
Liver Growth Factor (LGF) Upregulates Frataxin Protein Expression and Reduces Oxidative Stress in Friedreich’s Ataxia Transgenic Mice

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
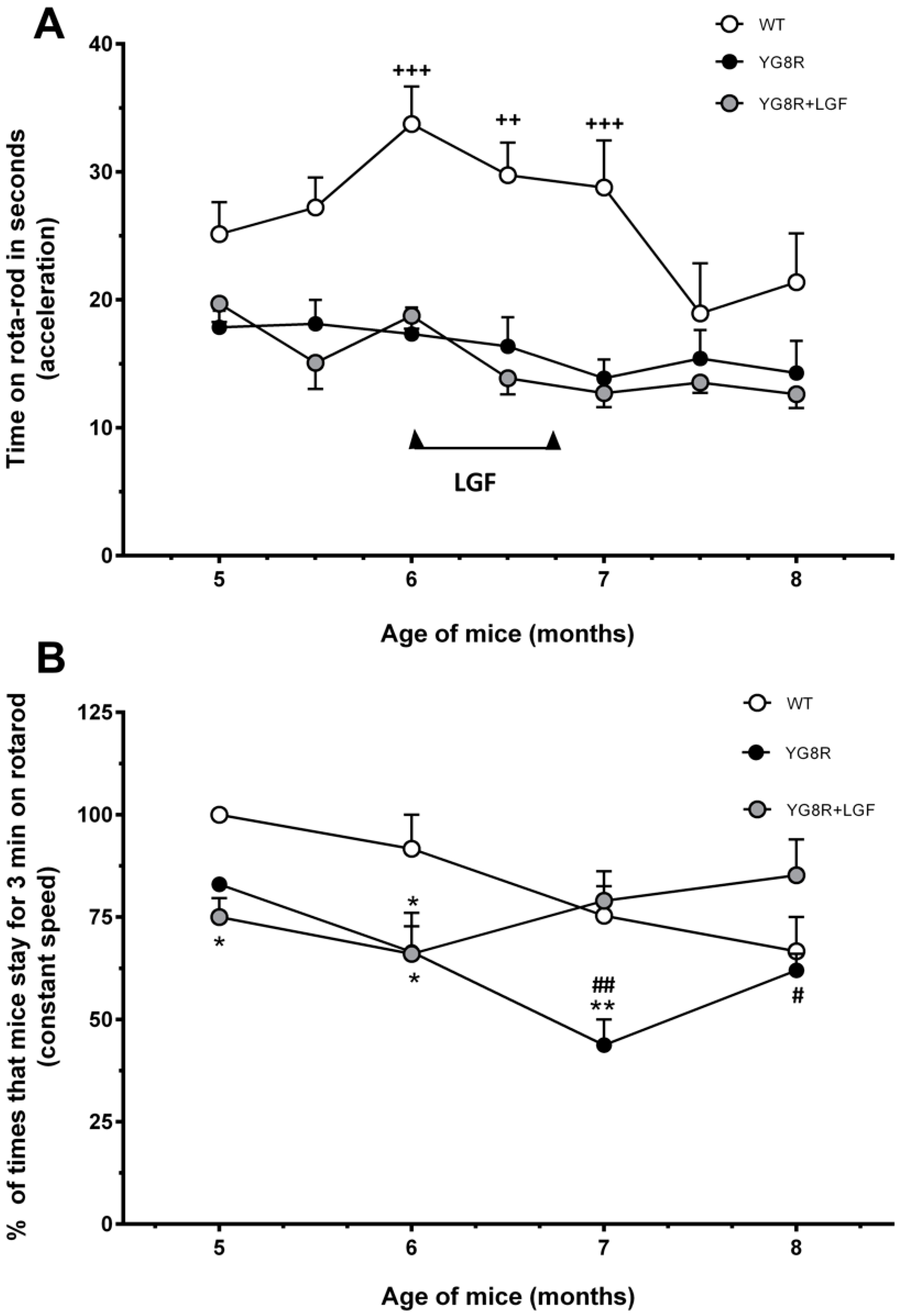
2.1. Liver Growth Factor (LGF) Ameliorates Motor Coordination in YG8R Mice
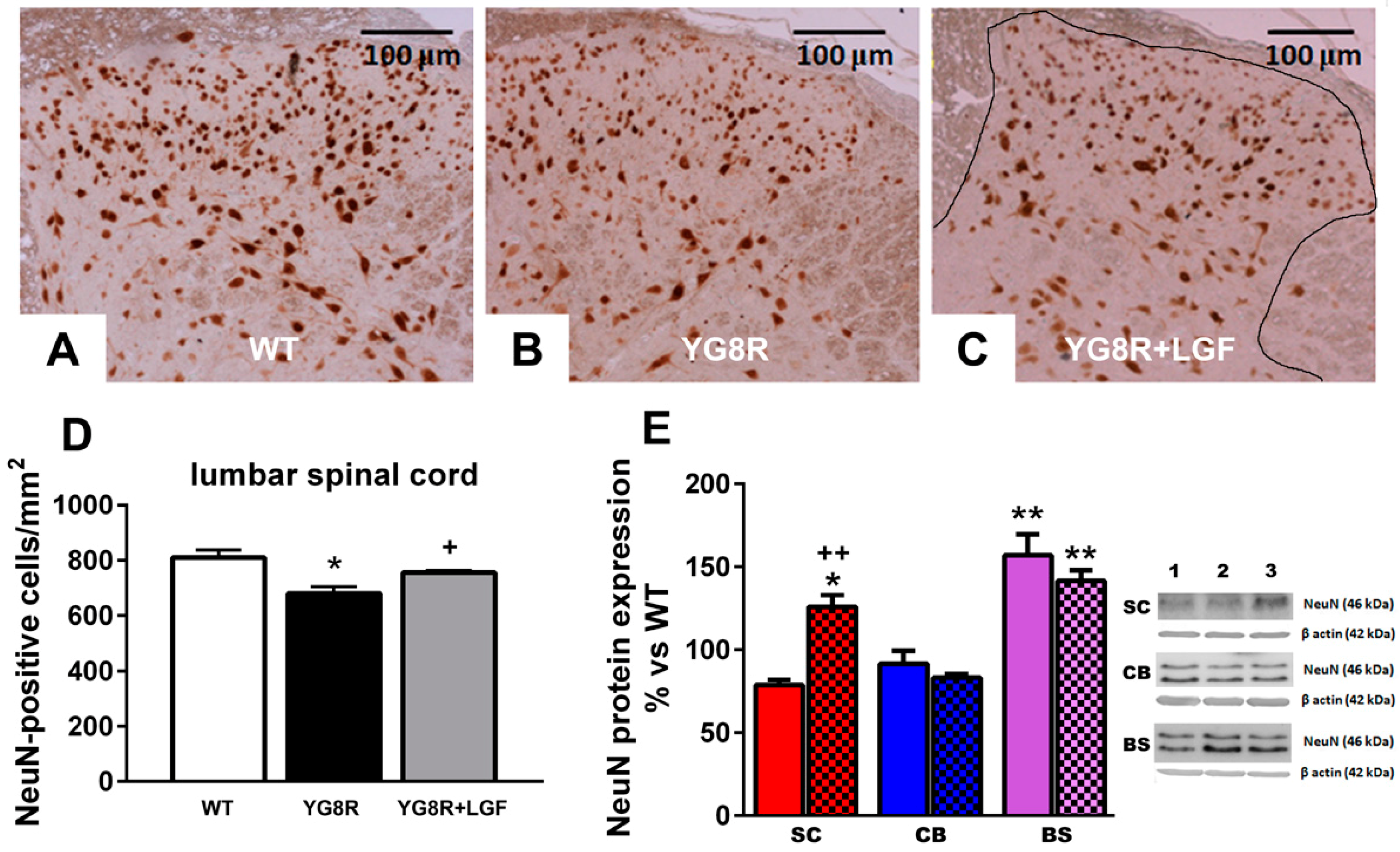
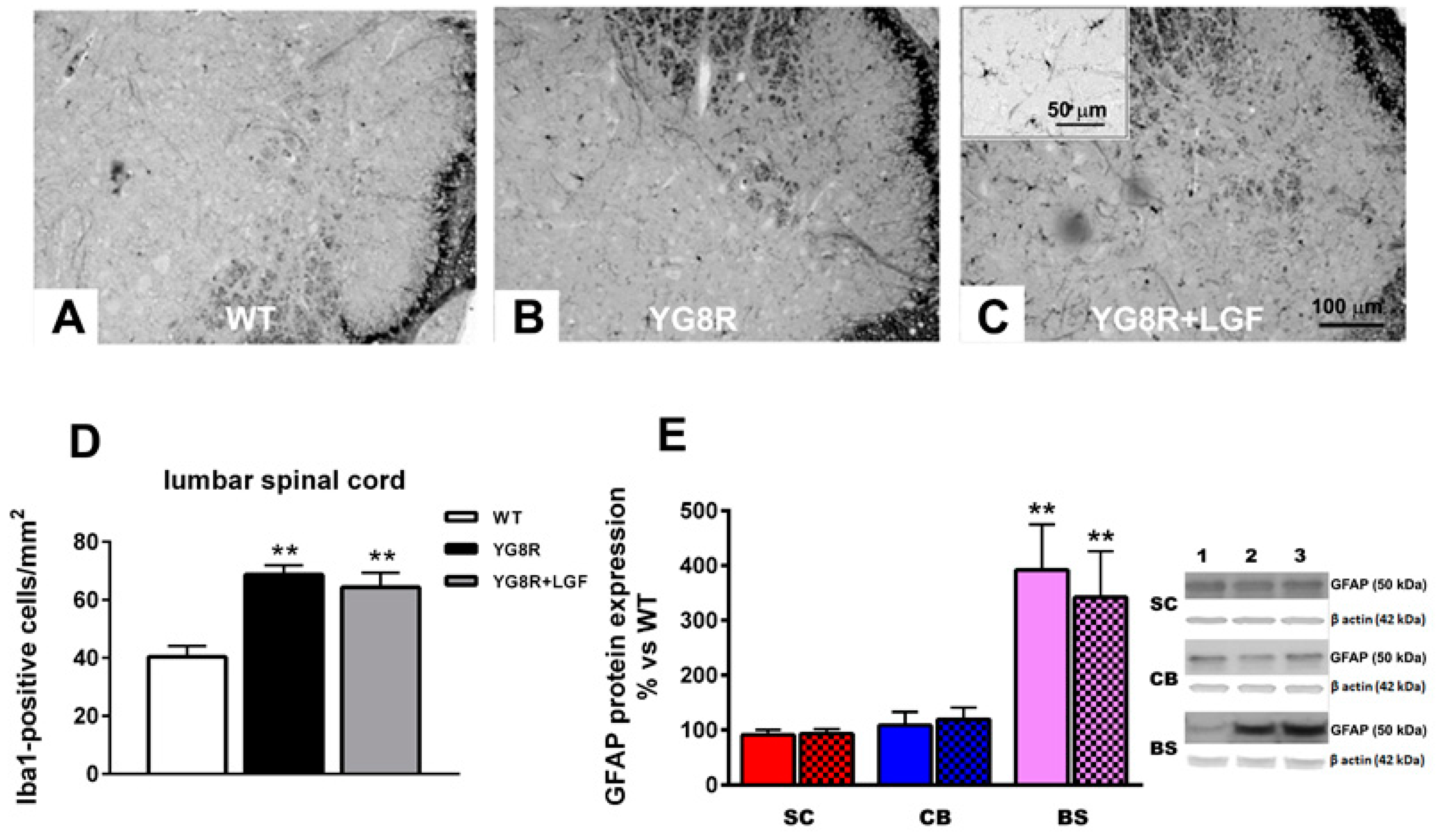
2.2. LGF Prevents Neuronal Lost in Spinal Cord of YG8R Mice
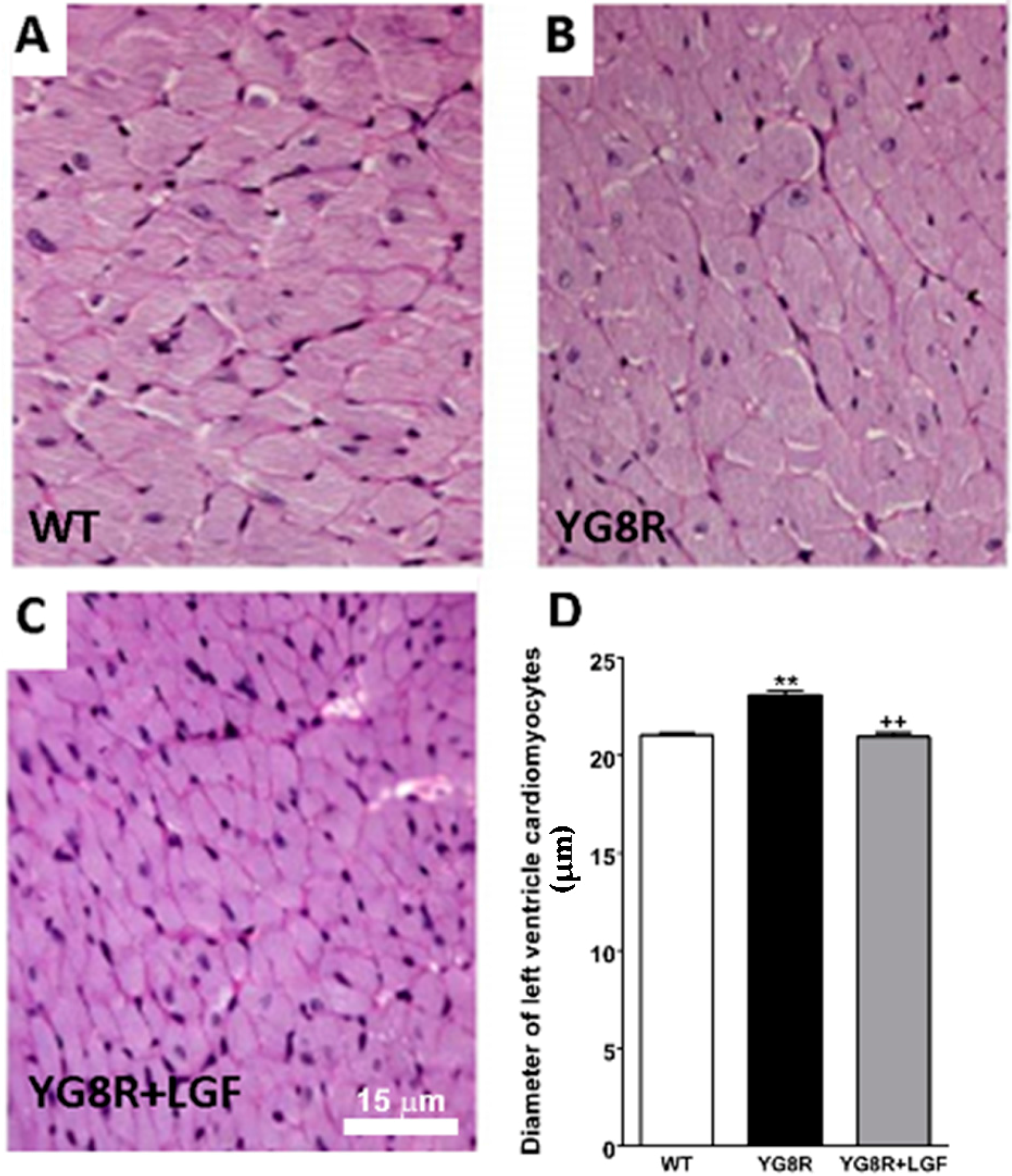
2.3. LGF Treatment Reverses Cardiac Hypertrophy inYG8R Mice
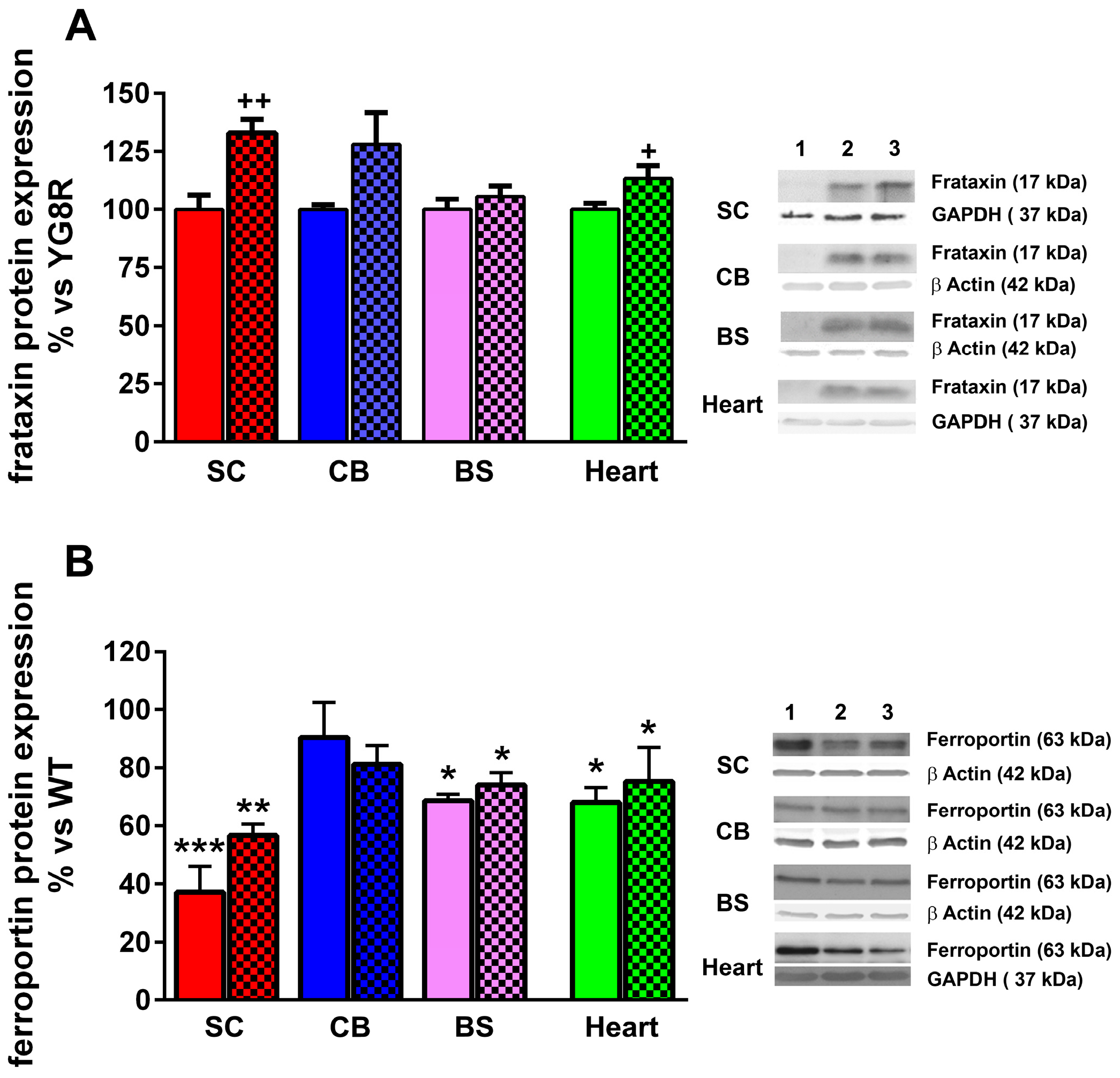
2.4. LGF Treatment Modulates Frataxin Expression in YG8R Mice
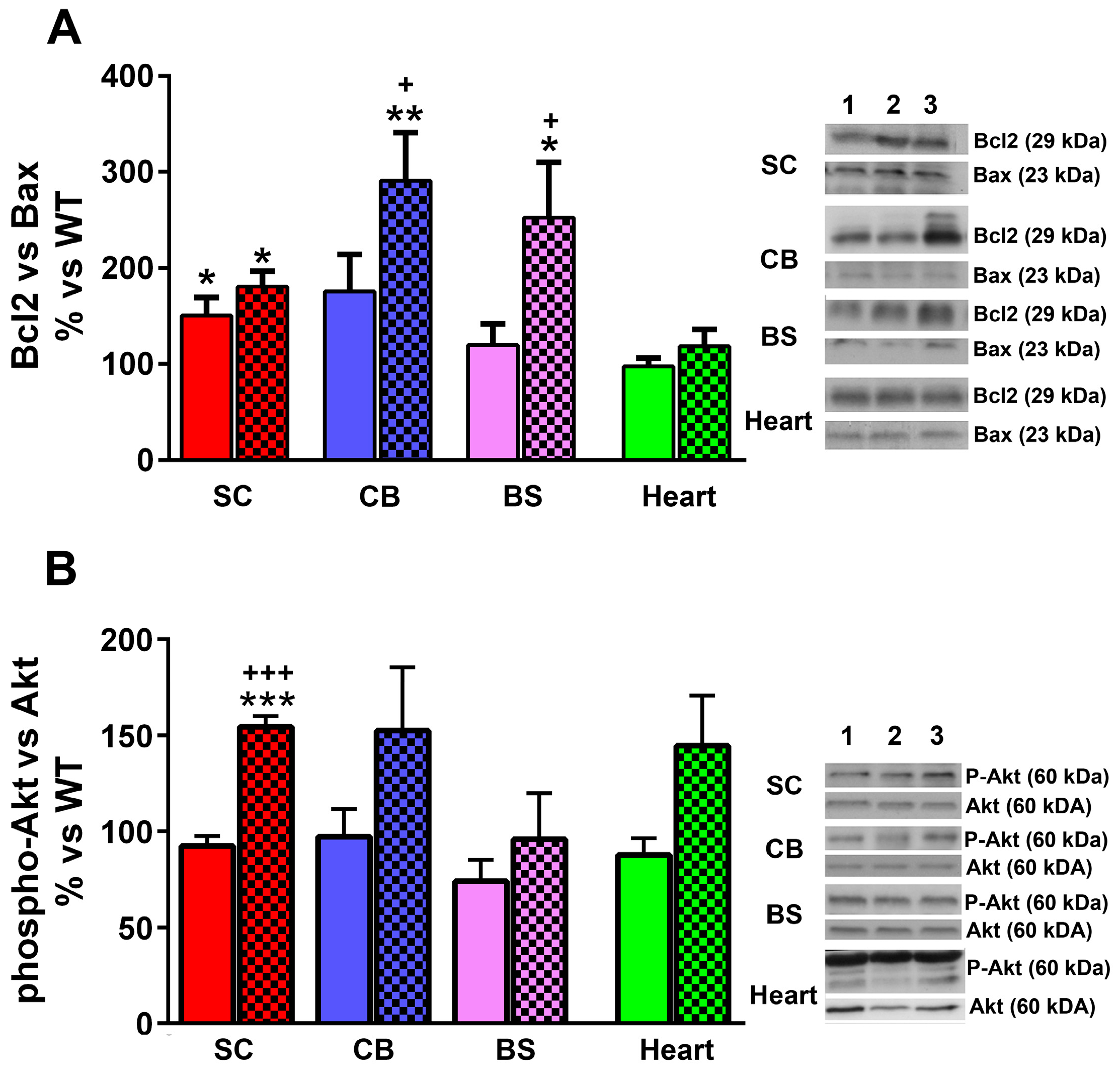
2.5. Effect of LGF Treatment in the Expression of Proteins Involved in Cell Survival
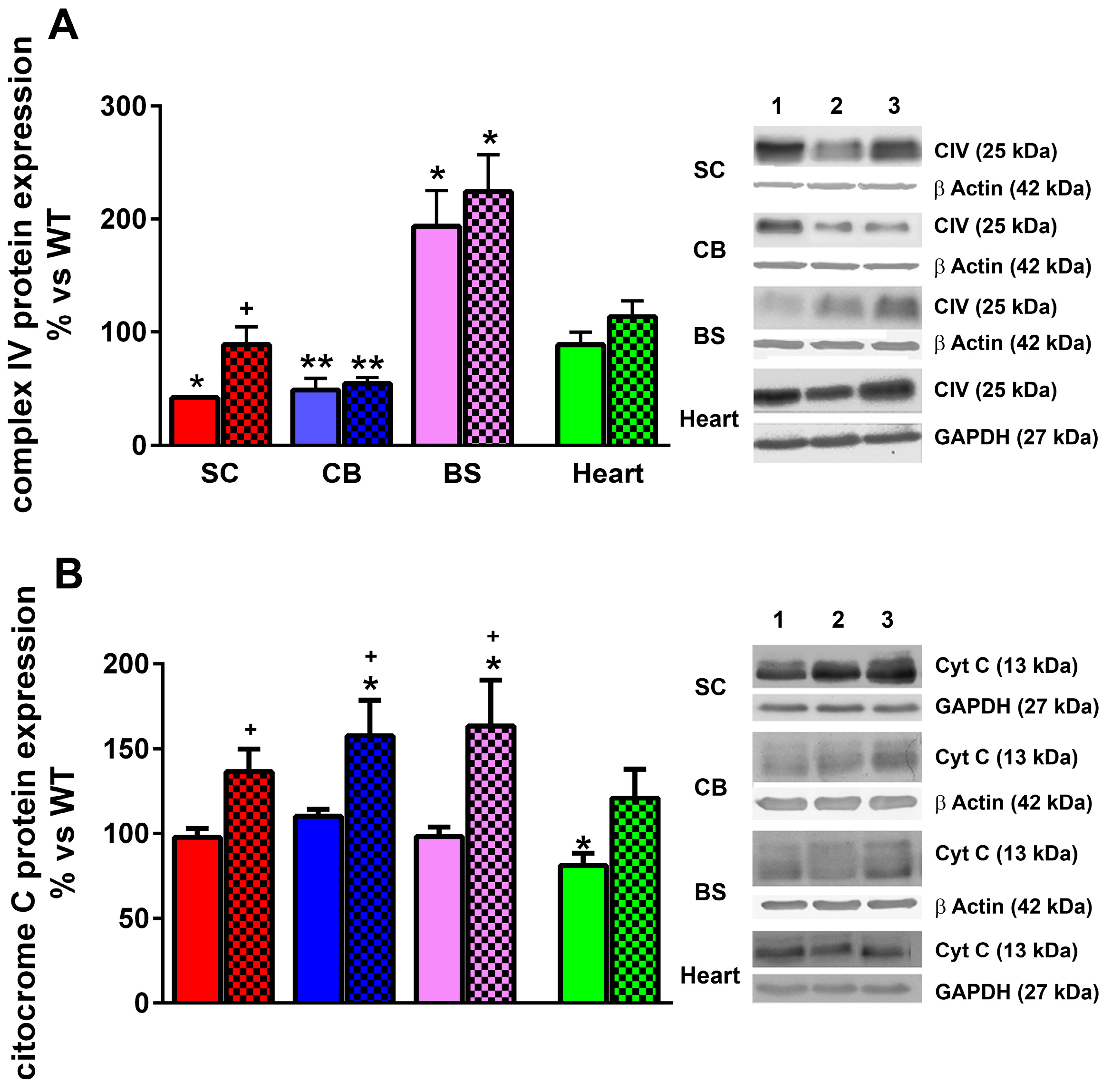
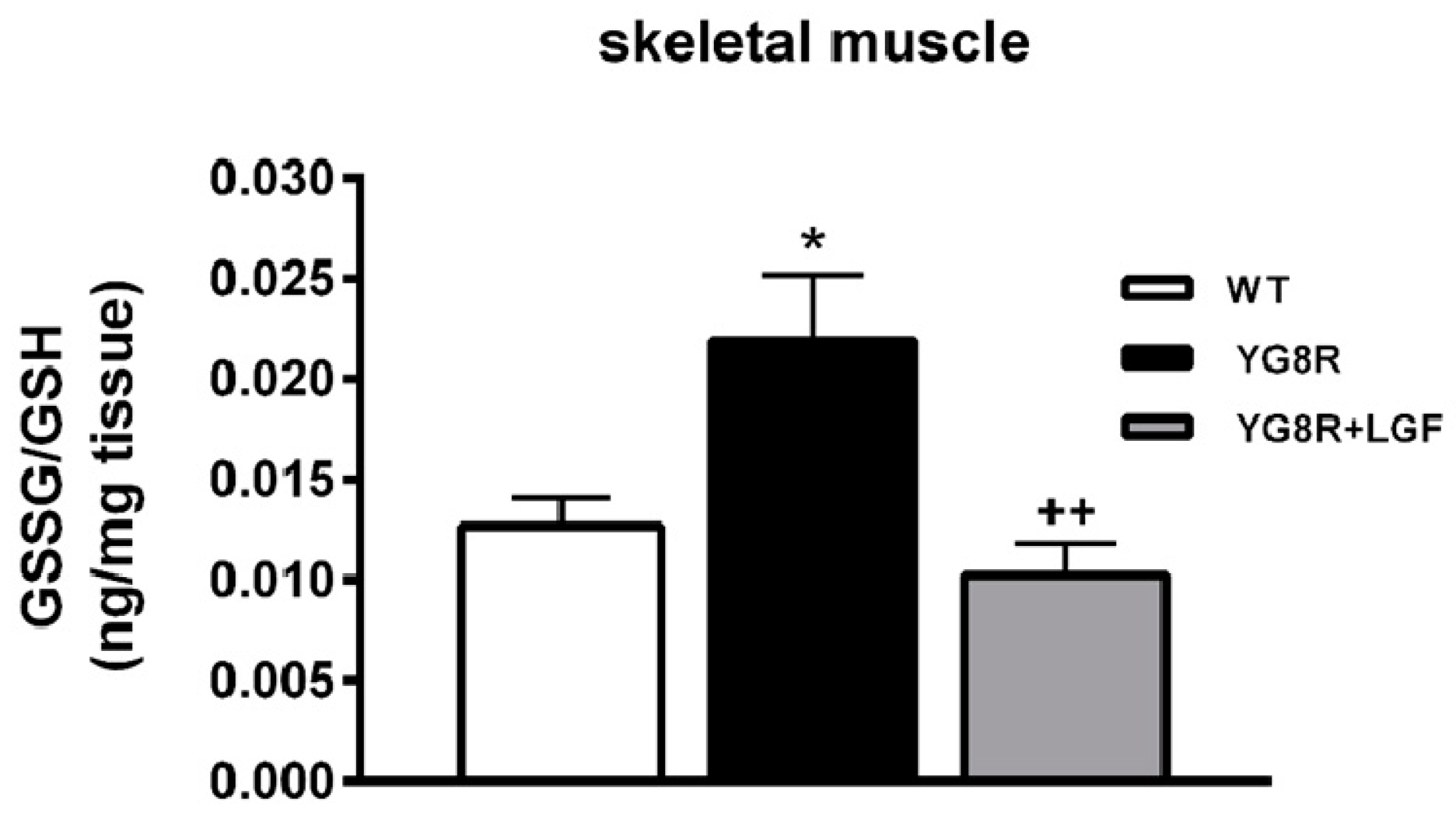
2.6. LGF Treatment Modulates Cytochrome C and Complex IV Protein Expression and Reduces Oxidative Stress in YG8R Mice
3. Discussion
4. Experimental Section
4.1. LGF Purification
4.2. Ethics Statement
4.3. Experimental Model of Friedreich’s Ataxia in Mice
4.4. Behavioral Testing
4.5. LGF Administration
4.6. Tissue Processing
4.7. Antibodies and Immunochemicals (Histology)
4.8. Immunohistochemistry and Morphometric Analysis
4.9. Heart Histological Analysis
4.10. Western Blotting Protein Analysis
4.11. Glutathione Determination
4.12. Data Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Babcock, M.; de Silva, D.; Oaks, R.; Davis-Kaplan, S.; Jiralerspong, S.; Montermini, L.; Pandolfo, M.; Kaplan, J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 1997, 276, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Rotig, A.; de Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 1997, 17, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Babady, N.E.; Carelle, N.; Wells, R.D.; Rouault, T.A.; Hirano, M.; Lynch, D.R.; Delatycki, M.B.; Wilson, R.B.; Isaya, G.; Puccio, H. Advancements in the pathophysiology of Friedreich’s ataxia and new prospects for treatments. Mol. Genet. Metab. 