
Toosendanin Exerts an Anti-Cancer Effect in Glioblastoma by Inducing Estrogen Receptor β- and p53-Mediated Apoptosis

Abstract
:1. Introduction
2. Results
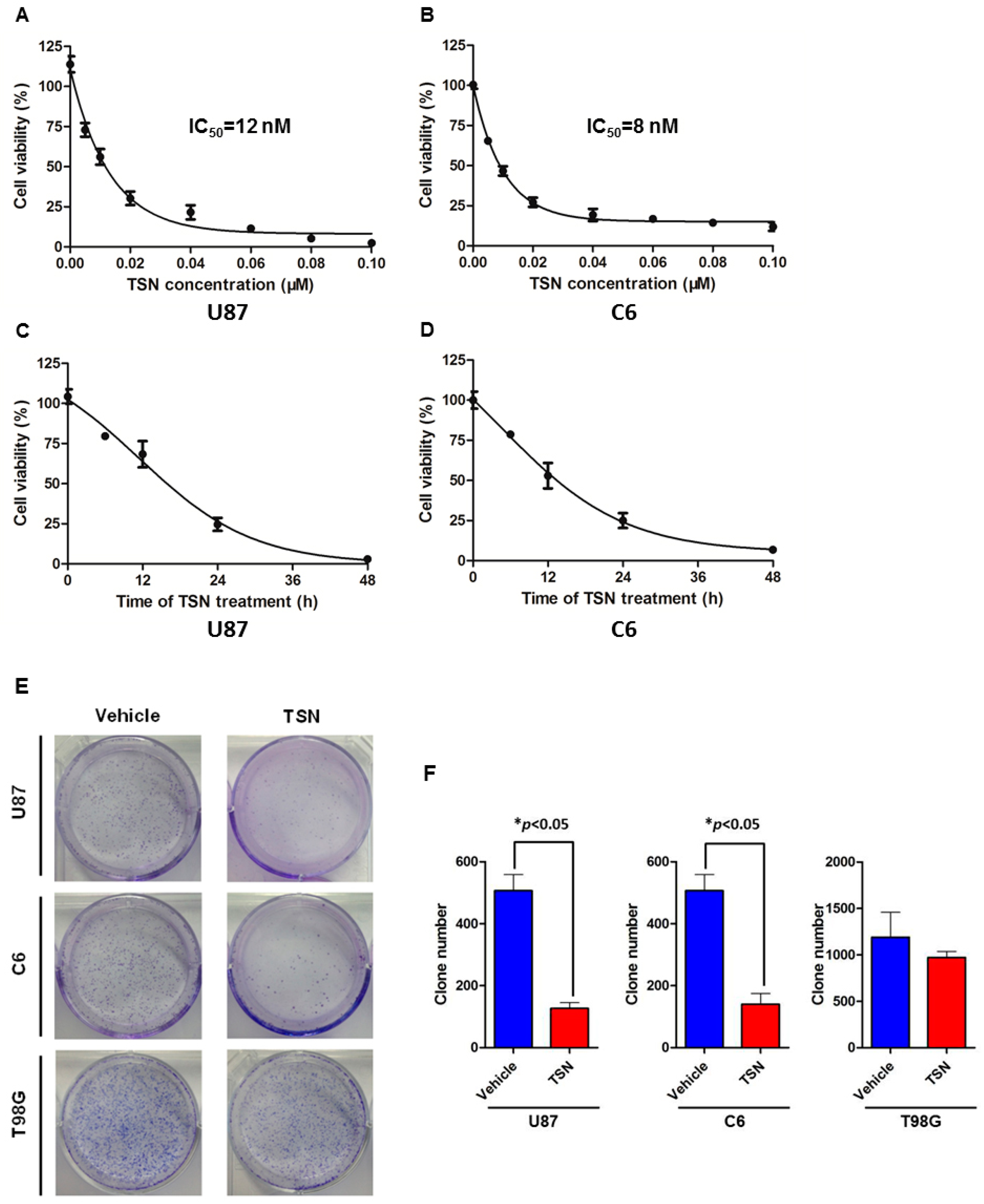
2.1. TSN (Toosendanin) Inhibits GBM (Glioblastoma) U87 and C6 Cell Proliferation
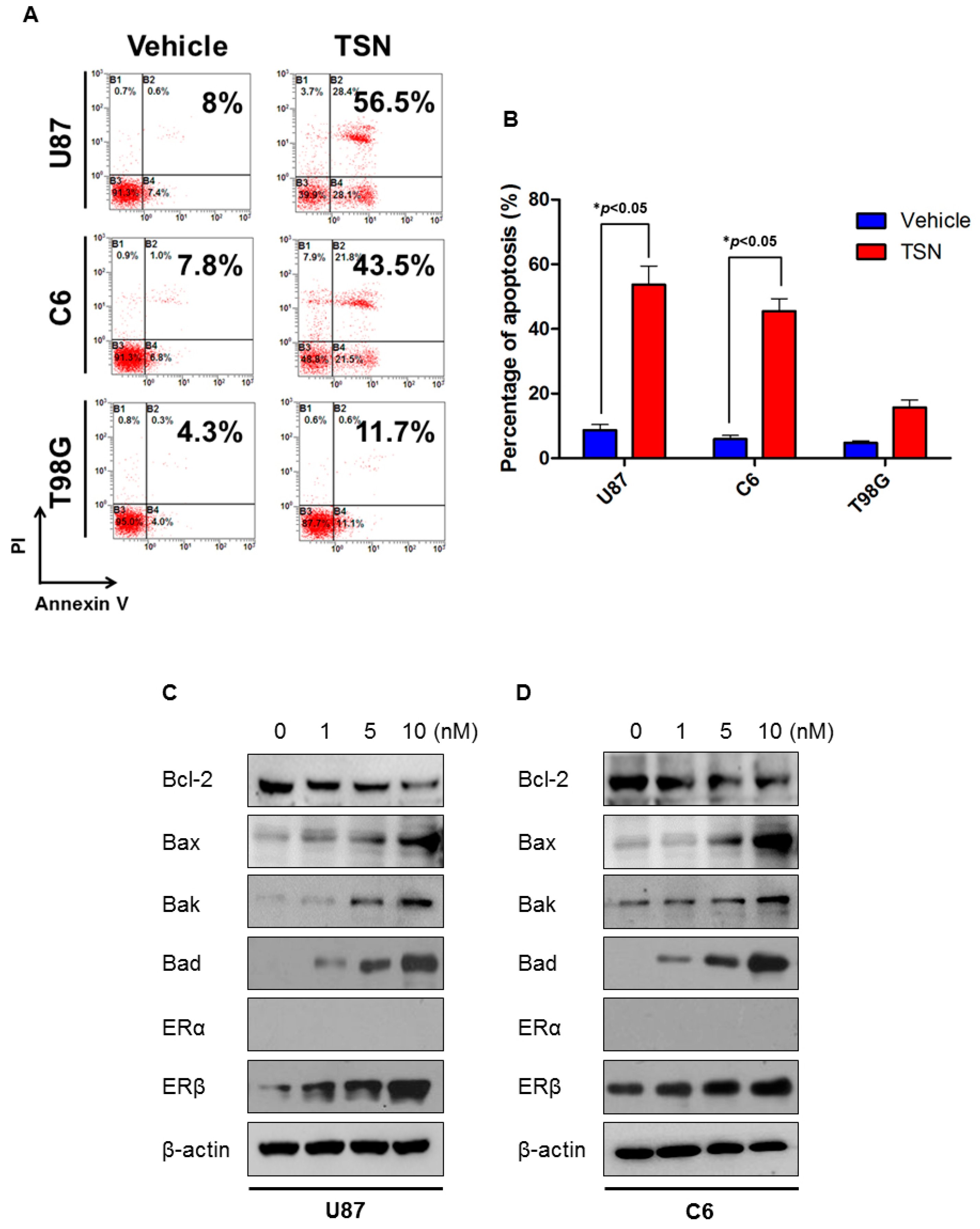
2.2. TSN Induces GBM U87 and C6 Cell Apoptosis In Vitro
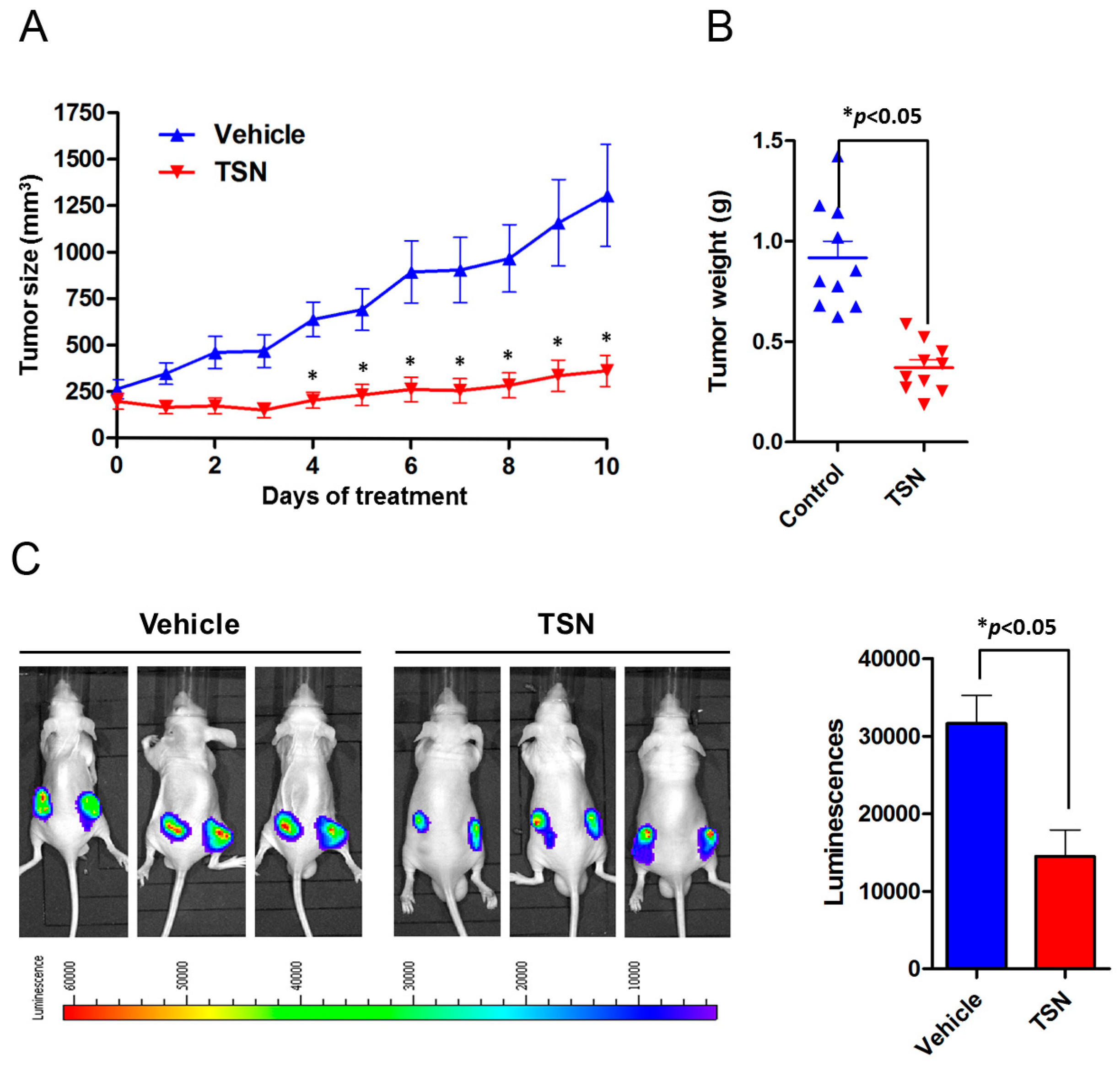
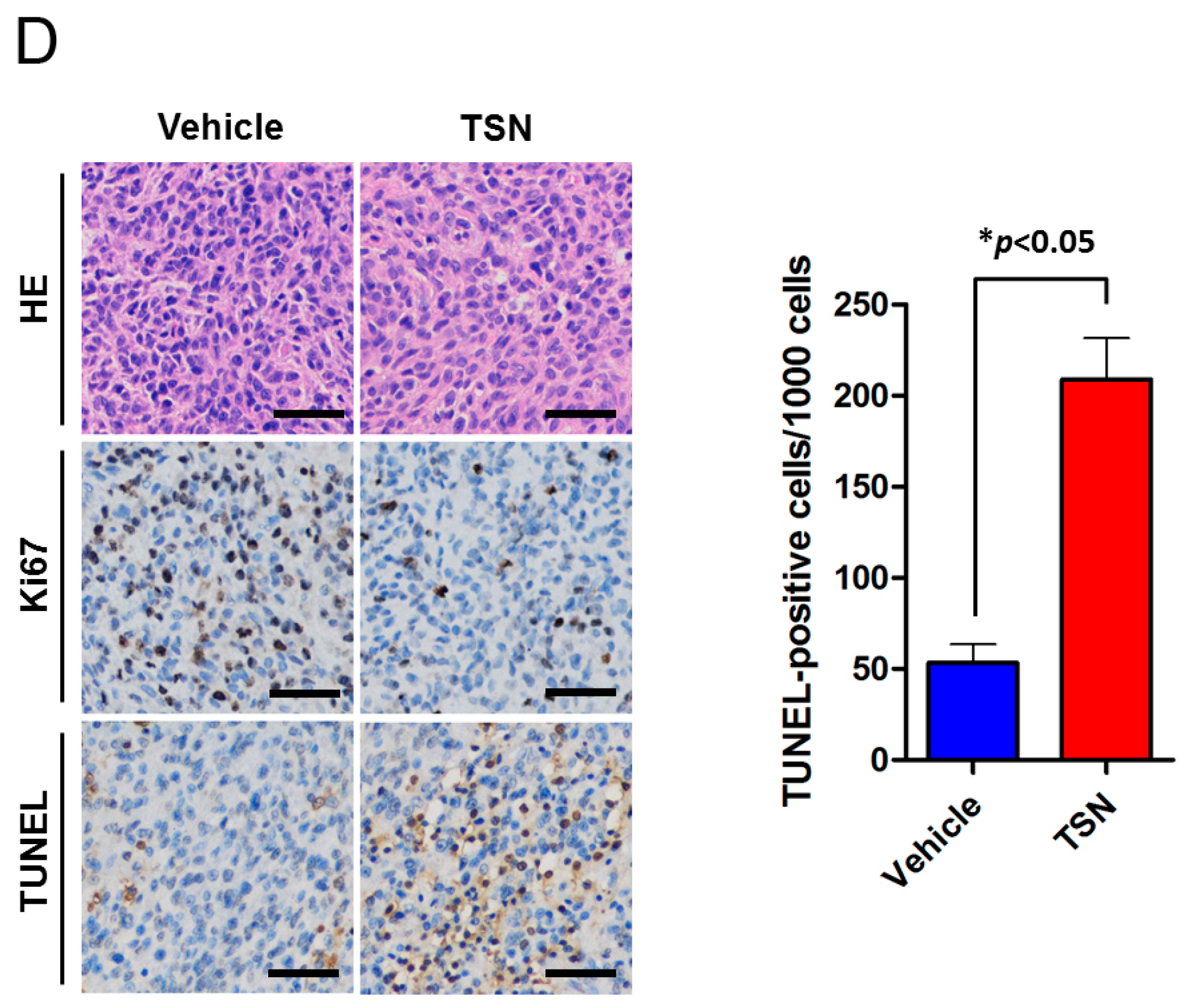
2.3. TSN Induces GBM U87 Cell Apoptosis In Vivo
2.4. TSN Does Not Exhibit Cytotoxicity in GBM T98G Cells
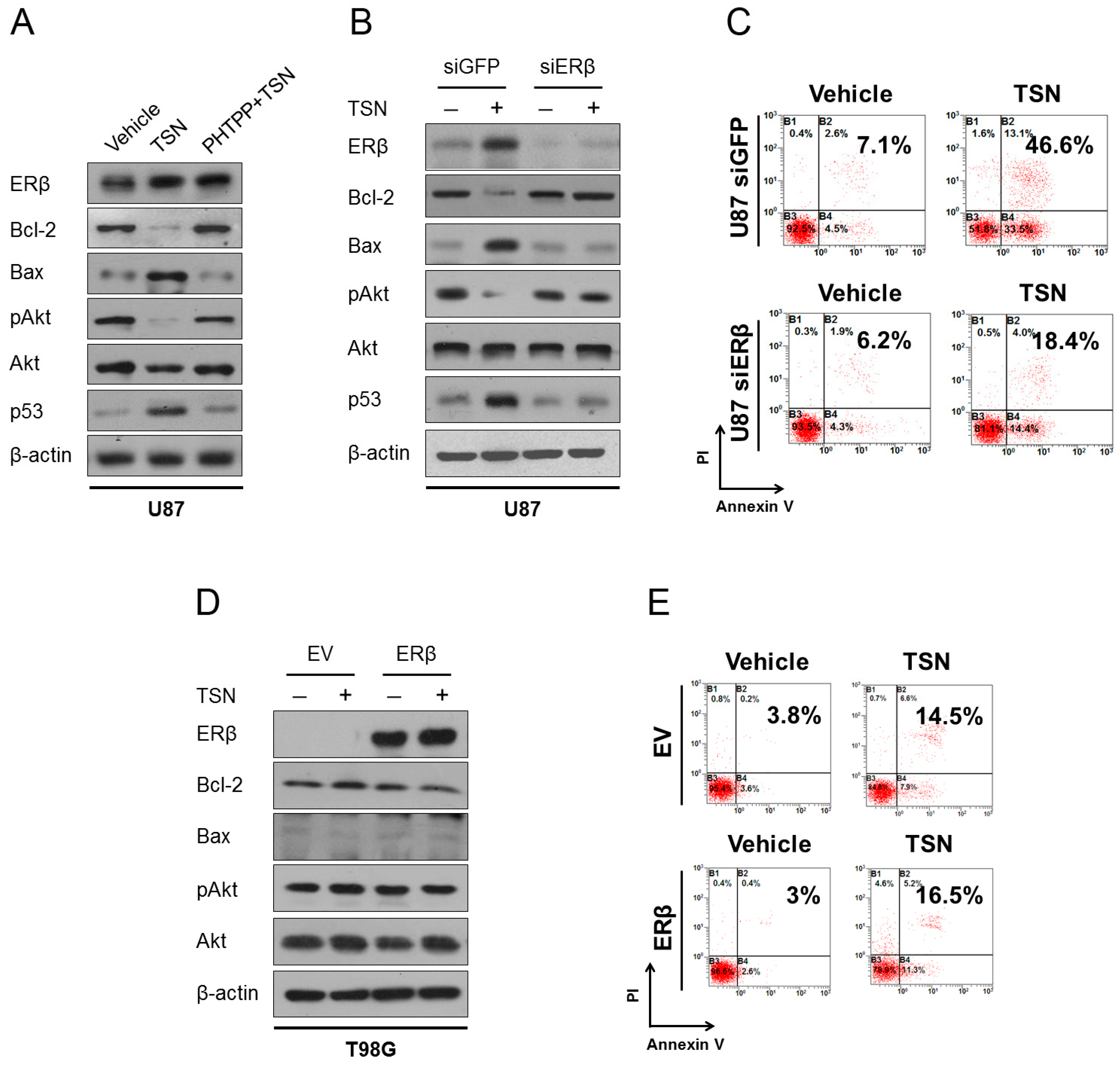
2.5. ERβ (Estrogen Receptor β) Is Required for the Cytotoxicity of TSN
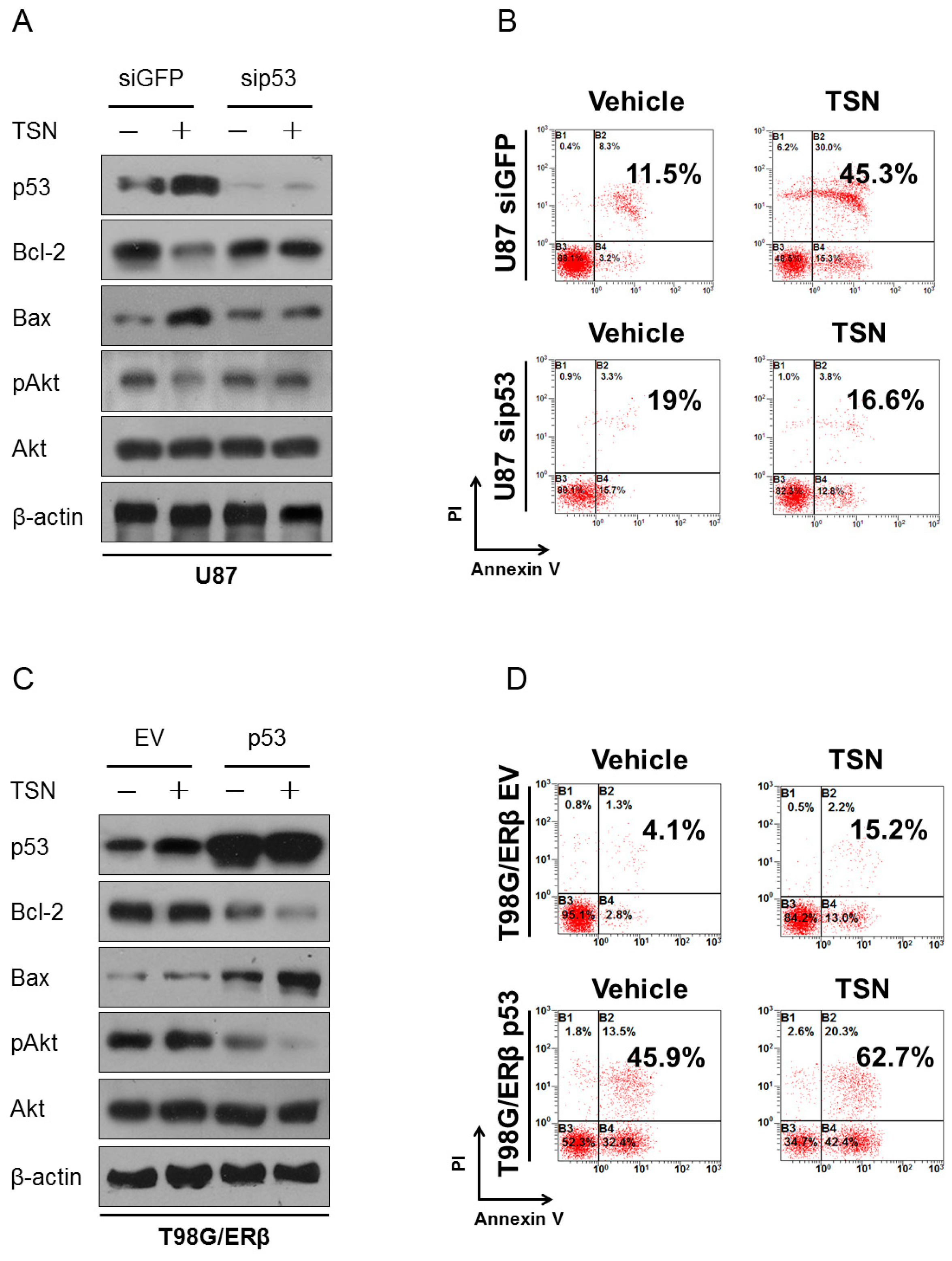
2.6. p53 as the Apoptotic Executor of TSN
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. MTT Assay
4.3. Colony Formation Assay
4.4. Flow Cytometry Analysis of Apoptosis
4.5. Western Blotting
4.6. Xenograft Models
4.7. Immunohistochemistry and TUNEL Assay
4.8. siRNAs, Constructs, and Transfection
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ATCC | American Type Culture Collection |
| DMSO | Dimethyl sulfoxide |
| ER | Estrogen receptor |
| FBS | Fetal bovine serum |
| GBM | Glioblastoma |
