
A Novel Pathogenic BRCA1 Splicing Variant Produces Partial Intron Retention in the Mature Messenger RNA

 and
and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
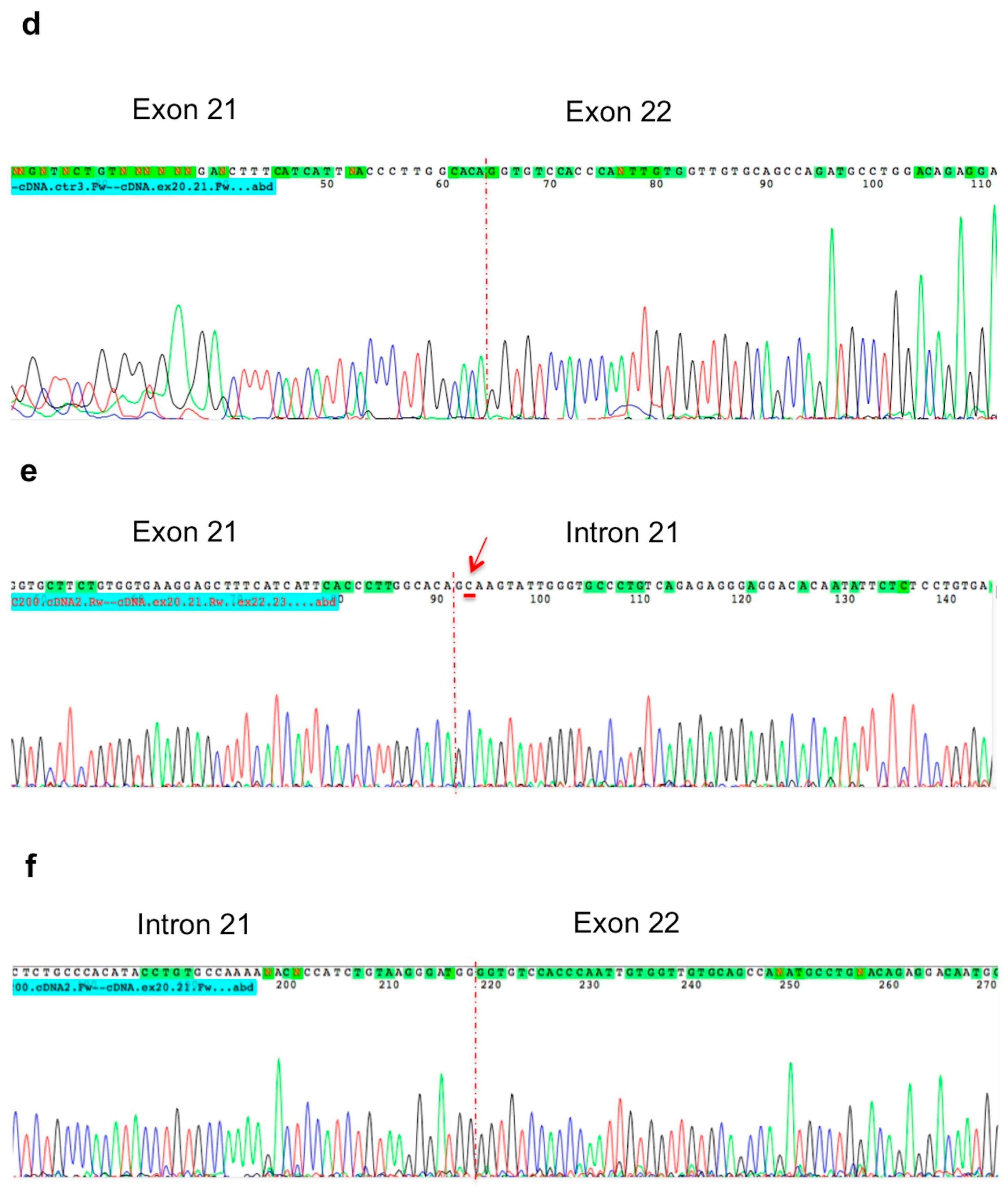
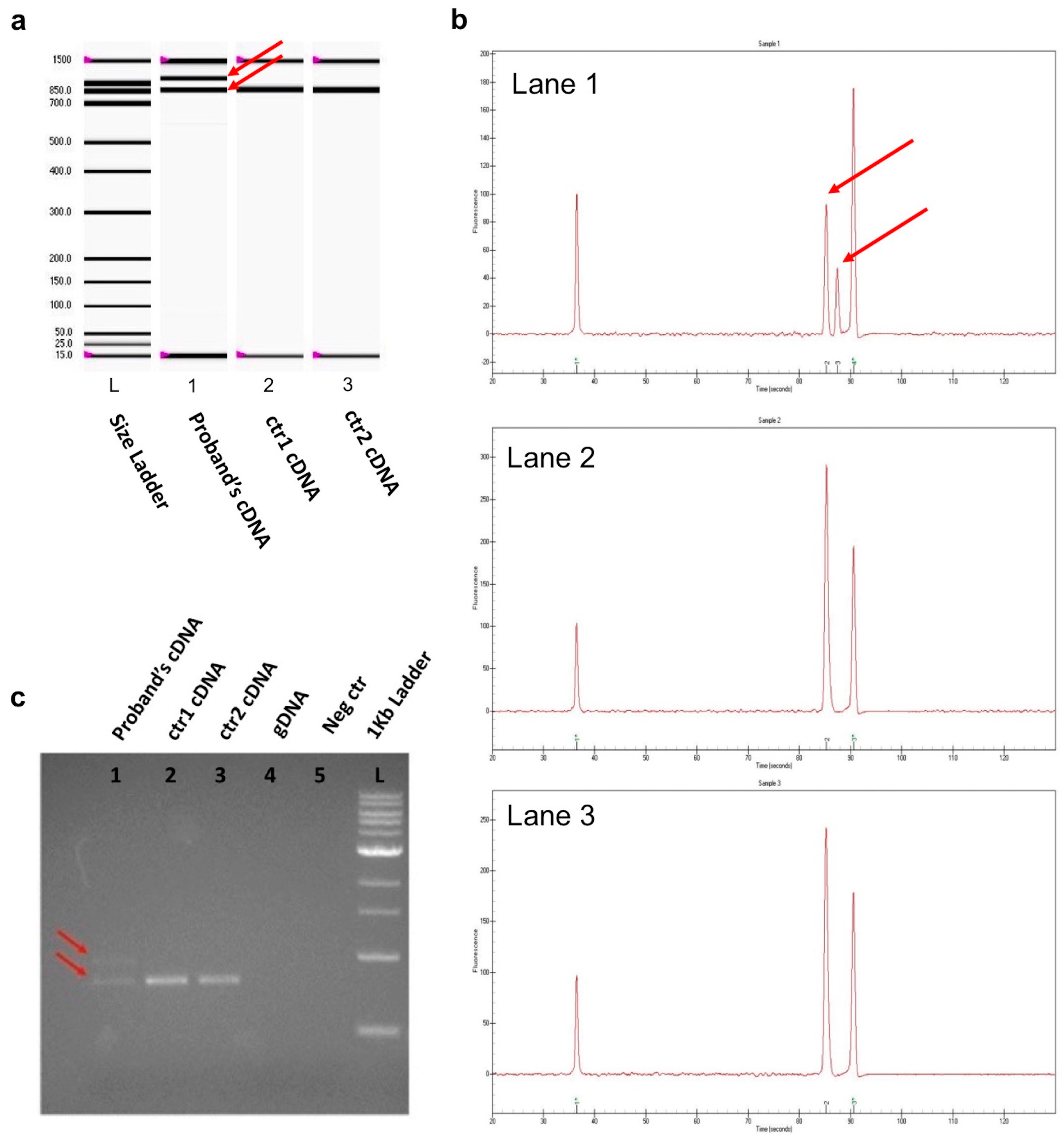
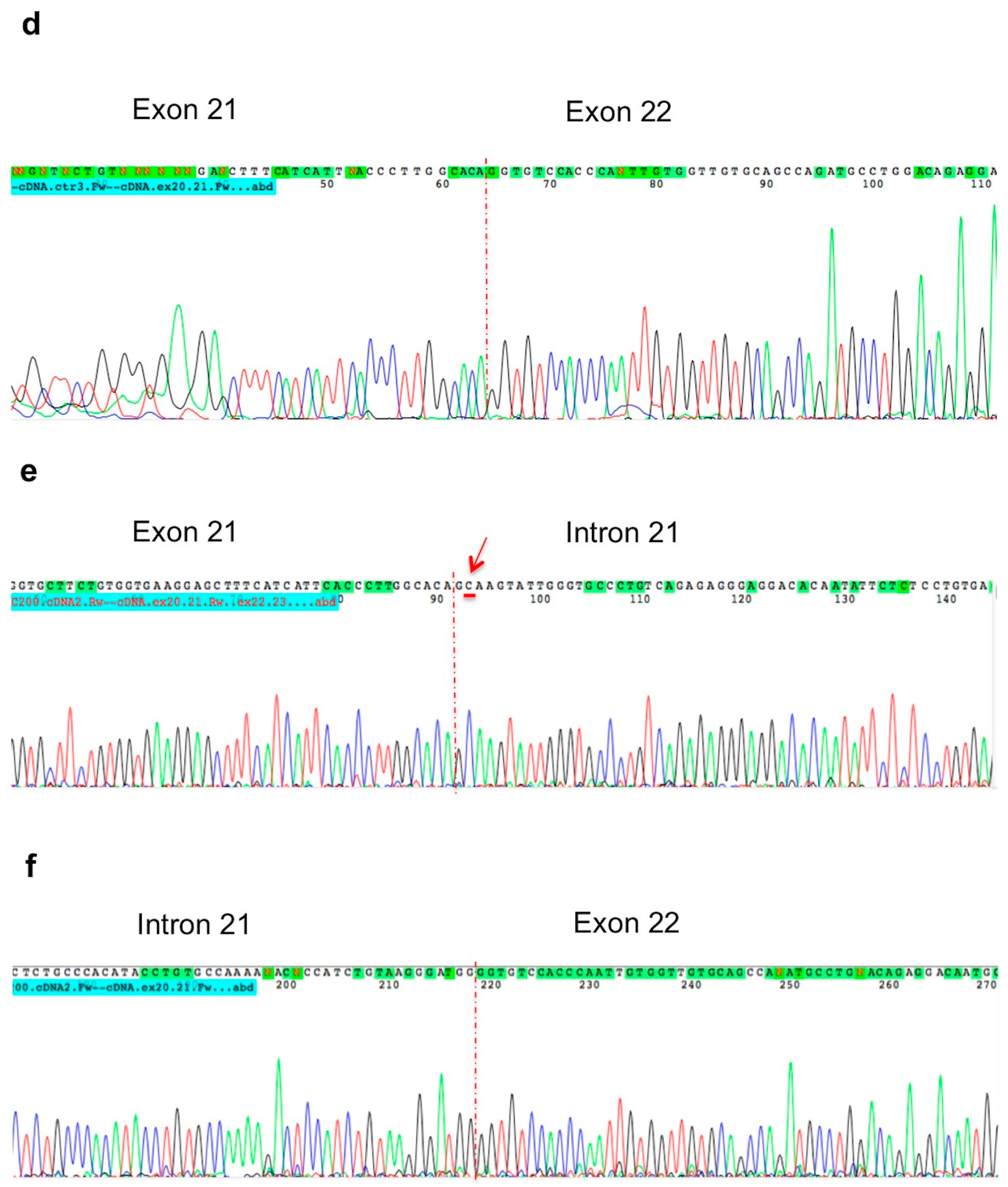
2. Results
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. DNA Extraction and Molecular Screening of the BRCA Genes
4.3. Bioinformatic Analysis
4.4. RNA Extraction and RT-PCR Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| HBOCs | Hereditary breast and ovarian cancers |
| BIC | Breast Cancer Information Core database |
| UV | Unclassified Variants |
| NGS | Next Generation Sequencing |
References
- Kobayashi, H.; Ohno, S.; Sasaki, Y.; Matsuura, M. Hereditary breast and ovarian cancer susceptibility genes. Oncol. Rep. 2013, 30, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.J.; Thomassen, M.; Gerdes, A.M.; Kruse, T.A. Hereditary breast cancer: Clinical, pathological and molecular characteristics. Breast Cancer 2014, 8, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Narod, S.A. BRCA mutations in the management of breast cancer: The state of the art. Nat. Rev. Clin. Oncol. 2010, 7, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Engel, C.; Fischer, C. Breast cancer risks and risk prediction models. Breast Care 2015, 10, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Cheon, J.Y.; Mozersky, J.; Cook-Deegan, R. Variants of uncertain significance in BRCA: A harbinger of ethical and policy issues to come? Genome Med. 2014, 6, 121. [Google Scholar] [CrossRef] [PubMed]