2007, 92, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, M. Drug insight: Antioxidant therapy in inherited ataxias. Nat. Clin. Pract. Neurol. 2008, 4, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Aranca, T.V.; Jones, T.M.; Shaw, J.D.; Staffetti, J.S.; Ashizawa, T.; Kuo, S.H.; Fogel, B.L.; Wilmot, G.R.; Perlman, S.L.; Onyike, C.U.; et al. Emerging therapies in Friedreich’s ataxia. Neurodegener. Dis. Manag. 2016, 6, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Marmolino, D.; Manto, M. Past, present and future therapeutics for cerebellar ataxias. Curr. Neuropharmacol. 2010, 8, 41–61. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Gil, J.J.; Escartin, P.; Garcia-Canero, R.; Trilla, C.; Veloso, J.J.; Sanchez, G.; Moreno-Caparros, A.; Enrique de Salamanca, C.; Lozano, R.; Gavilanes, J.G.; et al. Purification of a liver DNA-synthesis promoter from plasma of partially hepatectomized rats. Biochem. J. 1986, 235, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Gil, J.J.; Gavilanes, J.G.; Sanchez, G.; Garcia-Canero, R.; Garcia-Segura, J.M.; Santamaria, L.; Trilla, C.; Escartin, P. Identification of a liver growth factor as an albumin-bilirubin complex. Biochem. J. 1987, 243, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Gil, J.J.; Sanchez, G.; Trilla, C.; Escartin, P. Identification of biliprotein as a liver growth factor. Hepatology 1988, 8, 484–486. [Google Scholar] [CrossRef] [PubMed]
- Conde, M.V.; Gonzalez, M.C.; Quintana-Villamandos, B.; Abderrahim, F.; Briones, A.M.; Condezo-Hoyos, L.; Regadera, J.; Susin, C.; Gomez de Diego, J.J.; Delgado-Baeza, E.; et al. Liver growth factor treatment restores cell-extracellular matrix balance in resistance arteries and improves left ventricular hypertrophy in SHR. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1153–H1165. [Google Scholar] [CrossRef] [PubMed]
- Diaz Gil, J.J.; Rua, C.; Machin, C.; Cereceda, R.M.; Garcia-Canero, R.; de Foronda, M.; Perez de Diego, J.; Trilla, C.; Escartin, P. Hepatic growth induced by injection of the liver growth factor into normal rats. Growth Regul. 1994, 4, 113–122. [Google Scholar] [PubMed]
