
Cell Death in Chondrocytes, Osteoblasts, and Osteocytes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Chondrocyte Death
1.1. Chondrocyte Proliferation and Apoptosis
1.1.1. Runx2, Indian Hedgehog (Ihh), and Parathyroid Hormone-Related Peptide (Pthrp) Regulate Chondrocyte Proliferation and Differentiation
1.1.2. Cyclin D1 Plays an Important Role in Chondrocyte Proliferation
1.1.3. Can the Overexpression of Both Cdk6 and Cyclin D1 Induce Chondrocyte Proliferation?
1.1.4. p107 Plays a Key Role in Chondrocyte Proliferation and Apoptosis
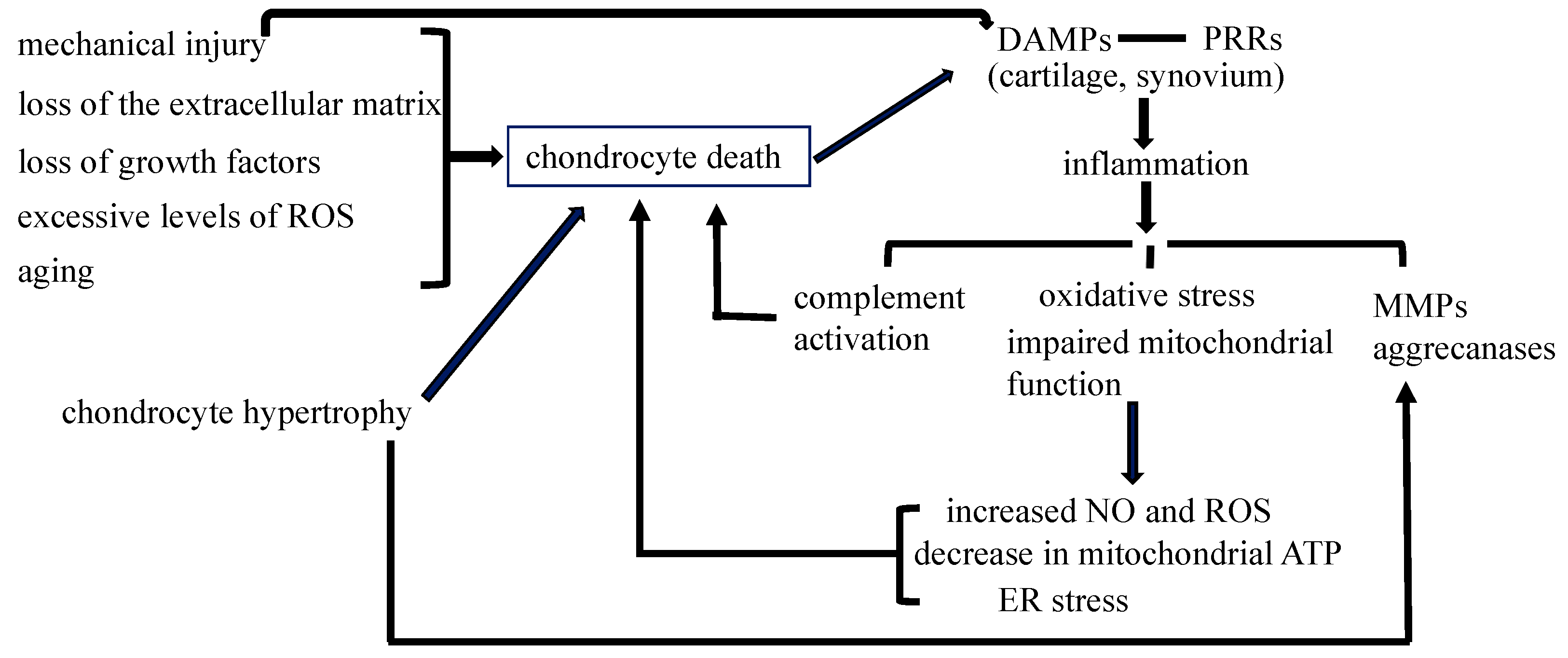
1.2. Chondrocyte Death in Osteoarthritis (OA)
1.2.1. Is Chondrocyte Death Involved in the Pathogenesis of OA?
1.2.2. Chondrocyte Death during Mechanical Injury May Contribute to the Development of OA
1.2.3. Non-Infectious Inflammatory Processes through PRRs Are Involved in the Pathogenesis of Posttraumatic OA
2. Osteoblast Death
2.1. Roles of Apoptosis in the Regulation of Bone Mass and Osteoblast Differentiation
Osteoblast Apoptosis Regulates Bone Mass
2.2. Osteoblast Apoptosis and Osteoblast Differentiation
2.2.1. Osteoblast Apoptosis Suppresses Osteoblast Differentiation In Vitro by Reducing Cell Density during Cultivation
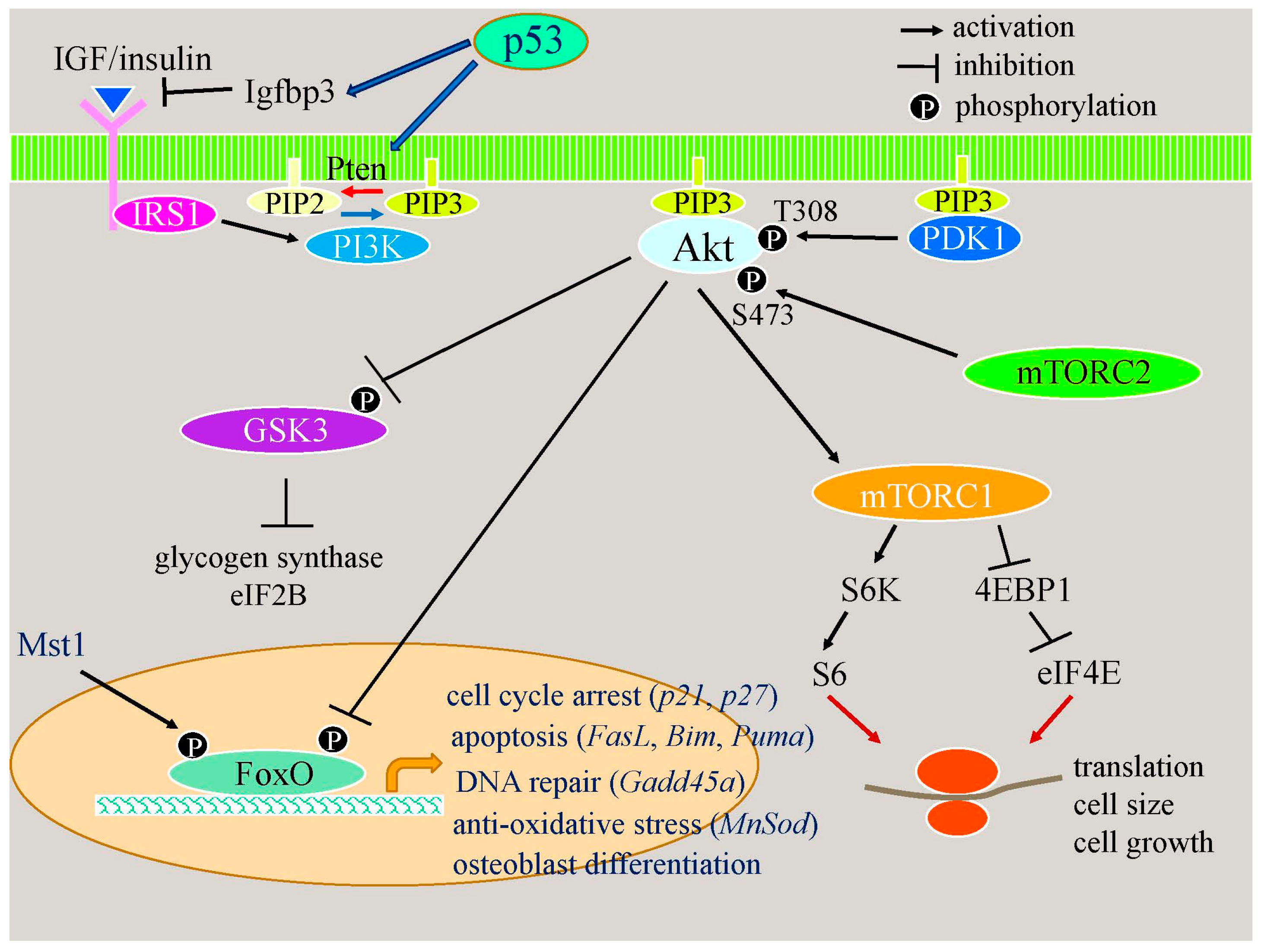
2.2.2. Apoptotic Responses Enhance Osteoblast Differentiation through the p53–Akt–FoxO Pathway
2.2.3. p53 Reduces Bone Formation by Inhibiting Osteoblast Proliferation and Enhancing Osteoblast Apoptosis