- D′Argenio, V.; Esposito, M.V.; Gilder, J.A.; Frisso, G.; Salvatore, F. Should a BRCA2 stop codon human variant, usually considered a polymorphism, be classified as a predisposing mutation? Cancer 2014, 12, 1594–1595. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.; Haroun, I.; Graham, T.C.; Eisen, A.; Kiss, A.; Warner, E. Variants of unknown significance in BRCA testing: Impact on risk perception, worry, prevention and counseling. Ann. Oncol. 2013, 24 (Suppl. 8), viii69–viii74. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.; Krieger, S.; Vezain, M.; Rousselin, A.; Tournier, I.; Martins, A.; Berthet, P.; Chevrier, A.; Dugast, C.; Layet, V.; et al. Screening BRCA1 and BRCA2 unclassified variants for splicing mutations using reverse transcription PCR on patient RNA and an ex vivo assay based on a splicing reporter minigene. J. Med. Genet. 2008, 45, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Acedo, A.; Sanz, D.J.; Durán, M.; Infante, M.; Pérez-Cabornero, L.; Miner, C.; Velasco, E.A. Comprehensive splicing functional analysis of DNA variants of the BRCA2 gene by hybrid minigenes. Breast Cancer Res. 2012, 14, R87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Garibay, G.R.; Acedo, A.; García-Casado, Z.; Gutiérrez-Enríquez, S.; Tosar, A.; Romero, A.; Garre, P.; Llort, G.; Thomassen, M.; Díez, O.; et al. Capillary electrophoresis analysis of conventional splicing assays: IARC analytical and clinical classification of 31 BRCA2 genetic variants. Hum. Mutat. 