- Diaz-Gil, J.J.; Garcia-Monzon, C.; Rua, C.; Martin-Sanz, P.; Cereceda, R.M.; Miquilena-Colina, M.E.; Machin, C.; Fernandez-Martinez, A.; Garcia-Canero, R. Liver growth factor antifibrotic activity in vivo is associated with a decrease in activation of hepatic stellate cells. Histol. Histopathol. 2009, 24, 473–479. [Google Scholar] [PubMed]
- Perez-Crespo, M.; Pericuesta, E.; Perez-Cerezales, S.; Arenas, M.I.; Lobo, M.V.; Diaz-Gil, J.J.; Gutierrez-Adan, A. Effect of liver growth factor on both testicular regeneration and recovery of spermatogenesis in busulfan-treated mice. Reprod. Biol. Endocrinol. 2011, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Somoza, B.; Abderrahim, F.; Gonzalez, J.M.; Conde, M.V.; Arribas, S.M.; Starcher, B.; Regadera, J.; Fernandez-Alfonso, M.S.; Diaz-Gil, J.J.; Gonzalez, M.C. Short-term treatment of spontaneously hypertensive rats with liver growth factor reduces carotid artery fibrosis, improves vascular function, and lowers blood pressure. Cardiovasc. Res. 2006, 69, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo-Gobernado, R.; Calatrava-Ferreras, L.; Perucho, J.; Reimers, D.; Casarejos, M.J.; Herranz, A.S.; Jimenez-Escrig, A.; Diaz-Gil, J.J.; Bazan, E. Liver growth factor as a tissue regenerating factor in neurodegenerative diseases. Recent Pat. CNS Drug Discov. 2014, 9, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo-Gobernado, R.; Calatrava-Ferreras, L.; Reimers, D.; Herranz, A.S.; Rodriguez-Serrano, M.; Miranda, C.; Jimenez-Escrig, A.; Diaz-Gil, J.J.; Bazan, E. Neuroprotective activity of peripherally administered liver growth factor in a rat model of Parkinson’s disease. PLoS ONE 2013, 8, e67771. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo-Gobernado, R.; Reimers, D.; Herranz, A.S.; Diaz-Gil, J.J.; Osuna, C.; Asensio, M.J.; Baena, S.; Rodriguez-Serrano, M.; Bazan, E. Mobilization of neural stem cells and generation of new neurons in 6-OHDA-lesioned rats by intracerebroventricular infusion of liver growth factor. J. Histochem. Cytochem. 