3. Osteocyte Death
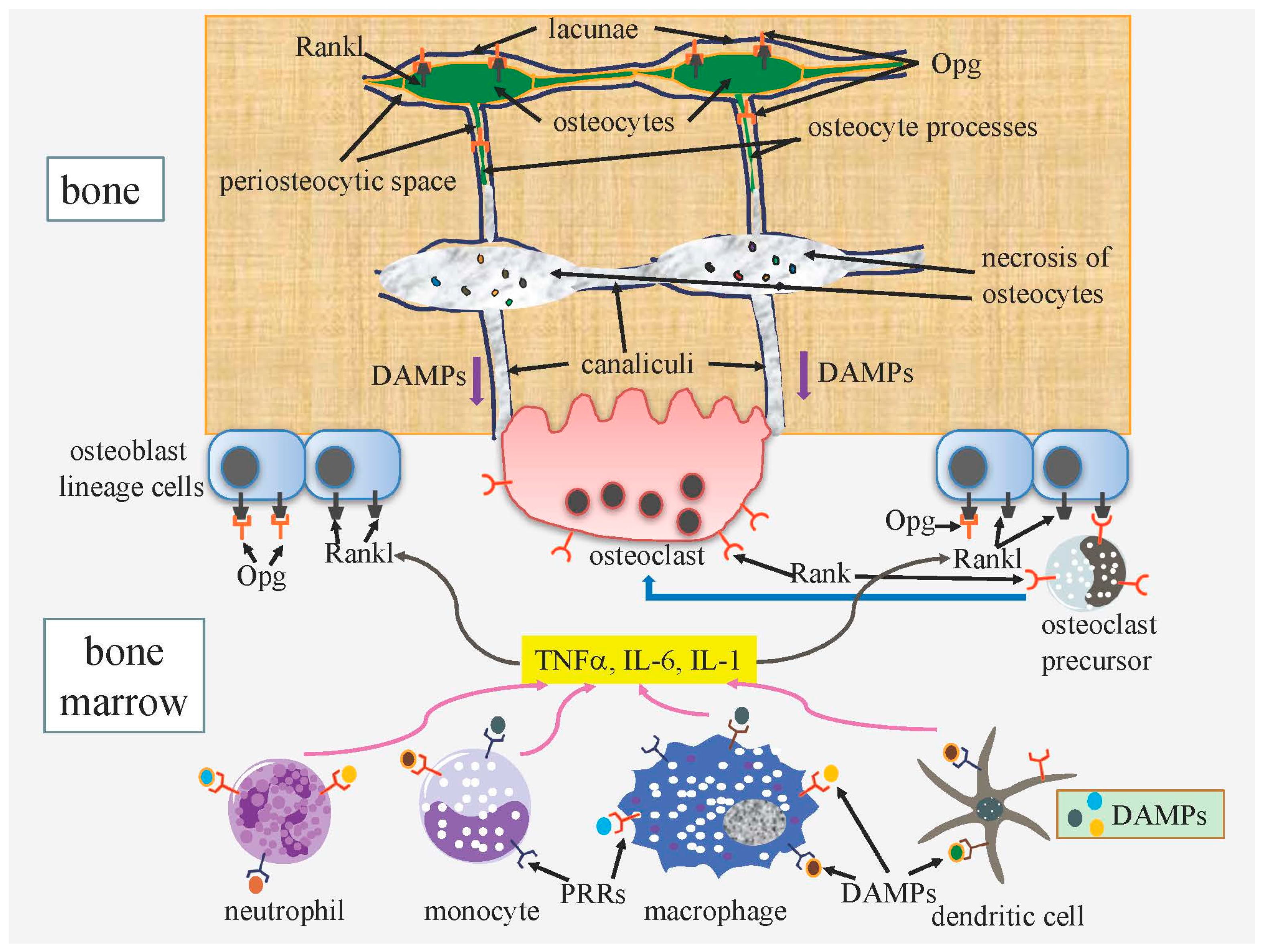
3.1. Osteocyte Death and Bone Remodeling
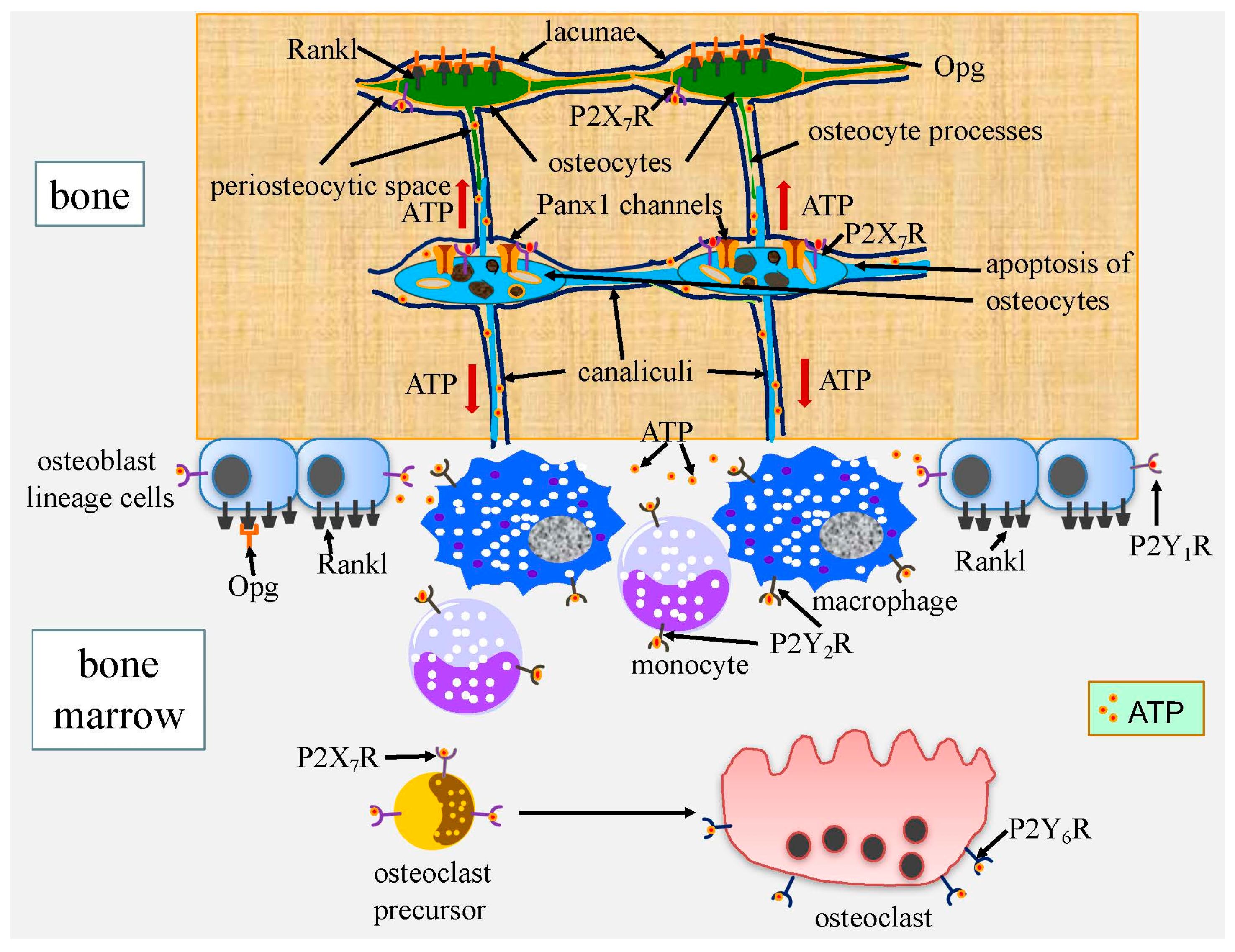
3.1.1. ATP Released from Apoptotic Cells through Pannexin Channels Enhances Bone Resorption
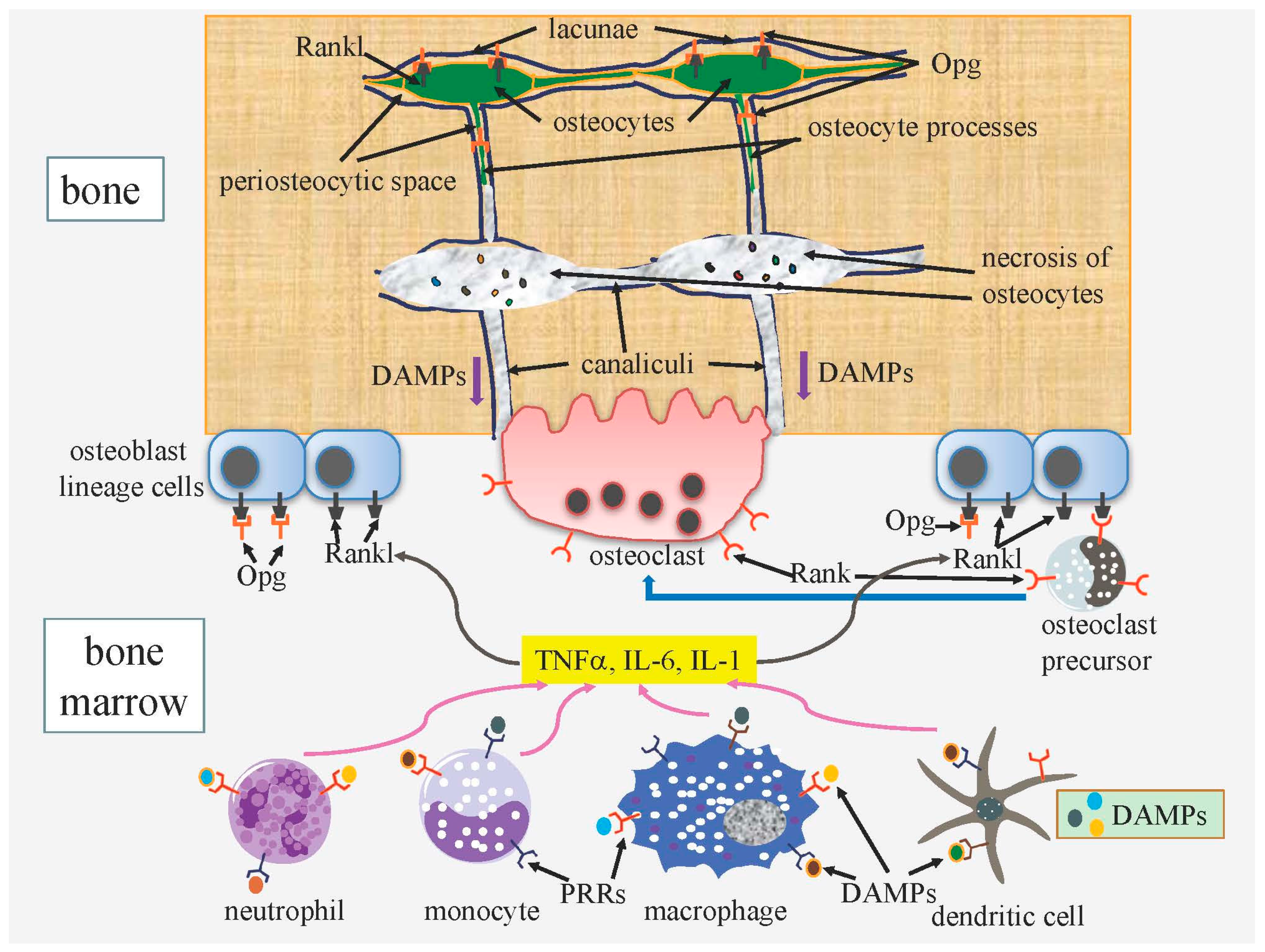
3.1.2. DAMPs Released from Necrotic Osteocytes Further Enhance Bone Resorption
3.2. Functions of Osteocytes
3.2.1. Do Live Osteocytes Inhibit Bone Resorption?
3.2.2. Regulation of the Release of Osteocyte-Derived Opg to the Bone Surface May Be a Major Role for Rankl on Osteocytes
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Yoshida, C.A.; Yamamoto, H.; Fujita, T.; Furuichi, T.; Ito, K.; Inoue, K.; Yamana, K.; Zanma, A.; Takada, K.; Ito, Y.; et al. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 2004, 18, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Inada, M.; Yasui, T.; Nomura, S.; Miyake, S.; Deguchi, K.; Himeno, M.; Sato, M.; Yamagiwa, H.; Kimura, T.; Yasui, N.; et al. Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev. Dyn. 1999, 214, 279–290. [Google Scholar] [CrossRef]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Kitagaki, J.; Tamamura, Y.; Gentili, C.; Koyama, E.; Enomoto, H.; Komori, T.; Pacifici, M.; Enomoto-Iwamoto, M. Runx2 expression and action in chondrocytes are regulated by retinoid signaling and parathyroid hormone-related peptide (PTHrP). Osteoarthr. Cartil. 2003, 11, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Li, T.F.; Dong, Y.; Ionescu, A.M.; Rosier, R.N.; Zuscik, M.J.; Schwarz, E.M.; O’Keefe, R.J.; Drissi, H. Parathyroid hormone-related peptide (PTHrP) inhibits Runx2 expression through the PKA signaling pathway. Exp. Cell Res. 2004, 299, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Vortkamp, A.; Lee, K.; Lanske, B.; Segre, G.V.; Kronenberg, H.M.; Tabin, C.J. Regulation of rate of cartilage differentiation by indian hedgehog and PTH-related protein. Science 1996, 273, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Himeno, M.; Enomoto, H.; Liu, W.; Ishizeki, K.; Nomura, S.; Kitamura, Y.; Komori, T. Impaired vascular invasion of Cbfa1-deficient cartilage engrafted in the spleen. J. Bone Miner. Res. 2002, 17, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Zelzer, E.; Glotzer, D.J.; Hartmann, C.; Thomas, D.; Fukai, N.; Soker, S.; Olsen, B.R. Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech. Dev. 2001, 106, 97–106. [Google Scholar] [CrossRef]
- Komori, T. Regulation of bone development and extracellular matrix protein genes by Runx2. Cell Tissue Res. 2010, 339, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Tsang, K.Y.; Tang, H.C.; Chan, D.; Cheah, K.S. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. USA 2014, 111, 12097–12102. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; von der Mark, K.; Henry, S.; Norton, W.; Adams, H.; de Crombrugghe, B. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 2014, 10, e1004820. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M. Revisiting the “Cdk-centric” view of the mammalian cell cycle. Cell Cycle 2005, 4, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Beier, F.; Ali, Z.; Mok, D.; Taylor, A.C.; Leask, T.; Albanese, C.; Pestell, R.G.; LuValle, P. TGFβ and PTHrP control chondrocyte proliferation by activating cyclin D1 expression. Mol. Biol. Cell 2001, 12, 3852–3863. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Zhang, X.M.; Karp, S.; Yang, Y.; McMahon, A.P. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 2001, 128, 5099–5108. [Google Scholar] [PubMed]
- Yang, Y.; Topol, L.; Lee, H.; Wu, J. Wnt5a and Wnt5b exhibit distinct activities in coordinating chondrocyte proliferation and differentiation. Development 2003, 130, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef] [PubMed]
- Fantl, V.; Stamp, G.; Andrews, A.; Rosewell, I.; Dickson, C. Mice lacking cyclin D1 are small and show defects in eye and mammary gland development. Genes Dev. 1995, 9, 2364–2372. [Google Scholar] [CrossRef] [PubMed]
