
Mesenchymal Stem Cells Preserve Working Memory in the 3xTg-AD Mouse Model of Alzheimer’s Disease

 and
and
Abstract
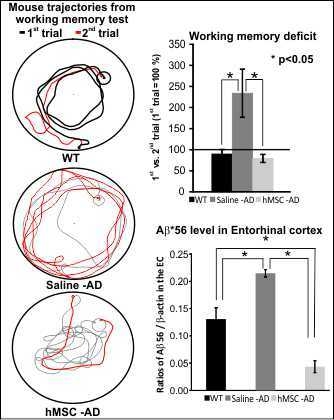
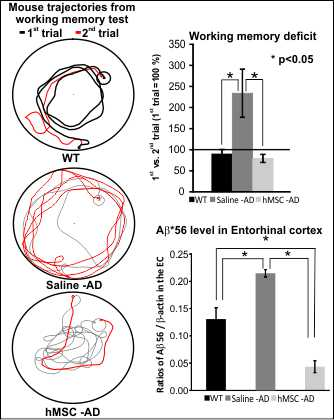
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
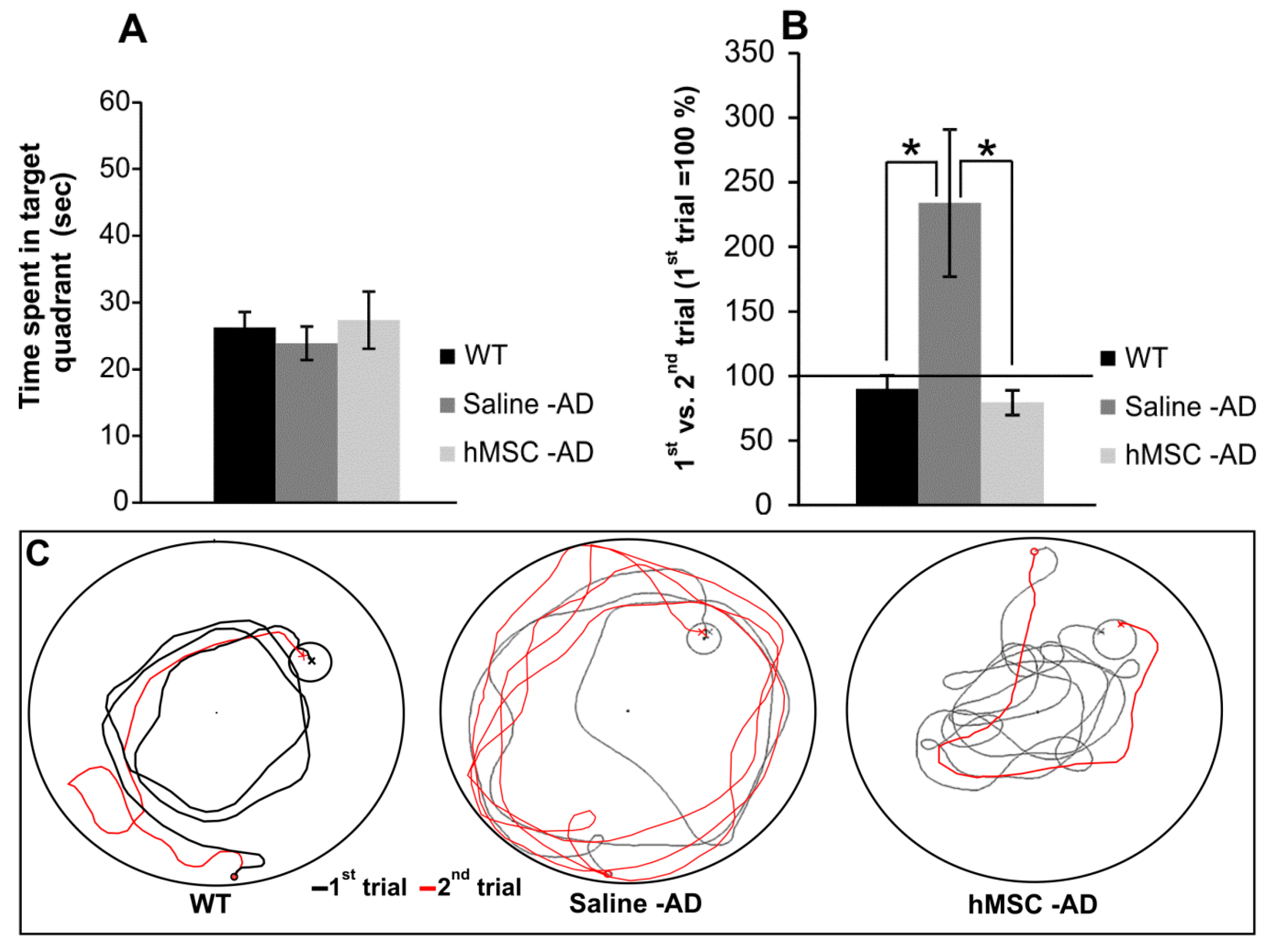
2.1. Reference and Working Memory

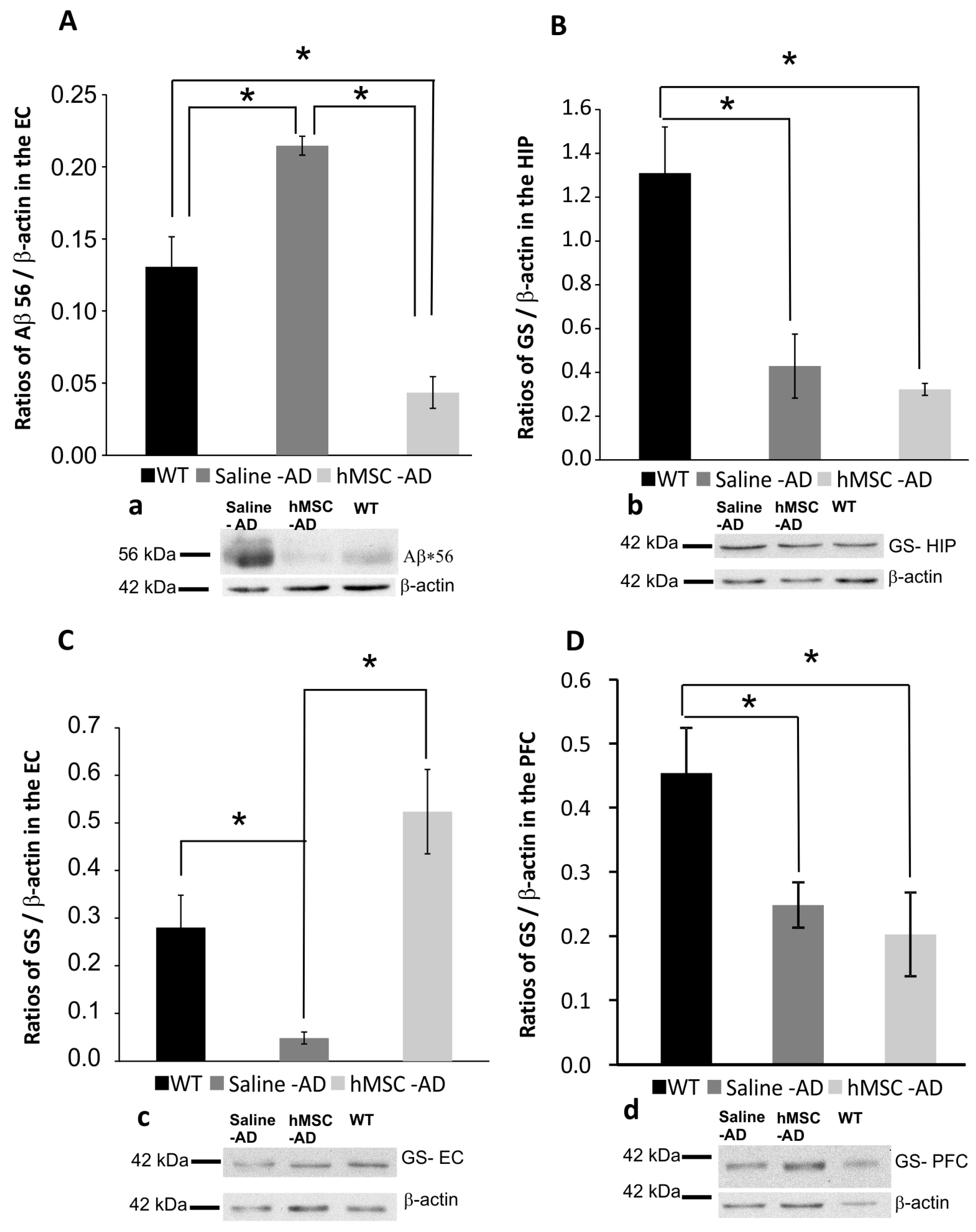