2009, 57, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Reimers, D.; Herranz, A.S.; Diaz-Gil, J.J.; Lobo, M.V.; Paino, C.L.; Alonso, R.; Asensio, M.J.; Gonzalo-Gobernado, R.; Bazan, E. Intrastriatal infusion of liver growth factor stimulates dopamine terminal sprouting and partially restores motor function in 6-hydroxydopamine-lesioned rats. J. Histochem. Cytochem. 2006, 54, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Reimers, D.; Osuna, C.; Gonzalo-Gobernado, R.; Herranz, A.S.; Diaz-Gil, J.J.; Jimenez-Escrig, A.; Asensio, M.J.; Miranda, C.; Rodriguez-Serrano, M.; Bazan, E. Liver growth factor promotes the survival of grafted neural stem cells in a rat model of Parkinson’s disease. Curr. Stem Cell Res. Ther. 2012, 7, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Condezo-Hoyos, L.; Abderrahim, F.; Conde, M.V.; Susin, C.; Diaz-Gil, J.J.; Gonzalez, M.C.; Arribas, S.M. Antioxidant activity of liver growth factor, a bilirubin covalently bound to albumin. Free Radic. Biol. Med. 2009, 46, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Condezo-Hoyos, L.; Arribas, S.M.; Abderrahim, F.; Somoza, B.; Gil-Ortega, M.; Diaz-Gil, J.J.; Conde, M.V.; Susin, C.; Gonzalez, M.C. Liver growth factor treatment reverses vascular and plasmatic oxidative stress in spontaneously hypertensive rats. J. Hypertens. 2012, 30, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Calatrava-Ferreras, L.; Gonzalo-Gobernado, R.; Reimers, D.; Herranz, A.S.; Jimenez-Escrig, A.; Diaz-Gil, J.J.; Casarejos, M.J.; Montero-Vega, M.T.; Bazan, E. Neuroprotective role of liver growth factor “LGF” in an experimental model of cerebellar ataxia. Int. J. Mol. Sci. 2014, 15, 19056–19073. [Google Scholar] [CrossRef] [PubMed]