2014, 35, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Houdayer, C.; Dehainault, C.; Mattler, C.; Michaux, D.; Caux-Moncoutier, V.; Pagès-Berhouet, S.; d’Enghien, C.D.; Laugé, A.; Castera, L.; Gauthier-Villars, M.; et al. Evaluation of in silico splice tools for decision-making in molecular diagnosis. Hum. Mutat. 2008, 29, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Vreeswijk, M.P.; Kraan, J.N.; van der Klift, H.M.; Vink, G.R.; Cornelisse, C.J.; Wijnen, J.T.; Bakker, E.; van Asperen, C.J.; Devilee, P. Intronic variants in BRCA1 and BRCA2 that affect RNA splicing can be reliably selected by splice-site prediction programs. Hum. Mutat. 2009, 30, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Théry, J.C.; Krieger, S.; Gaildrat, P.; Révillion, F.; Buisine, M.P.; Killian, A.; Duponchel, C.; Rousselin, A.; Vaur, D.; Peyrat, J.P.; et al. Contribution of bioinformatics predictions and functional splicing assays to the interpretation of unclassified variants of the BRCA genes. Eur. J. Hum. Genet. 2011, 19, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Pagani, F.; Baralle, F.E. Genomic variants in exons and introns: Identifying the splicing spoilers. Nat. Rev. Genet. 2004, 5, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.J.; Cooper, T.A. The pathobiology of splicing. J. Pathol. 2010, 220, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, A.Y.; Dandanell, M.; Jønson, L.; Ejlertsen, B.; Gerdes, A.M.; Nielsen, F.C.; Hansen, T.V. Functional characterization of BRCA1 gene variants by mini-gene splicing assay. Eur. J. Hum. Genet. 