1. Introduction
Neurodevelopmental disabilities, including developmental delay (DD), intellectual disability (ID), and autism spectrum disorder (ASD) affect up to 15% of children [
1]. However, in the vast majority of cases, a child’s clinical presentation does not allow for a definitive etiological diagnosis.
Autism spectrum disorder is characterized by impairment in three domains with onset of one or more of these before the age of 3 years: social interaction; communication skills; and restricted, repetitive, and stereotyped patterns of behavior, interests, and activities. About 40% of individuals with ASD also have a learning disability, and roughly 30% have other co-morbidities such as seizures [
1,
2,
3].
The etiology of ASD is complex and prominently involves genetic factors, including single gene changes, large genomic structural changes (i.e., deletions or duplications) known as copy number variants (CNV), and other polygenic conditions often influenced by the environment and epigenetic changes [
2,
3]. Genetic testing to pinpoint the underlying cause of ASD is critical to an individual’s clinical management. Further, chromosomal microarray analysis (CMA) has demonstrated the highest diagnostic yield for individuals with ASD as compared to other genetic tests. Therefore, along with previously recognized indications of DD, ID and multiple congenital anomalies (MCA), children and adults presenting with ASD should be offered CMA as a first tier genetic evaluation based on the clinical guidelines from multiple professional societies [
2,
3,
4,
5,
6,
7,
8,
9,
10].
Microarrays that employ a variety of designs and range of coverage for certain genomic regions have been applied to the clinical testing of individuals with these conditions. Diagnostic yield has increased over time as such arrays have evolved to include better coverage [
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21]. In 2011 the ACMG issued a guideline regarding optimal microarray design and recommended inclusion of additional probe content in areas of known clinical relevance [
22]. The following data summarizes our experience with real-world clinical CMA testing of individuals with a diagnosis of ASD in a CLIA-certified laboratory over a period of 4.2 years. The microarray platform utilized in this study was specifically designed to increase detection of CNVs in genomic regions of demonstrated relevance to DD/ID/ASD. We also compare our experience to a non-ASD population clinically tested in parallel in the same laboratory and on the same platform.
2. Results
A total of 10,351 custom, ultra-high resolution CMAs optimized for the detection of neurodevelopmental disorders (FirstStepDx PLUS
® (FSDX PLUS
®)) were performed over a period of four years. This testing population had a M:F ratio of 2.5:1 and a mean age of 7.0 years. Based on ICD-9 and ICD-10 codes at the time of referral, 55% of cases represented patients with a diagnosis of ASD with or without other features (ASD+ and ASD only, respectively).
Table 1 and
Table 2 show summary data of our neurodevelopmental patient cohort.
The mean age of testing was younger for the non-ASD group versus the ASD only group or the ASD+ group (
Table 2). Overall, neurologists were the most common referring physicians (36%), followed by developmental pediatricians (31%), pediatricians (16%), and medical geneticists (14%). Although psychiatrists referred only 2% of total cases, they had the highest percentage of their referrals for an indication of ASD (72%) with or without other features, while only 29% of the cases referred by geneticists had an indication including ASD. Of the total caseload, 74% of the ASD cases were referred by pediatric neurologists and developmental/behavioral pediatricians.
Overall, we observe a 28.0% diagnostic yield for potentially abnormal CNVs (
Table 3), with an average of 1.2 reportable CNVs detected per individual. Interestingly, the rate of pathogenic findings is significantly lower (4.4%) when the diagnostic indication is ASD only compared to diagnostic indication of DD/ID/MCA without a reported diagnosis of ASD (non-ASD cohort) (12.5%) (
p < 0.001). The pathogenic rate is only slightly higher in the ASD+ group (6.7%). However, VOUS rates are similar across the ASD only, ASD+, and non-ASD cohorts. The observation of lower rates of reportable CNVs for the ASD only cohort as compared to the ASD+ cohort, and the non-ASD cohort is maintained when looking at the overall yields for each group.
We stratified our cohort by age to determine whether there were differences in the rate of pathogenic findings when data were viewed this way, and found that rates were highest in the non-ASD cohort in the first year of life (18.9%), which then dropped to 10.7%–12.4% during childhood and adolescence (
Table 4). In the ASD cohort, the overall pathogenic rate was slightly higher for individuals with ASD+ as compared to the overall pathogenic rate for individuals with ASD only. The pathogenic rate in the ASD+ cohort started at 4.1% in the youngest group and rose to 8.5% in the 5.5–10 years range (
Table 5). The pathogenic rate in the ASD only cohort rose gradually with age from 3.4% in the youngest cohort (0–3.4 years) to a peak at 7.0% in adolescence (
Table 6).