- Sicinski, P.; Donaher, J.L.; Parker, S.B.; Li, T.; Fazeli, A.; Gardner, H.; Haslam, S.Z.; Bronson, R.T.; Elledge, S.J.; Weinberg, R.A. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell 1995, 82, 621–630. [Google Scholar] [CrossRef]
- Cobrinik, D.; Lee, M.H.; Hannon, G.; Mulligan, G.; Bronson, R.T.; Dyson, N.; Harlow, E.; Beach, D.; Weinberg, R.A.; Jacks, T. Shared role of the pRB-related p130 and p107 proteins in limb development. Genes Dev. 1996, 10, 1633–1644. [Google Scholar] [CrossRef] [PubMed]
- Rossi, F.; MacLean, H.E.; Yuan, W.; Francis, R.O.; Semenova, E.; Lin, C.S.; Kronenberg, H.M.; Cobrinik, D. p107 and p130 Coordinately regulate proliferation, Cbfa1 expression, and hypertrophic differentiation during endochondral bone development. Dev. Biol. 2002, 247, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Maruyama, Z.; Sakai, A.; Izumi, S.; Moriishi, T.; Yoshida, C.A.; Miyazaki, T.; Komori, H.; Takada, K.; Kawaguchi, H.; et al. Overexpression of Cdk6 and Ccnd1 in chondrocytes inhibited chondrocyte maturation and caused p53-dependent apoptosis without enhancing proliferation. Oncogene 2013, 33, 1862–1871. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.C.; Cardiff, R.D.; Zukerberg, L.; Lees, E.; Arnold, A.; Schmidt, E.V. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature 1994, 369, 669–671. [Google Scholar] [CrossRef] [PubMed]
- Mueller, A.; Odze, R.; Jenkins, T.D.; Shahsesfaei, A.; Nakagawa, H.; Inomoto, T.; Rustgi, A.K. A transgenic mouse model with cyclin D1 overexpression results in cell cycle, epidermal growth factor receptor, and p53 abnormalities. Cancer Res. 1997, 57, 5542–5549. [Google Scholar] [PubMed]
- Zhu, L.; Xie, E.; Chang, L.S. Differential roles of two tandem E2F sites in repression of the human p107 promoter by retinoblastoma and p107 proteins. Mol. Cell. Biol. 1995, 15, 3552–3562. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, D.L.; Wirt, S.E.; Zmoos, A.F.; Kareta, M.S.; Sage, J. Tandem E2F binding sites in the promoter of the p107 cell cycle regulator control p107 expression and its cellular functions. PLoS Genet. 2010, 6, e1001003. [Google Scholar] [CrossRef] [PubMed]
- Hurford, R.K., Jr.; Cobrinik, D.; Lee, M.H.; Dyson, N. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev. 1997, 11, 1447–1463. [Google Scholar] [CrossRef] [PubMed]
- Landman, A.S.; Danielian, P.S.; Lees, J.A. Loss of pRB and p107 disrupts cartilage development and promotes enchondroma formation. Oncogene 2012, 32, 4798–4805. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Regulation of Rb family proteins by Cdk6/Ccnd1 in growth plates. Cell Cycle 2013, 12, 2161–2162. [Google Scholar] [CrossRef] [PubMed]
- Calbo, J.; Parreno, M.; Sotillo, E.; Yong, T.; Mazo, A.; Garriga, J.; Grana, X. G1 cyclin/cyclin-dependent kinase-coordinated phosphorylation of endogenous pocket proteins differentially regulates their interactions with E2F4 and E2F1 and gene expression. J. Biol. Chem. 2002, 277, 50263–50274. [Google Scholar] [CrossRef] [PubMed]
- Liu-Bryan, R.; Terkeltaub, R. Emerging regulators of the inflammatory process in osteoarthritis. Nat. Rev. Rheumatol. 2015, 11, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Zamli, Z.; Sharif, M. Chondrocyte apoptosis: A cause or consequence of osteoarthritis? Int. J. Rheum. Dis. 2011, 14, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, K.; D’Lima, D.D.; Hashimoto, S.; Lotz, M. Cell death in cartilage. Osteoarthr. Cartil. 2004, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Functions of the osteocyte network in the regulation of bone mass. Cell Tissue Res. 2013, 352, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Roach, H.I.; Aigner, T.; Kouri, J.B. Chondroptosis: A variant of apoptotic cell death in chondrocytes? Apoptosis 2004, 9, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Glucocorticoid signaling and bone biology. Horm. Metab. Res. 2016, 48, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Bohensky, J.; Shapiro, I.M.; Leshinsky, S.; Watanabe, H.; Srinivas, V. PIM-2 is an independent regulator of chondrocyte survival and autophagy in the epiphyseal growth plate. J. Cell. Physiol. 