- Al-Mahdawi, S.; Pinto, R.M.; Varshney, D.; Lawrence, L.; Lowrie, M.B.; Hughes, S.; Webster, Z.; Blake, J.; Cooper, J.M.; King, R.; et al. Gaa repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics 2006, 88, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Huang, M.L.; Whitnall, M.; Becker, E.M.; Ponka, P.; Suryo Rahmanto, Y. The ins and outs of mitochondrial iron-loading: The metabolic defect in Friedreich’s ataxia. J. Mol. Med. (Berl.) 2010, 88, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Taroni, F.; Cortopassi, G.A. Frataxin, iron-sulfur clusters, heme, ROS, and aging. Antioxid. Redox Signal. 2006, 8, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Morin, D.; Bernhardt, R.; Buckpitt, A.; Cortopassi, G. Hemin rescues adrenodoxin, heme a and cytochrome oxidase activity in frataxin-deficient oligodendroglioma cells. Biochim. Biophys. Acta 2007, 1772, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; McLellan, L.I. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic. Res. 1999, 31, 273–300. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Schoenfeld, R.A.; Hayashi, G.; Napoli, E.; Akiyama, T.; Iodi Carstens, M.; Carstens, E.E.; Pook, M.A.; Cortopassi, G.A. Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich’s ataxia YG8R mouse model. Antioxid. Redox Signal. 2013, 19, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Mazurkiewicz, J.E. Friedreich ataxia: Neuropathology revised. J. Neuropathol. Exp. Neurol. 2013, 72, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Morral, J.A.; Davis, A.N.; Qian, J.; Petrocine, S.V.; Knutson, M.D.; Gibson, W.M.; Cusack, M.J.; Li, D. The dorsal root ganglion in Friedreich’s ataxia. Acta Neuropathol. 2009, 118, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Schoenfeld, R.; Shan, Y.; Tsai, H.J.; Hammock, B.; Cortopassi, G. Frataxin deficiency induces Schwann cell inflammation and death. Biochim. Biophys. Acta 2009, 1792, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H. Friedreich’s ataxia: Pathology, pathogenesis, and molecular genetics. J. Neurol. Sci. 2011, 303, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, M. Friedreich ataxia: The clinical picture. J. Neurol. 2009, 256 (Suppl. 1), 3–8. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Gil, J.J.; Munoz, J.; Albillos, A.; Rua, C.; Machin, C.; Garcia-Canero, R.; Cereceda, R.M.; Guijarro, M.C.; Trilla, C.; Escartin, P. Improvement in liver fibrosis, functionality and hemodynamics in CCl4-cirrhotic rats after injection of the liver growth factor. J. Hepatol. 1999, 30, 1065–1072. [Google Scholar] [CrossRef]