| HRP | Horseradish peroxidase |
| MAPK | Mitogen-activated protein kinase |
| MTT | 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide |
| PBS | Phosphate buffer saline |
| PCR | Polymerase chain reaction |
| PI3K | Phosphatidylinositol 3 kinase |
| siRNAs | Small interfering RNA |
| TCGA | The Cancer Genome Atlas |
| TSN | Toosendaninz |
| TUNEL | Minal deoxynucleotidyl transferase dUTP-mediated nicked end labeling |
References
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; di Patre, P.L.; Burkhard, C.; Schuler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic pathways to glioblastoma: A population-based study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Shah, U.; Morrison, T. A review of the symptomatic management of malignant gliomas in adults. J. Natl. Compr. Cancer Netw. 2013, 11, 424–429. [Google Scholar]
- Eyupoglu, I.Y.; Buchfelder, M.; Savaskan, N.E. Surgical resection of malignant gliomas—Role in optimizing patient outcome. Nat. Rev. Neurol. 2013, 9, 14–151. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.S.; Meyer, C.A.; Song, J.; Li, W.; Geistlinger, T.R.; Eeckhoute, J.; Brodsky, A.S.; Keeton, E.K.; Fertuck, K.C.; Hall, G.F.; et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006, 38, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef] [PubMed]