2007, 213, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Mistry, D.; Oue, Y.; Chambers, M.G.; Kayser, M.V.; Mason, R.M. Chondrocyte death during murine osteoarthritis. Osteoarthr. Cartil. 2004, 12, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M.; Fuller, C.J.; Whittles, C.E.; Sharif, M. Chondrocyte death by apoptosis is associated with cartilage matrix degradation. Osteoarthr. Cartil. 2007, 15, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Ochs, R.L.; Komiya, S.; Lotz, M. Linkage of chondrocyte apoptosis and cartilage degradation in human osteoarthritis. Arthritis Rheum. 1998, 41, 1632–1638. [Google Scholar] [CrossRef]
- Blanco, F.J.; Guitian, R.; Vazquez-Martul, E.; de Toro, F.J.; Galdo, F. Osteoarthritis chondrocytes die by apoptosis. A possible pathway for osteoarthritis pathology. Arthritis Rheum. 1998, 41, 284–289. [Google Scholar] [CrossRef]
- Sharif, M.; Whitehouse, A.; Sharman, P.; Perry, M.; Adams, M. Increased apoptosis in human osteoarthritic cartilage corresponds to reduced cell density and expression of caspase-3. Arthritis Rheum. 2004, 50, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Lee, Y.J.; Seong, S.C.; Choe, K.W.; Song, Y.W. Apoptotic chondrocyte death in human osteoarthritis. J. Rheumatol. 2000, 27, 455–462. [Google Scholar] [PubMed]
- Hashimoto, S.; Takahashi, K.; Amiel, D.; Coutts, R.D.; Lotz, M. Chondrocyte apoptosis and nitric oxide production during experimentally induced osteoarthritis. Arthritis Rheum. 1998, 41, 1266–1274. [Google Scholar] [CrossRef]
- Todd Allen, R.; Robertson, C.M.; Harwood, F.L.; Sasho, T.; Williams, S.K.; Pomerleau, A.C.; Amiel, D. Characterization of mature vs. aged rabbit articular cartilage: Analysis of cell density, apoptosis-related gene expression and mechanisms controlling chondrocyte apoptosis. Osteoarthr. Cartil. 2004, 12, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.S.; Horton, W.E., Jr. Chondrocyte apoptosis increases with age in the articular cartilage of adult animals. Anat. Rec. 1998, 250, 418–425. [Google Scholar] [CrossRef]
- D’Lima, D.; Hermida, J.; Hashimoto, S.; Colwell, C.; Lotz, M. Caspase inhibitors reduce severity of cartilage lesions in experimental osteoarthritis. Arthritis Rheum. 2006, 54, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Dang, A.C.; Warren, A.P.; Kim, H.T. Beneficial effects of intra-articular caspase inhibition therapy following osteochondral injury. Osteoarthr. Cartil. 2006, 14, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mani, S.B.; He, Y.; Hall, A.M.; Xu, L.; Li, Y.; Zurakowski, D.; Jay, G.D.; Warman, M.L. Induced superficial chondrocyte death reduces catabolic cartilage damage in murine posttraumatic osteoarthritis. J. Clin. Investig. 2016, 126, 2893–2902. [Google Scholar] [CrossRef] [PubMed]
- Del Carlo, M., Jr.; Loeser, R.F. Cell death in osteoarthritis. Curr. Rheumatol. Rep. 2008, 10, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Clements, K.M.; Bee, Z.C.; Crossingham, G.V.; Adams, M.A.; Sharif, M. How severe must repetitive loading be to kill chondrocytes in articular cartilage? Osteoarthr. Cartil. 2001, 9, 499–507. [Google Scholar] [CrossRef] [PubMed]
- D’Lima, D.D.; Hashimoto, S.; Chen, P.C.; Colwell, C.W., Jr.; Lotz, M.K. Human chondrocyte apoptosis in response to mechanical injury. Osteoarthr. Cartil. 2001, 9, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Clements, K.M.; Hollander, A.P.; Sharif, M.; Adams, M.A. Cyclic loading can denature type II collagen in articular cartilage. Connect. Tissue Res. 2004, 45, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Zemmyo, M.; Meharra, E.J.; Kuhn, K.; Creighton-Achermann, L.; Lotz, M. Accelerated, aging-dependent development of osteoarthritis in α1 integrin-deficient mice. Arthritis Rheum. 2003, 48, 2873–2880. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, S.W.; Helminen, H.J.; Khillan, J.S.; Bao, Y.; Prockop, D.J. Apoptosis of chondrocytes in transgenic mice lacking collagen II. Exp. Cell Res. 1997, 235, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Schelbergen, R.F.; Blom, A.B.; van den Bosch, M.H.; Sloetjes, A.; Abdollahi-Roodsaz, S.; Schreurs, B.W.; Mort, J.S.; Vogl, T.; Roth, J.; van den Berg, W.B.; et al. Alarmins S100A8 and S100A9 elicit a catabolic effect in human osteoarthritic chondrocytes that is dependent on Toll-like receptor 4. Arthritis Rheum. 2012, 64, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.; Jung, A.; Murphy, A.; Andreyev, A.; Dykens, J.; Terkeltaub, R. Mitochondrial oxidative phosphorylation is a downstream regulator of nitric oxide effects on chondrocyte matrix synthesis and mineralization. Arthritis Rheum. 2000, 43, 1560–1570. [Google Scholar] [CrossRef]
- Blanco, F.J.; Rego, I.; Ruiz-Romero, C. The role of mitochondria in osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Kurz, B.; Lemke, A.; Kehn, M.; Domm, C.; Patwari, P.; Frank, E.H.; Grodzinsky, A.J.; Schunke, M. Influence of tissue maturation and antioxidants on the apoptotic response of articular cartilage after injurious compression. Arthritis Rheum. 2004, 50, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Maneiro, E.; Lopez-Armada, M.J.; de Andres, M.C.; Carames, B.; Martin, M.A.; Bonilla, A.; del Hoyo, P.; Galdo, F.; Arenas, J.; Blanco, F.J. Effect of nitric oxide on mitochondrial respiratory activity of human articular chondrocytes. Ann. Rheum. Dis. 2005, 64, 388–395. [Google Scholar] [CrossRef] [PubMed]
- DelCarlo, M.; Loeser, R.F. Chondrocyte cell death mediated by reactive oxygen species-dependent activation of pkc-betai. Am. J. Physiol. Cell physiol. 2006, 290, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Rozelle, A.L.; Lepus, C.M.; Scanzello, C.R.; Song, J.J.; Larsen, D.M.; Crish, J.F.; Bebek, G.; Ritter, S.Y.; Lindstrom, T.M.; et al. Identification of a central role for complement in osteoarthritis. Nat. Med. 2011, 17, 1674–1679. [Google Scholar] [CrossRef] [PubMed]
- Bohana-Kashtan, O.; Ziporen, L.; Donin, N.; Kraus, S.; Fishelson, Z. Cell signals transduced by complement. Mol. Immunol. 2004, 41, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Uehara, Y.; Hirose, J.; Yamabe, S.; Okamoto, N.; Okada, T.; Oyadomari, S.; Mizuta, H. Endoplasmic reticulum stress-induced apoptosis contributes to articular cartilage degeneration via C/EBP homologous protein. Osteoarthr. Cartil. 2014, 22, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Ishida, K.; Matsushita, T.; Fujita, N.; Hayashi, S.; Sasaki, K.; Tei, K.; Kubo, S.; Matsumoto, T.; Fujioka, H.; et al. SIRT1 regulation of apoptosis of human chondrocytes. Arthritis Rheum. 2009, 60, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- Gagarina, V.; Gabay, O.; Dvir-Ginzberg, M.; Lee, E.J.; Brady, J.K.; Quon, M.J.; Hall, D.J. SIRT1 enhances survival of human osteoarthritic chondrocytes by repressing protein tyrosine phosphatase 1B and activating the insulin-like growth factor receptor pathway. Arthritis Rheum. 2010, 62, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Gabay, O.; Oppenhiemer, H.; Meir, H.; Zaal, K.; Sanchez, C.; Dvir-Ginzberg, M. Increased apoptotic chondrocytes in articular cartilage from adult heterozygous SIRT1 mice. Ann. Rheum. Dis. 2012, 71, 613–616. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Jia, D.; Plotkin, L.I.; Bellido, T.; Powers, C.C.; Stewart, S.A.; Manolagas, S.C.; Weinstein, R.S. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology 2004, 145, 1835–1841. [Google Scholar] [CrossRef] [PubMed]
- Kousteni, S.; Bellido, T.; Plotkin, L.I.; O’Brien, C.A.; Bodenner, D.L.; Han, L.; Han, K.; DiGregorio, G.B.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S.; et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: Dissociation from transcriptional activity. Cell 2001, 104, 719–730. [Google Scholar] [CrossRef]