- Surra, J.C.; Guillen, N.; Barranquero, C.; Arbones-Mainar, J.M.; Navarro, M.A.; Gascon, S.; Arnal, C.; Godino, J.; Guzman, M.A.; Diaz-Gil, J.J.; et al. Sex-dependent effect of liver growth factor on atherosclerotic lesions and fatty liver disease in apolipoprotein E knockout mice. Histol. Histopathol. 2010, 25, 609–618. [Google Scholar] [PubMed]
- Franco, C.; Fernandez, S.; Torres-Aleman, I. Frataxin deficiency unveils cell-context dependent actions of insulin-like growth factor I on neurons. Mol. Neurodegener. 2012, 7, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, R.L.; Qian, J.; Santambrogio, P.; Levi, S.; Koeppen, A.H. Relation of cytosolic iron excess to cardiomyopathy of Friedreich’s ataxia. Am. J. Cardiol. 2012, 110, 1820–1827. [Google Scholar] [CrossRef] [PubMed]
- Heidari, M.M.; Houshmand, M.; Hosseinkhani, S.; Nafissi, S.; Khatami, M. Complex I and ATP content deficiency in lymphocytes from Friedreich’s ataxia. Can. J. Neurol. Sci. 2009, 36, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Llorens, J.V.; Navarro, J.A.; Martinez-Sebastian, M.J.; Baylies, M.K.; Schneuwly, S.; Botella, J.A.; Molto, M.D. Causative role of oxidative stress in a drosophila model of Friedreich ataxia. FASEB J. 2007, 21, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Sutak, R.; Xu, X.; Whitnall, M.; Kashem, M.A.; Vyoral, D.; Richardson, D.R. Proteomic analysis of hearts from frataxin knockout mice: Marked rearrangement of energy metabolism, a response to cellular stress and altered expression of proteins involved in cell structure, motility and metabolism. Proteomics 2008, 8, 1731–1741. [Google Scholar] [CrossRef] [PubMed]