- Kushner, P.J.; Agard, D.A.; Greene, G.L.; Scanlan, T.S.; Shiau, A.K.; Uht, R.M.; Webb, P. Estrogen receptor pathways to AP-1. J. Steroid Biochem. Mol. Biol. 2000, 74, 311–317. [Google Scholar] [CrossRef]
- Tanaka, N.; Yonekura, H.; Yamagishi, S.; Fujimori, H.; Yamamoto, Y.; Yamamoto, H. The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-α through nuclear factor-κB, and by 17β-estradiol through SP-1 in human vascular endothelial cells. J. Biol. Chem. 2000, 275, 25781–25790. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R. Cell localization, physiology, and nongenomic actions of estrogen receptors. J. Appl. Physiol. 2001, 91, 1860–1867. [Google Scholar] [PubMed]
- Campbell, R.A.; Bhat-Nakshatri, P.; Patel, N.M.; Constantinidou, D.; Ali, S.; Nakshatri, H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor α: A new model for anti-estrogen resistance. J. Biol. Chem. 2001, 276, 9817–9824. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Ma, Y.X.; Wang, C.; Yuan, R.Q.; Meng, Q.; Wang, J.A.; Erdos, M.; Goldberg, I.D.; Webb, P.; Kushner, P.J.; et al. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene 2001, 20, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Lazennec, G. Estrogen receptor β, a possible tumor suppressor involved in ovarian carcinogenesis. Cancer Lett. 2006, 231, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Treeck, O.; Lattrich, C.; Springwald, A.; Ortmann, O. Estrogen receptor β exerts growth-inhibitory effects on human mammary epithelial cells. Breast Cancer Res. Treat. 2010, 120, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Pearce, S.T.; Jordan, V.C. The biological role of estrogen receptors α and β in cancer. Crit. Rev. Oncol. Hematol. 2004, 50, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.; Edvardsson, K.; Lewandowski, S.A.; Strom, A.; Gustafsson, J.A. A genome-wide study of the repressive effects of estrogen receptor β on estrogen receptor α signaling in breast cancer cells. Oncogene 2008, 27, 1019–1032. [Google Scholar] [CrossRef] [PubMed]
- Helguero, L.A.; Faulds, M.H.; Gustafsson, J.A.; Haldosen, L.A. Estrogen receptors α (ERα) and β (ERβ) differentially regulate proliferation and apoptosis of the normal murine mammary epithelial cell line HC11. Oncogene 2005, 24, 6605–6616. [Google Scholar] [CrossRef] [PubMed]
- Wade, C.B.; Robinson, S.; Shapiro, R.A.; Dorsa, D.M. Estrogen receptor (ER)α and ERβ exhibit unique pharmacologic properties when coupled to activation of the mitogen-activated protein kinase pathway. Endocrinology 2001, 142, 2336–2342. [Google Scholar] [PubMed]
- Jassam, N.; Bell, S.M.; Speirs, V.; Quirke, P. Loss of expression of oestrogen receptor β in colon cancer and its association with dukes’ staging. Oncol. Rep. 2005, 14, 17–21. [Google Scholar] [PubMed]
- Jordan, V.C. Selective estrogen receptor modulation: Concept and consequences in cancer. Cancer Cell 2004, 5, 207–213. [Google Scholar] [CrossRef]
- Fang, X.F.; Cui, Z.J. The anti-botulism triterpenoid toosendanin elicits calcium increase and exocytosis in rat sensory neurons. Cell. Mol. Neurobiol. 2011, 31, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.L.; Li, M.F. Biological effects of toosendanin, a triterpenoid extracted from chinese traditional medicine. Prog. Neurobiol. 2007, 82, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.Z.; Wang, Z.F.; Shi, Y.L. Involvement of cytochrome c release and caspase activation in toosendanin-induced PC12 cell apoptosis. Toxicology 2004, 201, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, Z.F.; Tang, M.Z.; Shi, Y.L. Growth inhibition and apoptosis-induced