While largely targeting a pediatric population, a subset of 383 patients comprised adults over 18 years of age at the time of testing (parental and sibling studies excluded). Interestingly, in the non-ASD cohort the pathogenic rate in adults tested was 18.1%, the highest for any age cohort in this population after the first year of life (
Table 4). While the percentage of pathogenic findings in adults with an indication of ASD (9.8% for ASD+; 5.6% for ASD only) was much lower than in the non-ASD population. It is worth noting that older age cohorts maintain high diagnostic yields with or without indications of ASD (
Table 4,
Table 5 and
Table 6).
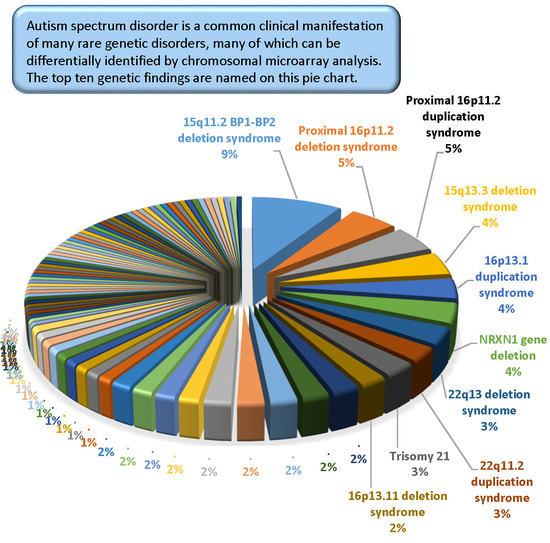
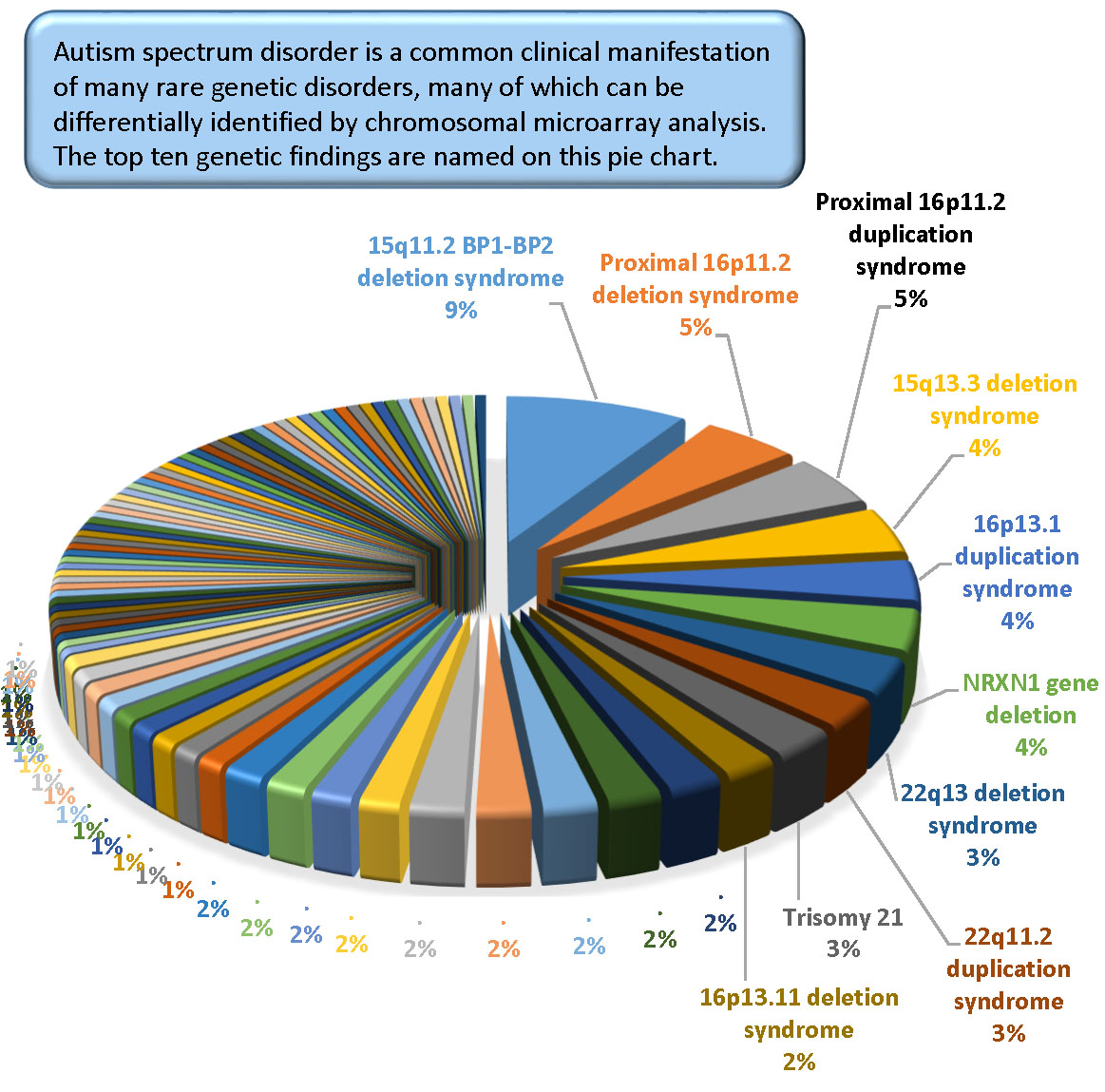
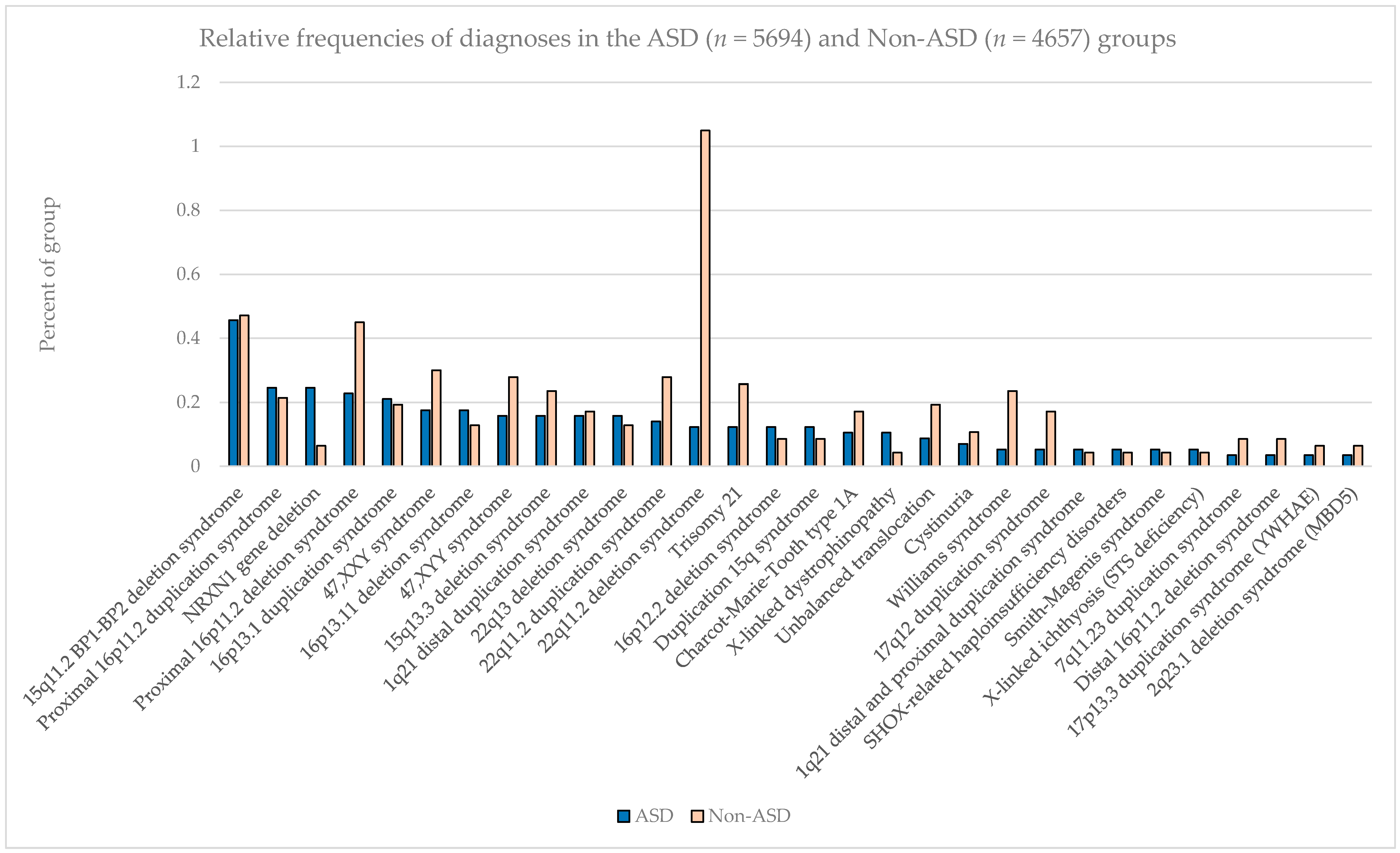
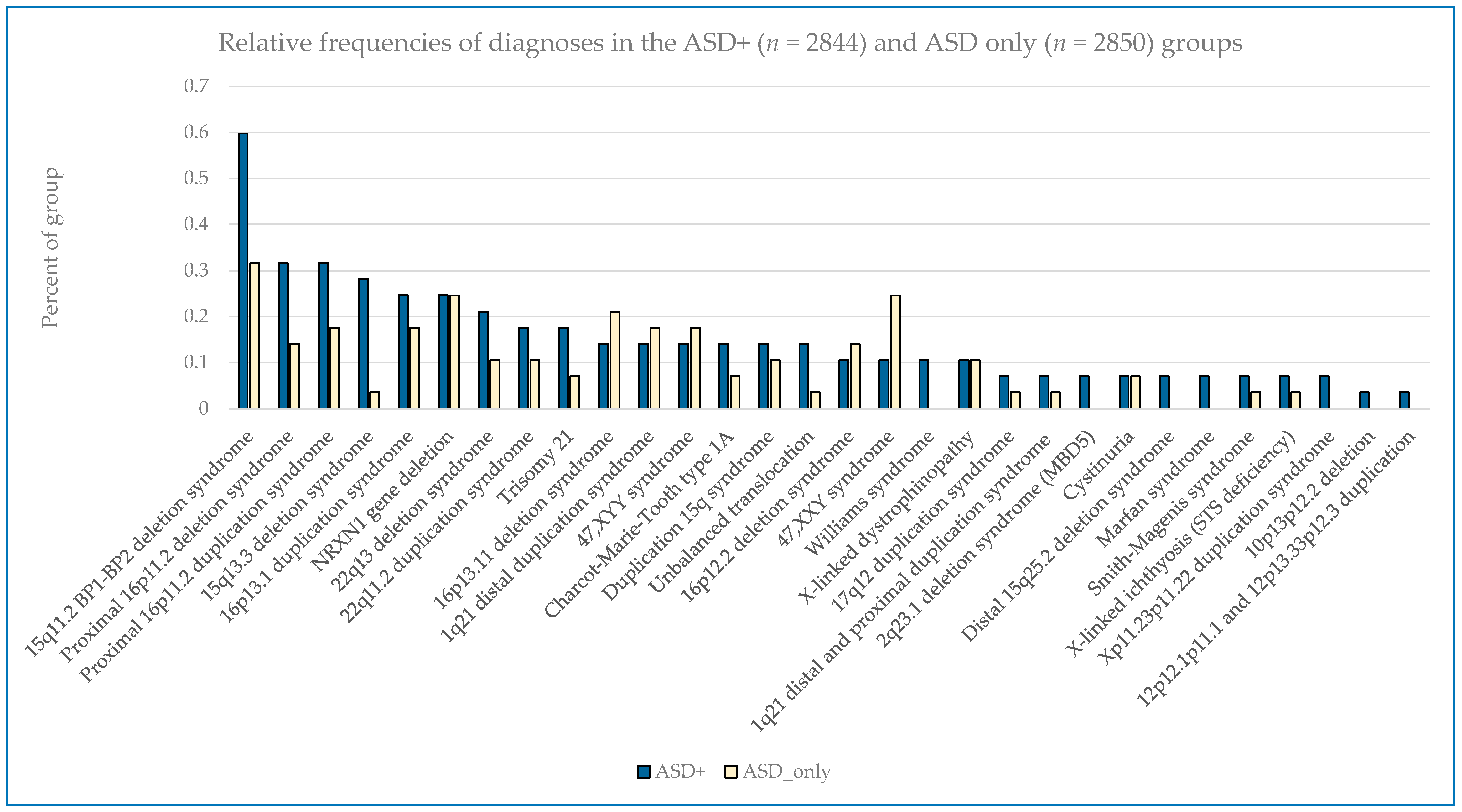
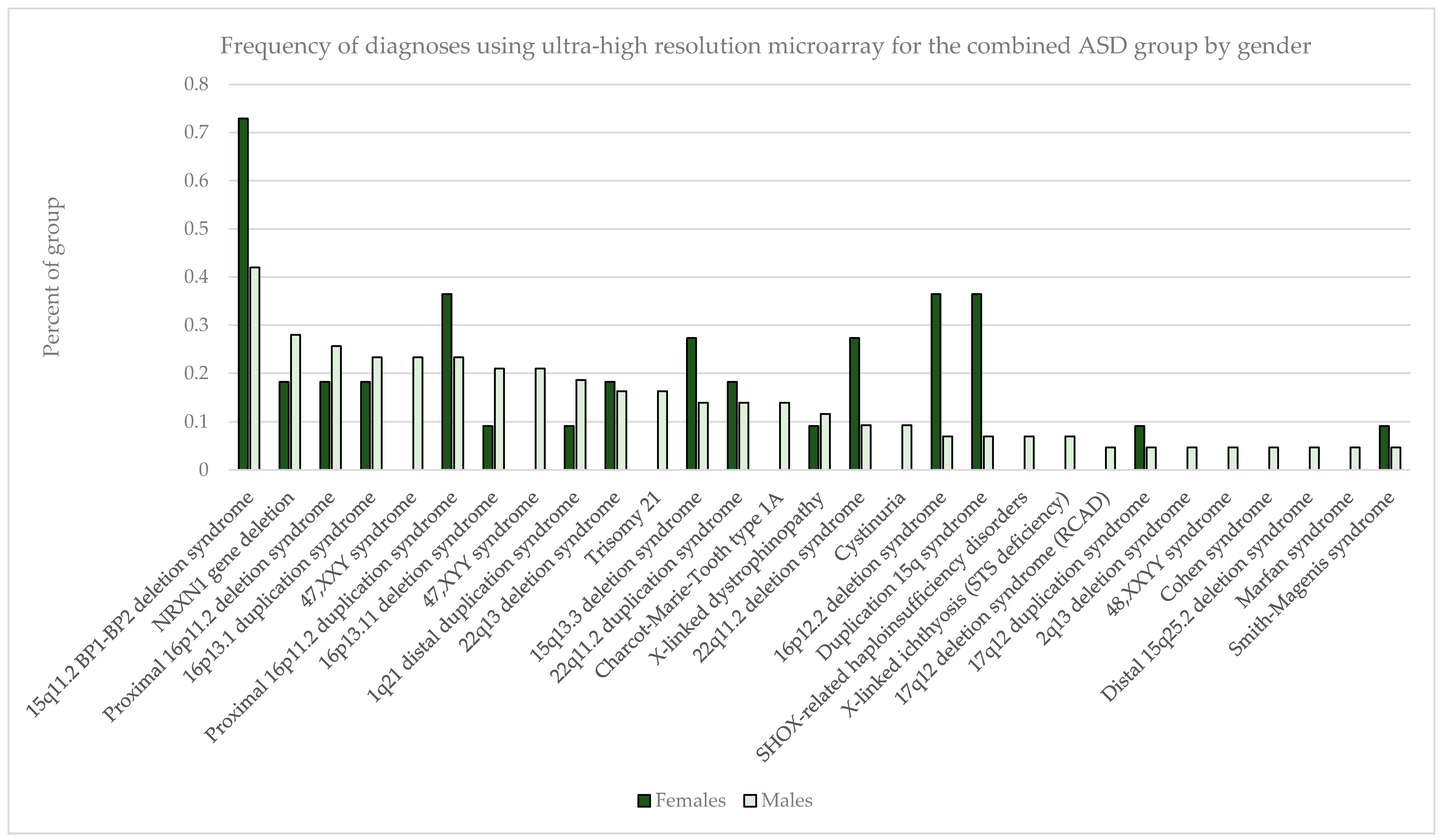
The most common pathogenic findings detected in this series of individuals evaluated by CMA are shown in
Figure 1,
Figure 2 and
Figure 3. We observed, in some cases, striking differences in the frequencies of pathogenic findings when patients are grouped by testing indications and/or gender. For example, the 22q11.2 deletion and, to a lesser extent, the proximal 16p11.2 deletion, were far more prevalent in the non-ASD group than in the combined ASD group, suggesting that indications other than ASD in these patients are common or that ASD is less readily diagnosed in these subgroups (
Figure 1). However, the 15q11.2 BP1–BP2 deletion (also known as the Burnside-Butler susceptibility locus) and the proximal 16p11.2 duplication were equally likely to be detected if ASD was indicated or not (
Figure 1). In contrast,
NRXN1 gene deletions were much more common when ASD was indicated in comparison to when it was not, by a factor of nearly 4-fold (
Figure 1).
