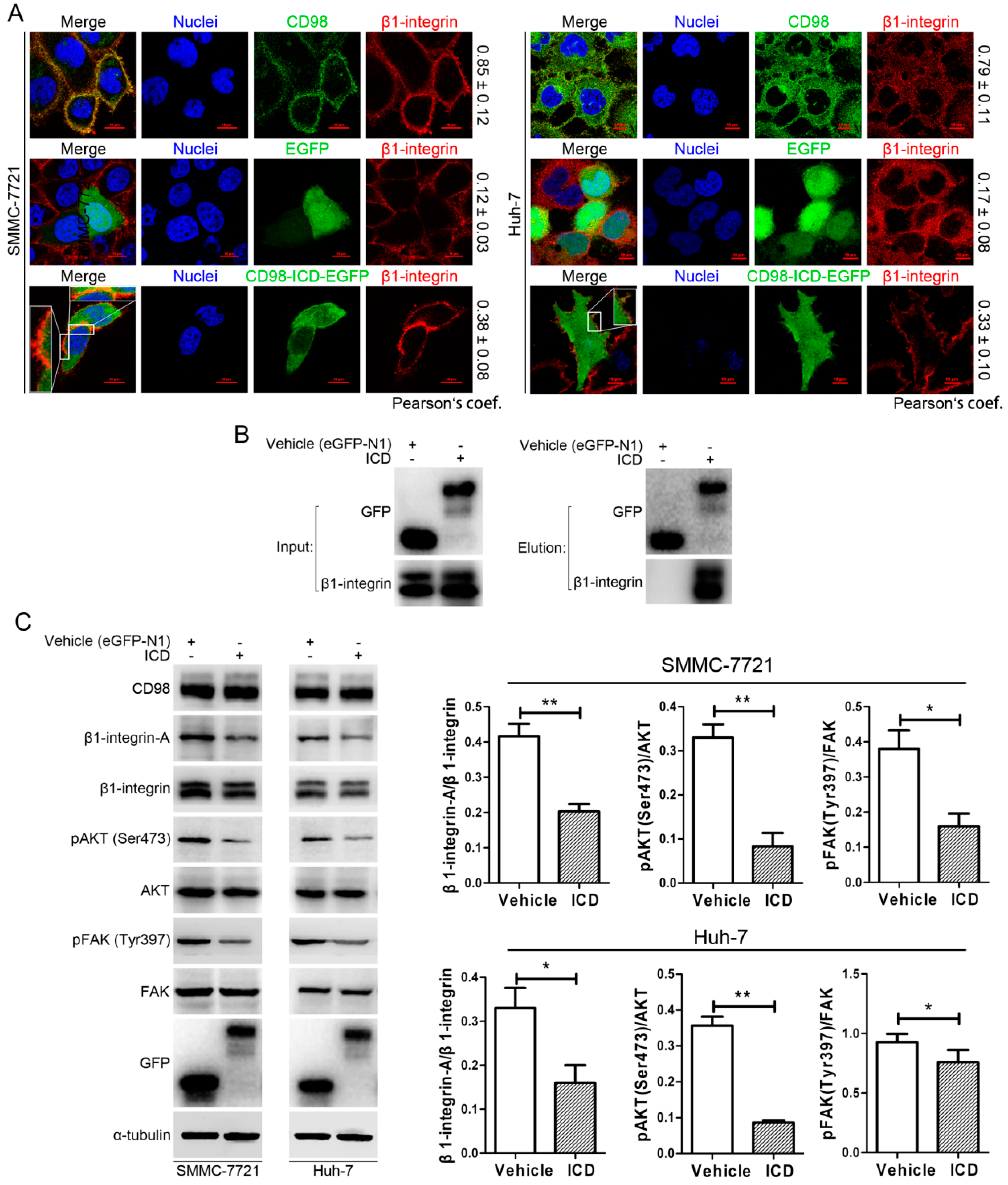
2.4. CD98-ICD Inhibits β1-Integrin Signaling
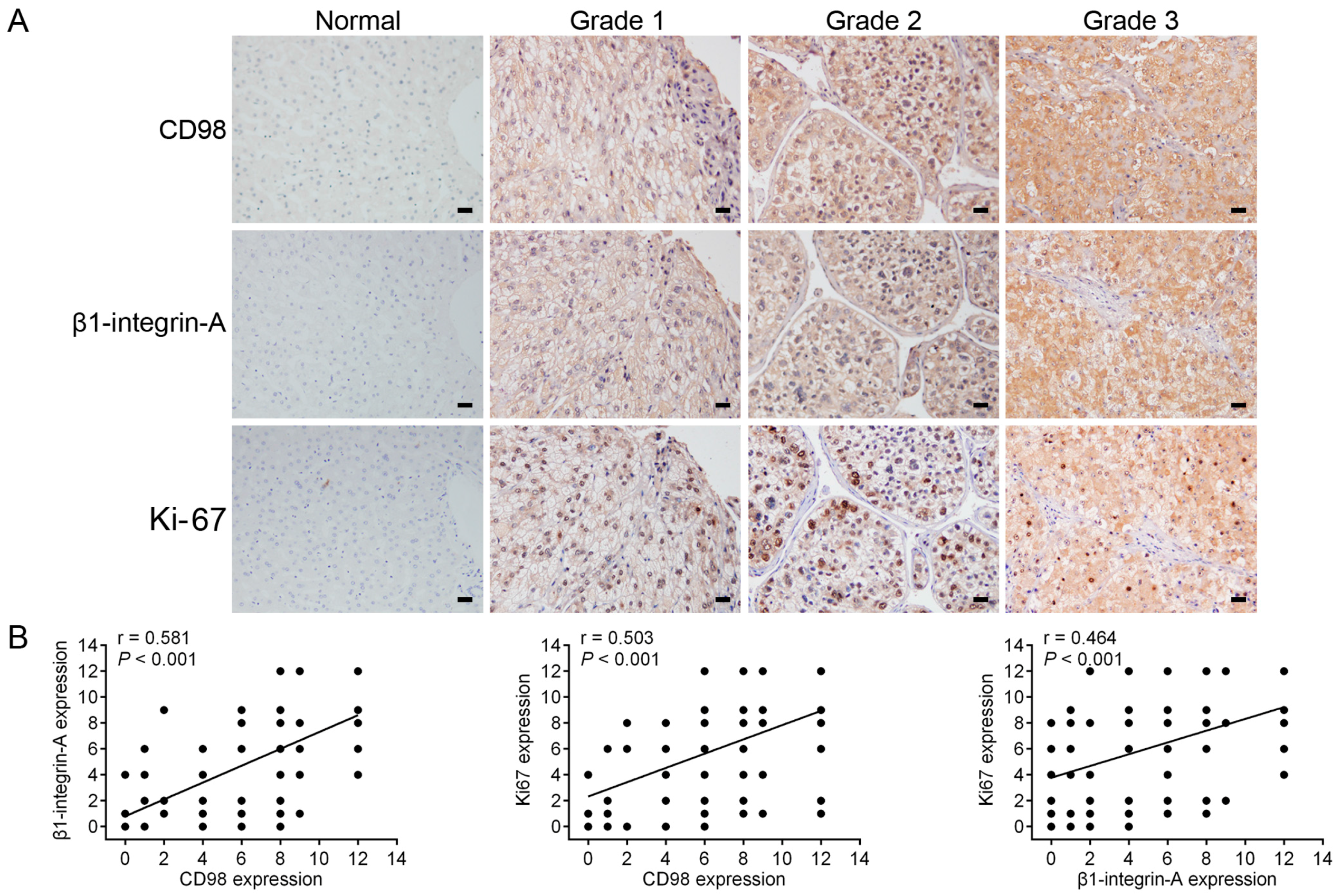
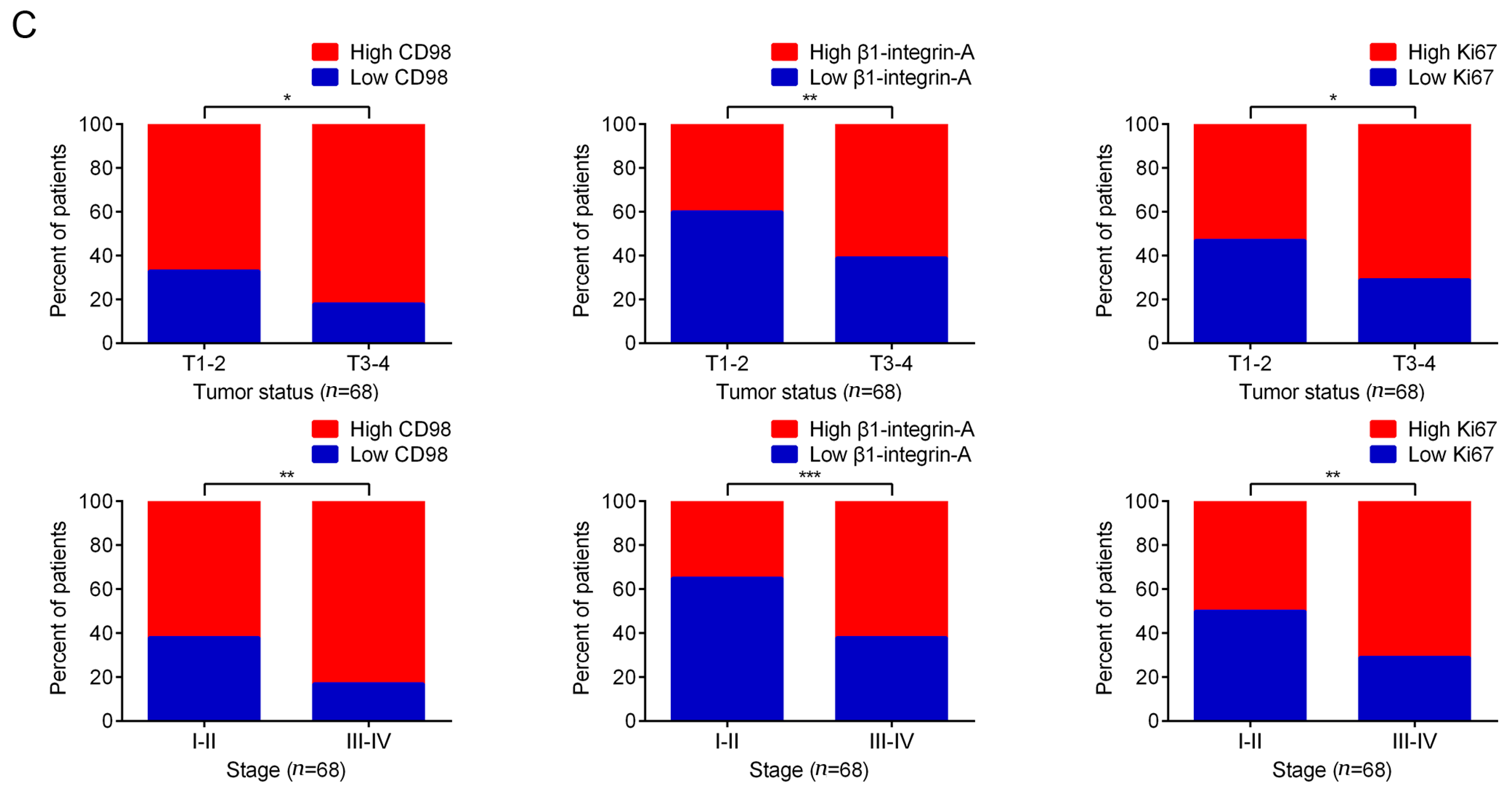
2.5. Immunohistochemical Detection of the Expression of CD98, β1-Integrin-A and Ki-67 in HCC Specimens
3. Discussion
4. Experimental Section
4.1. Cell Culture and Antibodies
4.2. Plasmids
4.3. Cell Spreading Assay
4.4. Adhesion Assay
4.5. Cell Proliferation Assay
4.6. Xenograft Model
4.7. FACS
4.8. Image Analysis
4.8.1. Immunofluorescence
4.8.2. Co-Localization Analysis
4.9. Co-IP and Western Blot
4.10. Immunohistochemistry
4.11. Clinical Specimens
4.12. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef]
- Qi, X.; Zhao, Y.; Li, H.; Guo, X.; Han, G. Management of hepatocellular carcinoma: An overview of major findings from meta-analyses. Oncotarget 2016, 7, 34703–34751. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Liang, Y.; Zheng, T.; Song, R.; Wang, J.; Shi, H.; Sun, B.; Xie, C.; Li, Y.; Han, J.; et al. FCN2 inhibits epithelial-mesenchymal transition-induced metastasis of hepatocellular carcinoma via TGF-β/smad signaling. Cancer Lett. 2016, 378, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.M.; Ginsberg, M.H. CD98 at the crossroads of adaptive immunity and cancer. J. Cell Sci. 2012, 125, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Gomez, A.; Barrasa, J.I.; Olmo, N.; Lecona, E.; Burghardt, H.; Palacin, M.; Lizarbe, M.A.; Turnay, J. 4F2hc-silencing impairs tumorigenicity of HeLa cells via modulation of galectin-3 and β-catenin signaling, and MMP-2 expression. Biochim. Biophys. Acta 2013, 1833, 2045–2056. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, E.; Sato, M.; Yang, H.; Miyagawa, F.; Harasaki, M.; Tomita, K.; Matsuoka, S.; Noma, A.; Iwai, K.; Minato, N. 4F2 (CD98) heavy chain is associated covalently with an amino acid transporter and controls intracellular trafficking and membrane topology of 4F2 heterodimer. J. Biol. Chem. 1999, 274, 3009–3016. [Google Scholar] [CrossRef] [PubMed]