effect on human cancer cells of toosendanin, a triterpenoid derivative from chinese traditional medicine. Investig. New Drugs 2005, 23, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Batistatou, A.; Kyzas, P.A.; Goussia, A.; Arkoumani, E.; Voulgaris, S.; Polyzoidis, K.; Agnantis, N.J.; Stefanou, D. Estrogen receptor β (ERβ) protein expression correlates with BAG-1 and prognosis in brain glial tumours. J. Neurooncol. 2006, 77, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Gallo, D.; de Stefano, I.; Grazia Prisco, M.; Scambia, G.; Ferrandina, G. Estrogen receptor β in cancer: An attractive target for therapy. Curr. Pharm. Des. 2012, 18, 2734–2757. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.H.; Cheng, S.F.; Wu, C.C.; Chu, C.H.; Weng, Y.J.; Lin, C.S.; Lee, S.D.; Wu, H.C.; Huang, C.Y.; Kuo, W.W. Apoptotic effects of over-expressed estrogen receptor-β on lovo colon cancer cell is mediated by p53 signalings in a ligand-dependent manner. Chin. J. Physiol. 2006, 49, 110–116. [Google Scholar] [PubMed]
- Lu, X.; Ji, C.; Tong, W.; Lian, X.; Wu, Y.; Fan, X.; Gao, Y. Integrated analysis of microrna and mrna expression profiles highlights the complex and dynamic behavior of toosendanin-induced liver injury in mice. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Networt. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Pravettoni, A.; Mornati, O.; Martini, P.G.; Marino, M.; Colciago, A.; Celotti, F.; Motta, M.; Negri-Cesi, P. Estrogen receptor β (ERβ) and inhibition of prostate cancer cell proliferation: Studies on the possible mechanism of action in du145 cells. Mol. Cell. Endocrinol. 2007, 263, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Arai, N.; Strom, A.; Rafter, J.J.; Gustafsson, J.A. Estrogen receptor β mRNA in colon cancer cells: Growth effects of estrogen and genistein. Biochem. Biophys. Res. Commun. 2000, 270, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Sareddy, G.R.; Li, X.; Liu, J.; Viswanadhapalli, S.; Garcia, L.; Gruslova, A.; Cavazos, D.; Garcia, M.; Strom, A.M.; Gustafsson, J.A.; et al. Selective estrogen receptor β agonist LY500307 as a novel therapeutic agent for glioblastoma. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, L.; Chen, J.; Ling, Q.; Wang, H.; Li, S.; Li, L.; Yang, S.; Xia, M.; Jing, L. Estrogen receptor β agonist enhances temozolomide sensitivity of glioma cells by inhibiting PI3K/AKT/mTOR pathway. Mol. Med. Rep. 2015, 11, 1516–1522. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Species | ERα | ERβ | P53 |