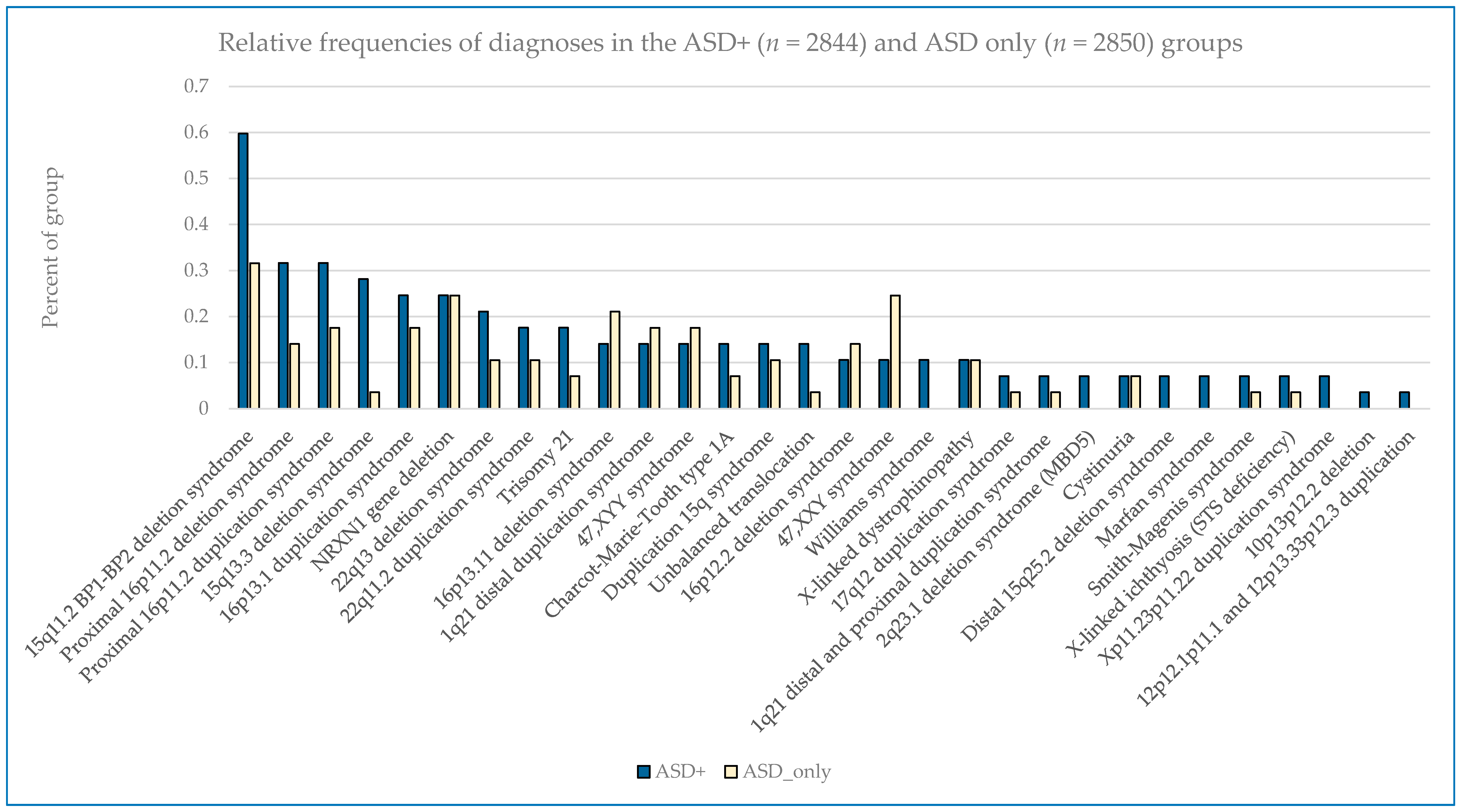
Figure 2 displays some similarities and differences between the ASD+ and ASD only populations; for example the 15q11.2 BP1–BP2 deletion is the most common finding for both cohorts, but detection frequencies vary significantly for other findings, with 47,XXY being the next most common for the ASD-only cohort while the proximal 16p11.2 deletions and duplications are the next most frequent finding in the ASD+ cohort.