2014, 22, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Pagani, F.; Buratti, E.; Stuani, C.; Bendix, R.; Dörk, T.; Baralle, F.E. A new type of mutation causes a splicing defect in ATM. Nat. Genet. 2002, 30, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, M.; de Conti, L.; Baralle, D.; Romano, M.; Ayala, Y.M.; Baralle, F.E. hnRNP H binding at the 5′ splice site correlates with the pathological effect of two intronic mutations in the NF-1 and TSHβ genes. Nucleic Acids Res. 2004, 32, 4224–4236. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Chun, J.; Powel, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.J.; Au, A.Y.; Ritchie, W.; Rasko, J.E. Intron retention in mRNA: No longer nonsense: Known and putative roles of intron retention in normal and disease biology. Bioessays 2016, 38, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Liede, A.; Karlan, B.Y.; Narod, S.A. Cancer risks for male carriers of germline mutations in BRCA1 or BRCA2: A review of the literature. J. Clin. Oncol. 2004, 22, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Eswaran, J.; Horvath, A.; Godbole, S.; Reddy, S.D.; Mudvari, P.; Ohshiro, K.; Cyanam, D.; Nair, S.; Fuqua, S.A.; Polyak, K.; et al. RNA sequencing of cancer reveals novel splicing alterations. Sci. Rep. 2013, 3, 1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Argenio, V.; Esposito, M.V.; Telese, A.; Precone, V.; Starnone, F.; Nunziato, M.; Cantiello, P.; Iorio, M.; Evangelista, E.; D’Aiuto, M.; et al. The molecular analysis of BRCA1 and BRCA2: Next-generation sequencing supersedes conventional