- Stanislaus, D.; Yang, X.; Liang, J.D.; Wolfe, J.; Cain, R.L.; Onyia, J.E.; Falla, N.; Marder, P.; Bidwell, J.P.; Queener, S.W.; et al. In vivo regulation of apoptosis in metaphyseal trabecular bone of young rats by synthetic human parathyroid hormone (1–34) fragment. Bone 2000, 27, 209–218. [Google Scholar] [CrossRef]
- Gohel, A.; McCarthy, M.B.; Gronowicz, G. Estrogen prevents glucocorticoid-induced apoptosis in osteoblasts in vivo and in vitro. Endocrinology 1999, 140, 5339–5347. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, R.S.; Jilka, R.L.; Parfitt, A.M.; Manolagas, S.C. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J. Clin. Investig. 1998, 102, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, L.I.; Weinstein, R.S.; Parfitt, A.M.; Roberson, P.K.; Manolagas, S.C.; Bellido, T. Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin. J. Clin. Investig. 1999, 104, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Jilka, R.L.; Weinstein, R.S.; Bellido, T.; Roberson, P.; Parfitt, A.M.; Manolagas, S.C. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J. Clin. Investig. 1999, 104, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Tomkinson, A.; Reeve, J.; Shaw, R.W.; Noble, B.S. The death of osteocytes via apoptosis accompanies estrogen withdrawal in human bone. J. Clin. Endocrinol. Metab. 1997, 82, 3128–3135. [Google Scholar] [CrossRef] [PubMed]
- Moriishi, T.; Fukuyama, R.; Miyazaki, T.; Furuichi, T.; Ito, M.; Komori, T. Overexpression of BCLXL in osteoblasts inhibits osteoblast apoptosis and increases bone volume and strength. J. Bone Miner. Res. 2016, 31, 1366–1380. [Google Scholar] [CrossRef] [PubMed]
- Jilka, R.L.; O’Brien, C.A.; Roberson, P.K.; Bonewald, L.F.; Weinstein, R.S.; Manolagas, S.C. Dysapoptosis of osteoblasts and osteocytes increases cancellous bone formation but exaggerates cortical porosity with age. J. Bone Miner. Res. 2014, 29, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, J.; Datta, N.S.; Chun, Y.-H.P.; Yang, D.-Y.; Carey, A.A.; Kreider, J.M.; Goldstein, S.A.; McCauley, L.K. Role of Bcl2 in osteoclastogenesis and PTH anabolic actions in bone. J. Bone Miner. Res. 2007, 23, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Nagase, Y.; Iwasawa, M.; Akiyama, T.; Kadono, Y.; Nakamura, M.; Oshima, Y.; Yasui, T.; Matsumoto, T.; Hirose, J.; Nakamura, H.; et al. Anti-apoptotic molecule Bcl-2 regulates the differentiation, activation, and survival of both osteoblasts and osteoclasts. J. Biol. Chem. 2009, 284, 36659–36669. [Google Scholar] [CrossRef] [PubMed]
- Moriishi, T.; Kawai, Y.; Komori, H.; Rokutanda, S.; Eguchi, Y.; Tsujimoto, Y.; Asahina, I.; Komori, T. Bcl2 deficiency activates foxo through akt inactivation and accelerates osteoblast differentiation. PLoS ONE 2014, 9, e86629. [Google Scholar] [CrossRef] [PubMed]
- Moriishi, T.; Maruyama, Z.; Fukuyama, R.; Ito, M.; Miyazaki, T.; Kitaura, H.; Ohnishi, H.; Furuichi, T.; Kawai, Y.; Masuyama, R.; et al. Overexpression of Bcl2 in osteoblasts inhibits osteoblast differentiation and induces osteocyte apoptosis. PLoS ONE 2011, 6, e27487. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.C.; Liu, Y.; Thant, L.M.; Pang, J.; Palmer, G.; Alikhani, M. Foxo1, a novel regulator of osteoblast differentiation and skeletogenesis. J. Biol. Chem. 2010, 285, 31055–31065. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, M.F.; Flowers, S.; Bhattacharya, R.; Faibish, D.; Behl, Y.; Kotton, D.N.; Gerstenfeld, L.; Moran, E.; Graves, D.T. FOXO1 modulates osteoblast differentiation. Bone 2011, 48, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Ambrogini, E.; Almeida, M.; Martin-Millan, M.; Paik, J.-H.; DePinho, R.A.; Han, L.; Goellner, J.; Weinstein, R.S.; Jilka, R.L.; O’Brien, C.A. FoxO-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell Metab. 2010, 11, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Tzivion, G.; Dobson, M.; Ramakrishnan, G. FoxO transcription factors; regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta 2011, 1813, 1938–1945. [Google Scholar] [CrossRef] [PubMed]