Males outnumbered females in our study population, and the rate of abnormality differed significantly with females having higher rates of pathogenic findings across all diagnostic groupings (
p < 0.001 for non-ASD and ASD+ groups as well as overall); ASD only detection rates in females vs. males were not statistically different (
p = 0.22) (
Table 7). In the combined ASD group, in addition to specific sex-limited diagnoses like 47,XXY and 47,XYY, there were excesses by gender in the prevalence of several common abnormalities, notably with the 15q11.2 BP1–BP2 deletion, 15q duplication, 15q13.3 deletion, proximal 16p11.2 duplication, 16p12.2 deletion, and 22q11.2 deletion all skewed toward a female preponderance by up to 2–3-fold (
Figure 3).
3. Discussion
CMA is the guideline-recognized first-tier test in the evaluation of individuals with DD/ID, MCA, and most recently ASD [
4,
6,
7,
8,
9,
10,
15]. CMA yields significant rates of pathogenic or potentially pathogenic (VOUS) results [
2,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20], which have clinical utility for the case-by-case clinical management of individuals with these individually rare disorders [
23,
24,
25,
26,
27,
28,
29,
30,
31,
32,
33,
34].
Since the introduction of CMA technology, the total genomic content with probe coverage has progressively increased leading to higher diagnostic yields and better resolution of chromosomal abnormalities. Collectively this trend has resulted in corresponding increases in the clinical value of CMA testing [
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
24,
25,
26,
27,
28,
29,
30,
31,
32,
33,
34]. In addition to guidelines on the clinical indications for CMA, the American College of Medical Genetics and Genomics (ACMG) has issued guidance on the appropriate content and design of such arrays and specifically opined that, “It is desirable to have enrichment of probes targeting dosage-sensitive genes known to result in phenotypes consistent with common indications for a genomic screen (e.g., intellectual disability, developmental delays, autism, and congenital anomalies)” [
22]. We report here on over four years of clinical experience with a real-world referral base for testing on an ultra-high resolution chromosomal microarray specifically designed to extend the scope of detection for individuals with ASD and other neurodevelopmental disorders. This microarray was optimized through the addition of probes targeting genomic regions more recently identified to have pathogenic relevance for DD, ID, and ASD [
21,
35,
36].
The overall detection rate in this series for clinically established pathogenic CNVs of 8.6% is comparable to other reported series/platforms [
2,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20], despite the inherent bias toward lower rates based on the real-world referral base and higher percentage of individuals with ASD in this population. When ASD is not among the testing indications, the rate of pathogenic findings is 12.5% and the overall diagnostic yield is 32.6%, both of which are at the upper end of reported diagnostic rates. We have previously shown that diagnostic yield varies significantly on a multivariate basis including but not limited to: referring physician specialty, age of patient at testing, patient gender, and referring indication or combination of indications for testing [
20]. When we look in this study at the influence of an ASD diagnosis on pathogenic diagnosis rates by practitioners, geneticists have the highest detection rate when there is no indication of ASD for testing, but also the lowest rate for individuals with ASD only.
The overall detection rate in this series for clinically established pathogenic CNVs of 8.6% is comparable to other reported series/platforms [
2,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20], despite the inherent bias toward lower rates based on the real-world referral base and higher percentage of individuals with ASD in this population. When ASD is not among the testing indications, the rate of pathogenic findings is 12.5% and the overall diagnostic yield is 32.6%, both of which are at the upper end of reported diagnostic rates. We have previously shown that diagnostic yield varies significantly on a multivariate basis including but not limited to: referring physician specialty, age of patient at testing, patient gender, and referring indication or combination of indications for testing [
20]. When we look in this study at the influence of an ASD diagnosis on pathogenic diagnosis rates by practitioners, geneticists have the highest detection rate when there is no indication of ASD for testing, but also the lowest rate for individuals with ASD only.
While significantly lower than both the overall population and the ASD-excluded sub-population (
p < 0.0001), the diagnostic yield in all cases including ASD (ASD only and ASD+) are 5.4% pathogenic, 19.0% VOUS, and 24.4% overall. This overall rate exceeds those previously reported [
3,
14,
15,
16,
17] and this supports the value of incremental targeted content for CMA design.
We detected VOUS at an overall rate of 19.4%. Although earlier literature did not typically consider VOUS in the diagnostic yield, this was due to inconsistent criteria for reporting, lack of established databases of normal population variants, and limited sharing of data [
10]. Today, it is common and reasonable to consider VOUS in the overall diagnostic yield [
3,
14,
15]. Many VOUS results will evolve into clearly pathogenic findings based on emerging clinical evidence [
37]. The excess of males to females in our overall and combined ASD cohorts are consistent with previous reports. In addition, we confirmed the higher rates of abnormalities in the tested female populations in comparison to males, which has been previously observed [
3,
14].