approaches. Clin. Chim. Acta 2015, 446, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Precone, V.; del Monaco, V.; Esposito, M.V.; de Palma, F.D.; Ruocco, A.; Salvatore, F.; D’Argenio, V. Cracking the Code of Human Diseases Using Next-Generation Sequencing: Applications, Challenges, and Perspectives. BioMed Res. Int. 2015, 2015, 161648. [Google Scholar] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esposito, M.V.; Nunziato, M.; Starnone, F.; Telese, A.; Calabrese, A.; D’Aiuto, G.; Pucci, P.; D’Aiuto, M.; Baralle, F.; D’Argenio, V.; et al. A Novel Pathogenic BRCA1 Splicing Variant Produces Partial Intron Retention in the Mature Messenger RNA. Int. J. Mol. Sci. 2016, 17, 2145. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122145
Esposito MV, Nunziato M, Starnone F, Telese A, Calabrese A, D’Aiuto G, Pucci P, D’Aiuto M, Baralle F, D’Argenio V, et al. A Novel Pathogenic BRCA1 Splicing Variant Produces Partial Intron Retention in the Mature Messenger RNA. International Journal of Molecular Sciences. 2016; 17(12):2145. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122145
Chicago/Turabian StyleEsposito, Maria Valeria, Marcella Nunziato, Flavio Starnone, Antonella Telese, Alessandra Calabrese, Giuseppe D’Aiuto, Pietro Pucci, Massimiliano D’Aiuto, Francisco Baralle, Valeria D’Argenio, and et al. 2016. "A Novel Pathogenic BRCA1 Splicing Variant Produces Partial Intron Retention in the Mature Messenger RNA" International Journal of Molecular Sciences 17, no. 12: 2145. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122145