- Cormerais, Y.; Giuliano, S.; LeFloch, R.; Front, B.; Durivault, J.; Tambutte, E.; Massard, P.A.; de la Ballina, L.R.; Endou, H.; Wempe, M.F.; et al. Genetic disruption of the multifunctional CD98/LAT1 complex demonstrates the key role of essential amino acid transport in the control of mTORC1 and tumor growth. Cancer Res. 2016, 76, 4481–4492. [Google Scholar] [CrossRef] [PubMed]
- Rosell, A.; Meury, M.; Alvarez-Marimon, E.; Costa, M.; Perez-Cano, L.; Zorzano, A.; Fernandez-Recio, J.; Palacin, M.; Fotiadis, D. Structural bases for the interaction and stabilization of the human amino acid transporter LAT2 with its ancillary protein 4F2hc. Proc. Natl. Acad. Sci. USA 2014, 111, 2966–2971. [Google Scholar] [CrossRef] [PubMed]
- Fenczik, C.A.; Zent, R.; Dellos, M.; Calderwood, D.A.; Satriano, J.; Kelly, C.; Ginsberg, M.H. Distinct domains of CD98hc regulate integrins and amino acid transport. J. Biol. Chem. 2001, 276, 8746–8752. [Google Scholar] [CrossRef] [PubMed]
- Feral, C.C.; Nishiya, N.; Fenczik, C.A.; Stuhlmann, H.; Slepak, M.; Ginsberg, M.H. CD98hc (SLC3A2) mediates integrin signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Fenczik, C.A.; Sethi, T.; Ramos, J.W.; Hughes, P.E.; Ginsberg, M.H. Complementation of dominant suppression implicates CD98 in integrin activation. Nature 1997, 390, 81–85. [Google Scholar] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.D.; Cheresh, D.A. Role of integrins in cell invasion and migration. Nat. Rev. Cancer 2002, 2, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, R.; Alahari, S.K. Important role of integrins in the cancer biology. Cancer Metastasis Rev. 2010, 29, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Estrach, S.; Lee, S.A.; Boulter, E.; Pisano, S.; Errante, A.; Tissot, F.S.; Cailleteau, L.; Pons, C.; Ginsberg, M.H.; Feral, C.C. CD98hc (SLC3A2) loss protects against ras-driven tumorigenesis by modulating integrin-mediated mechanotransduction. Cancer Res. 2014, 74, 6878–6889. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-M.; Hahn, J.-H. CD98 activation increases surface expression and clustering of β1 integrins in MCF-7 cells through FAK/Src- and cytoskeleton-independent mechanisms. Exp. Mol. Med. 2008, 40, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Feral, C.C.; Zijlstra, A.; Tkachenko, E.; Prager, G.; Gardel, M.L.; Slepak, M.; Ginsberg, M.H. CD98hc (SLC3A2) participates in fibronectin matrix assembly by mediating integrin signaling. J. Cell Biol. 2007, 178, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Poettler, M.; Unseld, M.; Braemswig, K.; Haitel, A.; Zielinski, C.C.; Prager, G.W. CD98hc (SLC3A2) drives integrin-dependent renal cancer cell behavior. Mol. Cancer 2013, 12, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Yarden, Y. Endocytosis and cancer. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, T. Cell adhesion and its endocytic regulation in cell migration during neural development and cancer metastasis. Int. J. Mol. Sci. 2012, 13, 4564–4590. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Wang, Y.; Yang, X.M.; Xu, B.Q.; Feng, F.; Wang, B.; Liang, Q.; Li, Y.; Zhou, Y.; Jiang, J.L.; et al. Basigin-mediated redistribution of CD98 promotes cell spreading and tumorigenicity in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2015, 34, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Baez, L.; Donaldson, J.G. Hook1, microtubules, and Rab22: Mediators of selective sorting of clathrin-independent endocytic cargo proteins on endosomes. Bioarchitecture 2013, 3, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Baez, L.; Cole, N.B.; Kramer, H.; Donaldson, J.G. Microtubule-dependent endosomal sorting of clathrin-independent cargo by hook1. J. Cell Biol. 2013, 201, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.; Browne, C.D.; Ruppert, R.; Feral, C.C.; Fassler, R.; Rickert, R.C.; Ginsberg, M.H. CD98hc facilitates B cell proliferation and adaptive humoral immunity. Nat. Immunol. 2009, 10, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Ablack, J.N.; Metz, P.J.; Chang, J.T.; Cantor, J.M.; Ginsberg, M.H. Ubiquitylation of CD98 limits cell proliferation and clonal expansion. J. Cell Sci. 2015, 128, 4273–4278. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Collis, E.A.; Mackinnon, A.C.; Simpson, K.J.; Haslett, C.; Zent, R.; Ginsberg, M.; Sethi, T. CD98hc (SLC3A2) interaction with β1 integrins is required for transformation. J. Biol. Chem. 2004, 279, 54731–54741. [Google Scholar] [CrossRef] [PubMed]
- Prager, G.W.; Feral, C.C.; Kim, C.; Han, J.; Ginsberg, M.H. CD98hc (SLC3A2) interaction with the integrin β subunit cytoplasmic domain mediates adhesive signaling. J. Biol. Chem. 2007, 282, 24477–24484. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Kudoh, H.; Enomoto, T.; Hashimoto, Y.; Masuko, T. Enhanced tumorigenicity caused by truncation of the extracellular domain of GP125/CD98 heavy chain. Oncogene 2000, 19, 6209–6215. [Google Scholar] [CrossRef] [PubMed]
- Goldenring, J.R. A central role for vesicle trafficking in epithelial neoplasia: Intracellular highways to carcinogenesis. Nat. Rev. Cancer 2013, 13, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Eaton, S.; Martin-Belmonte, F. Cargo sorting in the endocytic pathway: A key regulator of cell polarity and tissue dynamics. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Goksoy, E.; Ma, Y.Q.; Wang, X.; Kong, X.; Perera, D.; Plow, E.F.; Qin, J. Structural basis for the autoinhibition of talin in regulating integrin activation. Mol. Cell 2008, 31, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Wegener, K.L.; Partridge, A.W.; Han, J.; Pickford, A.R.; Liddington, R.C.; Ginsberg, M.H.; Campbell, I.D. Structural basis of integrin activation by talin. Cell 2007, 128, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Pasqualon, T.; Pruessmeyer, J.; Jankowski, V.; Babendreyer, A.; Groth, E.; Schumacher, J.; Koenen, A.; Weidenfeld, S.; Schwarz, N.; Denecke, B.; et al. A cytoplasmic C-terminal fragment of Syndecan-1 is generated by sequential proteolysis and antagonizes syndecan-1 dependent lung tumor cell migration. Oncotarget 2015, 6, 31295–31312. [Google Scholar] [PubMed]
- Palmer, T.D.; Ashby, W.J.; Lewis, J.D.; Zijlstra, A. Targeting tumor cell motility to prevent metastasis. Adv. Drug Deliv. Rev. 2011, 63, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Li, T.; Bononi, F.C.; Lac, D.; Kekessie, I.A.; Liu, Y.; Sanchez, E.; Mazloom, A.; Ma, A.H.; Lin, J.; et al. Discovery and characterization of a high-affinity and high-specificity peptide ligand LXY30 for in vivo targeting of α3 integrin-expressing human tumors. EJNMMI Res. 2016, 6, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E.; Pierschbacher, M.D. New perspectives in cell adhesion: RGD and integrins. Science 1987, 238, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.M.; Ginsberg, M.H.; Rose, D.M. Integrin-associated proteins as potential therapeutic targets. Immunol. Rev. 2008, 223, 236–251. [Google Scholar] [CrossRef] [PubMed]
- Ku, X.M.; Liao, C.G.; Li, Y.; Yang, X.M.; Yang, B.; Yao, X.Y.; Wang, L.; Kong, L.M.; Zhao, P.; Chen, Z.N. Epitope mapping of series of monoclonal antibodies against the hepatocellular carcinoma-associated antigen HAb18G/CD147. Scand. J. Immunol. 