- Bulteau, A.L.; Planamente, S.; Jornea, L.; Dur, A.; Lesuisse, E.; Camadro, J.M.; Auchere, F. Changes in mitochondrial glutathione levels and protein thiol oxidation in yfh1 yeast cells and the lymphoblasts of patients with Friedreich’s ataxia. Biochim. Biophys. Acta 2012, 1822, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.M.; Wilson-Delfosse, A.L.; Mieyal, J.J. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients 2012, 4, 1399–1440. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.M.; Santos, R. Neurodegeneration in Friedreich’s ataxia: From defective frataxin to oxidative stress. Oxid. Med. Cell. Longev. 2013, 2013, 487534. [Google Scholar] [CrossRef] [PubMed]
- Auchere, F.; Santos, R.; Planamente, S.; Lesuisse, E.; Camadro, J.M. Glutathione-dependent redox status of frataxin-deficient cells in a yeast model of Friedreich’s ataxia. Hum. Mol. Genet. 2008, 17, 2790–2802. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Tozzi, G.; Gaeta, L.M.; Bertini, E.; Serafini, V.; di Cesare, S.; Bonetto, V.; Casoni, F.; Carrozzo, R.; Federici, G.; et al. Actin glutathionylation increases in fibroblasts of patients with Friedreich’s ataxia: A potential role in the pathogenesis of the disease. J. Biol. Chem. 2003, 278, 42588–42595. [Google Scholar] [CrossRef] [PubMed]
- Piemonte, F.; Pastore, A.; Tozzi, G.; Tagliacozzi, D.; Santorelli, F.M.; Carrozzo, R.; Casali, C.; Damiano, M.; Federici, G.; Bertini, E. Glutathione in blood of patients with Friedreich’s ataxia. Eur. J. Clin. Investig. 2001, 31, 1007–1011. [Google Scholar] [CrossRef]
- Giron-Martinez, A.; Perez-Rial, S.; Terron-Exposito, R.; Diaz-Gil, J.J.; Gonzalez-Mangado, N.; Peces-Barba, G. Proliferative activity of liver growth factor is associated with an improvement of cigarette smoke-induced emphysema in mice. PLoS ONE 2014, 9, e112995. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Malik, A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006, 86, 279–367. [Google Scholar] [CrossRef] [PubMed]