A recent report by one of us (MGB) on CMA use in individuals with DD and ASD at a single midwest genetics center using relatively low resolution (<180 K) oligonucleotide arrays found 6 of 65 patients with ASD (9%) to have a pathogenic finding and 20% overall (13/65) had a reportable CNV. This is higher than the rate of pathogenic findings in our ASD population (6.7% ASD+; 4.4% ASD only); however, this was a much smaller total population and in addition was a closely-studied cohort within an academic medical center where all patients had complete clinical genetics evaluations [
3]. The overall diagnostic yield of 20% in that study was similar to rates reported by Shen et al. [
14] (18.2% in 932 patients with ASD) and Schaefer et al. [
15] (21% or 14 patients 68 with ASD) but is lower than the 24.4% observed with the optimized array in this series.
Numerous studies have now demonstrated the clinical actionability and utility of CMA testing [
23,
24,
25,
26,
27,
28,
38,
39], and increased yield as described here will extend the range and scope of this utility. The increased rate of CNVs classified as VOUS is therefore of potential clinical importance in this setting and consideration of location, gene content, and other factors may help clinicians with such patients despite the complexities of interpreting them and counseling families as to their potential significance. Of critical importance is the ongoing evaluation of novel methods to assess the potential role of VOUS in the underlying pathology of individual patients. This process will allow us as a community to realize the maximum benefit of the increased detection rate achieved through array and interpretation optimization. Furthermore, VOUS results have been clearly demonstrated to be of great importance to parents of patients with DD/ID/ASD [
34,
40,
41,
42,
43].
It is estimated that at least 20% of individuals with ASD have an underlying genetic syndrome, but a survey of a large autism center showed that less than 10% of their population had received any form of genetic evaluation [
44]. The evidence here also supports that patients with a diagnosis of ASD remain under-tested overall. The fact that the age of CMA diagnosis in those with ASD is a full standard deviation greater than the potential age of clinical diagnosis speaks to the delay or reticence in taking critical steps to better medically manage these patients. The direct correlation between higher rate of detected abnormalities and age in the ASD cohort suggests that earlier use of CMA and perhaps other genetic testing methods may be important for early intervention.
While still a relatively small sub-cohort, it is remarkable that adults (>18 years old) tested also have the highest pathogenic CNV rate of all diagnostic groups examined. This could be reflective of severity in that particular age group. For example, clinicians/families might believe that testing isn’t as valuable for adults but perform it anyway when the individual is considered to be relatively severely impaired. In addition, this may also reflect the desire for adults (or adult siblings of the individual with clinical features) to define recurrence risk to their potential offspring.
Even before prior CMA was introduced into clinical use, the most common chromosome abnormalities associated with apparently isolated ASD were duplication of the 15q11–q13 region (typically of maternal origin) [
45] and large microdeletions in the chromosome 16p11.2 and 22q regions reportedly accounting for as many as 1%–5% of ASD related abnormalities each [
3,
23]. Although these well-described recurrent abnormalities were prominent and relatively more abundant in our ASD cohorts, their prevalence was not as high as predicted by the literature; however, some of the reports suggesting high rates may have been biased by multiplex families.
Partial deletions involving the
NRXN1 gene are now well-described abnormalities, and impairment of the function of the synaptic adhesion protein it encodes, leading to a potential loss of synaptic integrity, is thought to be central to the pathogenesis of ASD [
46,
47].
NRXN1 gene deletions were significantly over-represented in both our ASD+ and ASD only groups but were also observed at least occasionally in the non-ASD group. The latter may be due to either early testing for some other clinical feature prior to the formal recognition of ASD-related features, the indubitable inadequacies of relying on physician coding on test requisitions for phenotypic data, or true overlap into other neurodevelopmental conditions without ASD.
The observational study of a large consecutive series of genetic testing for neurodevelopmental disorders, over half of which had feature of ASD highlights the significant value of CMA in defining not merely the underlying etiology but in directing future research into the underlying pathophysiology for improved and ultimately targeted treatments. While it is not ideal to rely on ICD-9/10 coding on test requisitions to define the phenotypic sub-groups of this population and parse the results relevant to ASD, the comparability and trends of this large data set suggest that the conclusions are neither random nor merely directional, and likely reflects a reasonable picture of the scope of abnormalities in these populations. Improved diagnostic tools will lead to increased clinical utility and in the end better clinical management.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}