2007, 65, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Li, H.W.; Yang, X.M.; Tang, J.; Wang, S.J.; Chen, Z.N.; Jiang, J.L. Effects of HAb18G/CD147 knockout on hepatocellular carcinoma cells in vitro using a novel zinc-finger nuclease-targeted gene knockout approach. Cell Biochem. Biophys. 2014, 71, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; Zhang, K.; Lv, M.; Miao, J.; Chen, Z.; Zhu, P. CD147 and CD98 complex-mediated homotypic aggregation attenuates the CypA-induced chemotactic effect on jurkat T cells. Mol. Immunol. 2015, 63, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wen, L.; Zhao, S.H.; Ai, Z.H.; Guo, J.Z.; Liu, W.C. FoxM1 expression is significantly associated with cisplatin-based chemotherapy resistance and poor prognosis in advanced non-small cell lung cancer patients. Lung Cancer 2013, 79, 173–179. [Google Scholar] [CrossRef] [PubMed]






© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, B.; Zhou, Y.; Wang, Y.; Yang, X.-M.; Liu, Z.-Y.; Li, J.-H.; Feng, F.; Chen, Z.-N.; Jiang, J.-L. Dominant Suppression of β1 Integrin by Ectopic CD98-ICD Inhibits Hepatocellular Carcinoma Progression. Int. J. Mol. Sci. 2016, 17, 1882. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17111882
Wu B, Zhou Y, Wang Y, Yang X-M, Liu Z-Y, Li J-H, Feng F, Chen Z-N, Jiang J-L. Dominant Suppression of β1 Integrin by Ectopic CD98-ICD Inhibits Hepatocellular Carcinoma Progression. International Journal of Molecular Sciences. 2016; 17(11):1882. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17111882
Chicago/Turabian StyleWu, Bo, Yang Zhou, Yu Wang, Xiang-Min Yang, Zhen-Yu Liu, Jiang-Hua Li, Fei Feng, Zhi-Nan Chen, and Jian-Li Jiang. 2016. "Dominant Suppression of β1 Integrin by Ectopic CD98-ICD Inhibits Hepatocellular Carcinoma Progression" International Journal of Molecular Sciences 17, no. 11: 1882. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17111882