- Tyner, S.D.; Venkatachalam, S.; Choi, J.; Jones, S.; Ghebranious, N.; Igelmann, H.; Lu, X.; Soron, G.; Cooper, B.; Brayton, C.; et al. p53 mutant mice that display early ageing-associated phenotypes. Nature 2002, 415, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Lengner, C.J.; Steinman, H.A.; Gagnon, J.; Smith, T.W.; Henderson, J.E.; Kream, B.E.; Stein, G.S.; Lian, J.B.; Jones, S.N. Osteoblast differentiation and skeletal development are regulated by Mdm2-p53 signaling. J. Cell Biol. 2006, 172, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Sakai, A.; Sakata, T.; Tanaka, S.; Okazaki, R.; Kunugita, N.; Norimura, T.; Nakamura, T. Disruption of the p53 gene results in preserved trabecular bone mass and bone formation after mechanical unloading. J. Bone Miner. Res. 2002, 17, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, R.; Sakai, A.; Ootsuyama, A.; Sakata, T.; Nakamura, T.; Norimura, T. Trabecular bone mass and bone formation are preserved after limb immobilisation in p53 null mice. Ann. Rheum. Dis. 2004, 63, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. p53 functions as a negative regulator of osteoblastogenesis, osteoblast-dependent osteoclastogenesis, and bone remodeling. J. Cell Biol. 2006, 172, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, K.A.; Lanciloti, N.J.; Moore, M.K.; Campione, A.L.; Chandar, N. p53 transactivity during in vitro osteoblast differentiation in a rat osteosarcoma cell line. Mol. Carcinog. 1999, 25, 132–138. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, foxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Marks, S.C.; Odgren, P.R. Structure and Development of the Skeleton; Academic Press: London, UK, 2002; Volume 1, pp. 3–15. [Google Scholar]
- Scemes, E.; Spray, D.C.; Meda, P. Connexins, pannexins, innexins: Novel roles of “hemi-channels”. Pflugers Arch. 2009, 457, 1207–1226. [Google Scholar] [CrossRef] [PubMed]
- Penuela, S.; Gehi, R.; Laird, D.W. The biochemistry and function of pannexin channels. Biochim. Biophys. Acta 2013, 1828, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Chekeni, F.B.; Elliott, M.R.; Sandilos, J.K.; Walk, S.F.; Kinchen, J.M.; Lazarowski, E.R.; Armstrong, A.J.; Penuela, S.; Laird, D.W.; Salvesen, G.S.; et al. Pannexin 1 channels mediate ′find-me′ signal release and membrane permeability during apoptosis. Nature 2010, 467, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, J.A. ATP P2 receptors and regulation of bone effector cells. J. Musculoskelet. Neuronal Interact. 2004, 4, 125–127. [Google Scholar] [PubMed]
- Buckley, K.A.; Hipskind, R.A.; Gartland, A.; Bowler, W.B.; Gallagher, J.A. Adenosine triphosphate stimulates human osteoclast activity via upregulation of osteoblast-expressed receptor activator of nuclear factor-kappa B ligand. Bone 2002, 31, 582–590. [Google Scholar] [CrossRef]
- Korcok, J.; Raimundo, L.N.; Du, X.; Sims, S.M.; Dixon, S.J. P2Y6 nucleotide receptors activate NF-κB and increase survival of osteoclasts. J. Biol. Chem. 2005, 280, 16909–16915. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, I.; Falzoni, S.; Zhang, B.; Pellegatti, P.; di Virgilio, F. The P2X7 receptor and Pannexin-1 are both required for the promotion of multinucleated macrophages by the inflammatory cytokine GM-CSF. J. Immunol. 2011, 187, 3878–3887. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, R.; Locovei, S.; Roque, A.; Alberto, A.P.; Dahl, G.; Spray, D.C.; Scemes, E. P2X7 receptor-pannexin1 complex: Pharmacology and signaling. Am. J. Physiol. Cell Physiol. 2008, 295, C752–C760. [Google Scholar] [CrossRef] [PubMed]
- Noble, B.S.; Reeve, J. Osteocyte function, osteocyte death and bone fracture resistance. Mol. Cell. Endocrinol. 2000, 159, 7–13. [Google Scholar] [CrossRef]