- Predescu, D.; Vogel, S.M.; Malik, A.B. Functional and morphological studies of protein transcytosis in continuous endothelia. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L895–L901. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Cell activation by glycated proteins. Age receptors, receptor recognition factors and functional classification of ages. Cell. Mol. Biol. (Noisy-le-grand) 1998, 44, 1013–1023. [Google Scholar]
- Carlsson, A.; Lindqvist, M. Effect of ethanol on the hydroxylation of tyrosine and tryptophan in rat brain in vivo. J. Pharm. Pharmacol. 1973, 25, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Tietze, F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: Applications to mammalian blood and other tissues. Anal. Biochem. 1969, 27, 502–522. [Google Scholar] [CrossRef]








© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calatrava-Ferreras, L.; Gonzalo-Gobernado, R.; Reimers, D.; Herranz, A.S.; Casarejos, M.J.; Jiménez-Escrig, A.; Regadera, J.; Velasco-Martín, J.; Vallejo-Muñoz, M.; Díaz-Gil, J.J.; et al. Liver Growth Factor (LGF) Upregulates Frataxin Protein Expression and Reduces Oxidative Stress in Friedreich’s Ataxia Transgenic Mice. Int. J. Mol. Sci. 2016, 17, 2066. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122066
Calatrava-Ferreras L, Gonzalo-Gobernado R, Reimers D, Herranz AS, Casarejos MJ, Jiménez-Escrig A, Regadera J, Velasco-Martín J, Vallejo-Muñoz M, Díaz-Gil JJ, et al. Liver Growth Factor (LGF) Upregulates Frataxin Protein Expression and Reduces Oxidative Stress in Friedreich’s Ataxia Transgenic Mice. International Journal of Molecular Sciences. 2016; 17(12):2066. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122066
Chicago/Turabian StyleCalatrava-Ferreras, Lucía, Rafael Gonzalo-Gobernado, Diana Reimers, Antonio S. Herranz, María J. Casarejos, Adriano Jiménez-Escrig, Javier Regadera, Juan Velasco-Martín, Manuela Vallejo-Muñoz, Juan José Díaz-Gil, and et al. 2016. "Liver Growth Factor (LGF) Upregulates Frataxin Protein Expression and Reduces Oxidative Stress in Friedreich’s Ataxia Transgenic Mice" International Journal of Molecular Sciences 17, no. 12: 2066. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122066