|---|---|---|---|---|
| U87 | Human | - | + | Wild Type |
| C6 | Rat | - | + | Wild Type |
| T98G | Human | + | - | Mutated |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, L.; Qu, D.; Wang, H.; Zhang, S.; Jia, C.; Shi, Z.; Wang, Z.; Zhang, J.; Ma, J. Toosendanin Exerts an Anti-Cancer Effect in Glioblastoma by Inducing Estrogen Receptor β- and p53-Mediated Apoptosis. Int. J. Mol. Sci. 2016, 17, 1928. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17111928
Cao L, Qu D, Wang H, Zhang S, Jia C, Shi Z, Wang Z, Zhang J, Ma J. Toosendanin Exerts an Anti-Cancer Effect in Glioblastoma by Inducing Estrogen Receptor β- and p53-Mediated Apoptosis. International Journal of Molecular Sciences. 2016; 17(11):1928. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17111928
Chicago/Turabian StyleCao, Liang, Dingding Qu, Huan Wang, Sha Zhang, Chenming Jia, Zixuan Shi, Zongren Wang, Jian Zhang, and Jing Ma. 2016. "Toosendanin Exerts an Anti-Cancer Effect in Glioblastoma by Inducing Estrogen Receptor β- and p53-Mediated Apoptosis" International Journal of Molecular Sciences 17, no. 11: 1928. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17111928