- Cardoso, L.; Herman, B.C.; Verborgt, O.; Laudier, D.; Majeska, R.J.; Schaffler, M.B. Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue. J. Bone Miner. Res. 2009, 24, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Emerton, K.B.; Hu, B.; Woo, A.A.; Sinofsky, A.; Hernandez, C.; Majeska, R.J.; Jepsen, K.J.; Schaffler, M.B. Osteocyte apoptosis and control of bone resorption following ovariectomy in mice. Bone 2010, 46, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Verborgt, O.; Gibson, G.J.; Schaffler, M.B. Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. J. Bone Miner. Res. 2000, 15, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Noble, B. Bone microdamage and cell apoptosis. Eur. Cells Mater. 2003, 6, 46–55; discusssion 55. [Google Scholar] [PubMed]
- Tatsumi, S.; Ishii, K.; Amizuka, N.; Li, M.; Kobayashi, T.; Kohno, K.; Ito, M.; Takeshita, S.; Ikeda, K. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007, 5, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Bursch, W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001, 8, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, O.D.; Laudier, D.M.; Majeska, R.J.; Sun, H.B.; Schaffler, M.B. Osteocyte apoptosis is required for production of osteoclastogenic signals following bone fatigue in vivo. Bone 2014, 64, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.Y.; Fritton, J.C.; Morgan, S.A.; Seref-Ferlengez, Z.; Basta-Pljakic, J.; Thi, M.M.; Suadicani, S.O.; Spray, D.C.; Majeska, R.J.; Schaffler, M.B. Pannexin-1 and P2X7-receptor are required for apoptotic osteocytes in fatigued bone to trigger rankl production in neighboring bystander osteocytes. J. Bone Miner. Res. 2016, 31, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.X. Necrotic death as a cell fate. Genes Dev. 2006, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Messmer, D.; Yang, H.; Telusma, G.; Knoll, F.; Li, J.; Messmer, B.; Tracey, K.J.; Chiorazzi, N. High mobility group box protein 1: An endogenous signal for dendritic cell maturation and Th1 polarization. J. Immunol. 2004, 173, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Svetkauskaite, D.; He, Q.; Kim, J.Y.; Strassheim, D.; Ishizaka, A.; Abraham, E. Involvement of Toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 2004, 279, 7370–7377. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Z.A.; Armour, C.L.; Phipps, S.; Sukkar, M.B. RAGE and TLRs: Relatives, friends or neighbours? Mol. Immunol. 2013, 56, 739–744. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A. Control of RANKL gene expression. Bone 2010, 46, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Bivi, N.; Condon, K.W.; Allen, M.R.; Farlow, N.; Passeri, G.; Brun, L.R.; Rhee, Y.; Bellido, T.; Plotkin, L.I. Cell autonomous requirement of connexin 43 for osteocyte survival: Consequences for endocortical resorption and periosteal bone formation. J. Bone Miner. Res. 2012, 27, 374–389. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Gu, S.; Riquelme, M.A.; Burra, S.; Callaway, D.; Cheng, H.; Guda, T.; Schmitz, J.; Fajardo, R.J.; Werner, S.L.; et al. Connexin 43 channels are essential for normal bone structure and osteocyte viability. J. Bone Miner. Res. 2015, 30, 436–448. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Parron, V.I.; Reece, J.; Zhang, J.Y.; Akiyama, S.K.; French, J.E. BCL2 inhibits cell adhesion, spreading, and motility by enhancing actin polymerization. Cell Res. 2010, 20, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Moriishi, T.; Fukuyama, R.; Ito, M.; Miyazaki, T.; Maeno, T.; Kawai, Y.; Komori, H.; Komori, T. Osteocyte network; a negative regulatory system for bone mass augmented by the induction of rankl in osteoblasts and sost in osteocytes at unloading. PLoS ONE 2012, 7, e40143. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komori, T. Cell Death in Chondrocytes, Osteoblasts, and Osteocytes. Int. J. Mol. Sci. 2016, 17, 2045. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122045
Komori T. Cell Death in Chondrocytes, Osteoblasts, and Osteocytes. International Journal of Molecular Sciences. 2016; 17(12):2045. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122045
Chicago/Turabian StyleKomori, Toshihisa. 2016. "Cell Death in Chondrocytes, Osteoblasts, and Osteocytes" International Journal of Molecular Sciences 17, no. 12: 2045. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122045