2.2. Influence of Human Mesenchymal Stem Cells (hMSCs) on Amyloid β (Aβ*56) and Glutamine Synthetase (GS) Levels

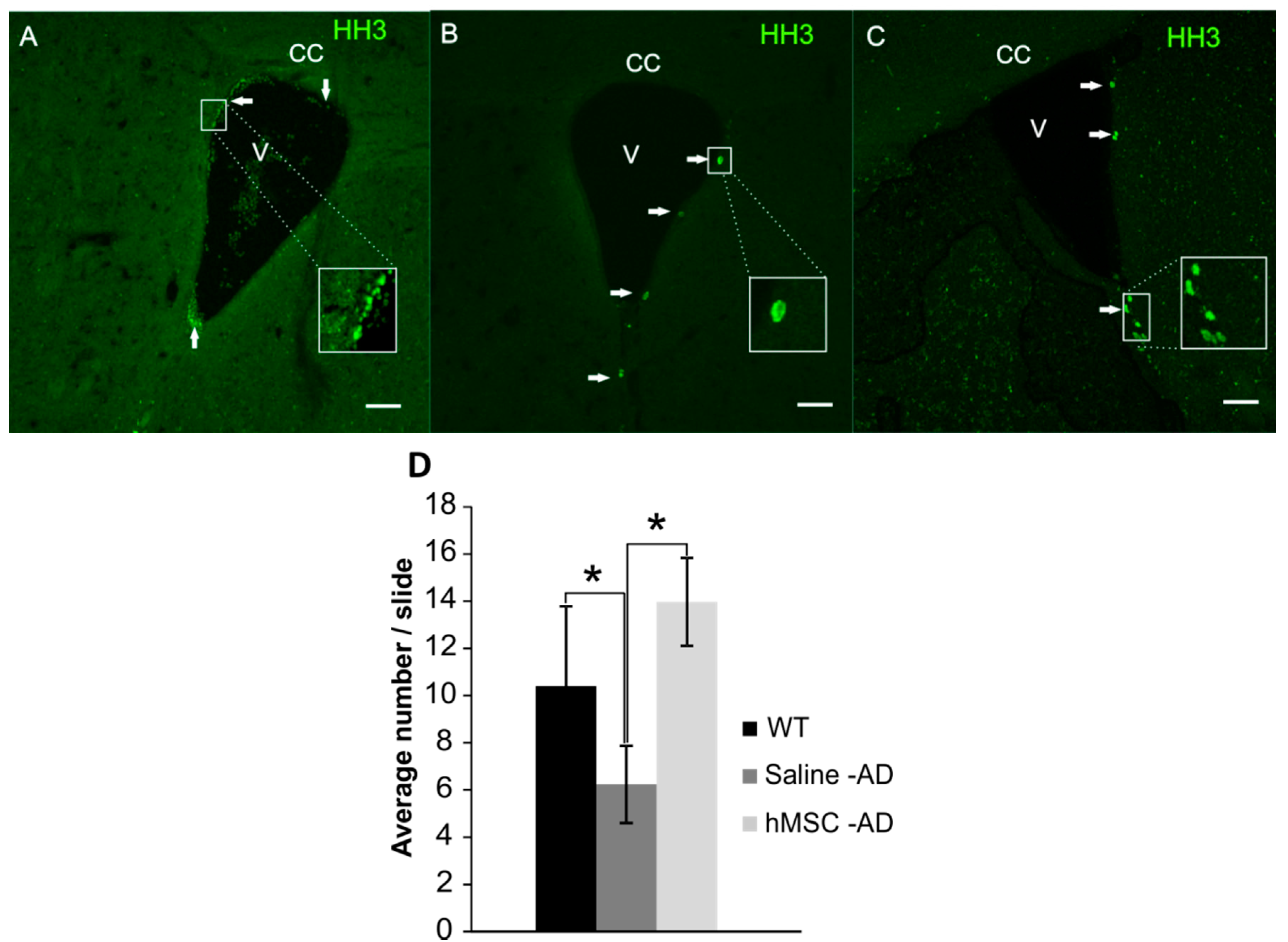
2.3. Neurogenesis
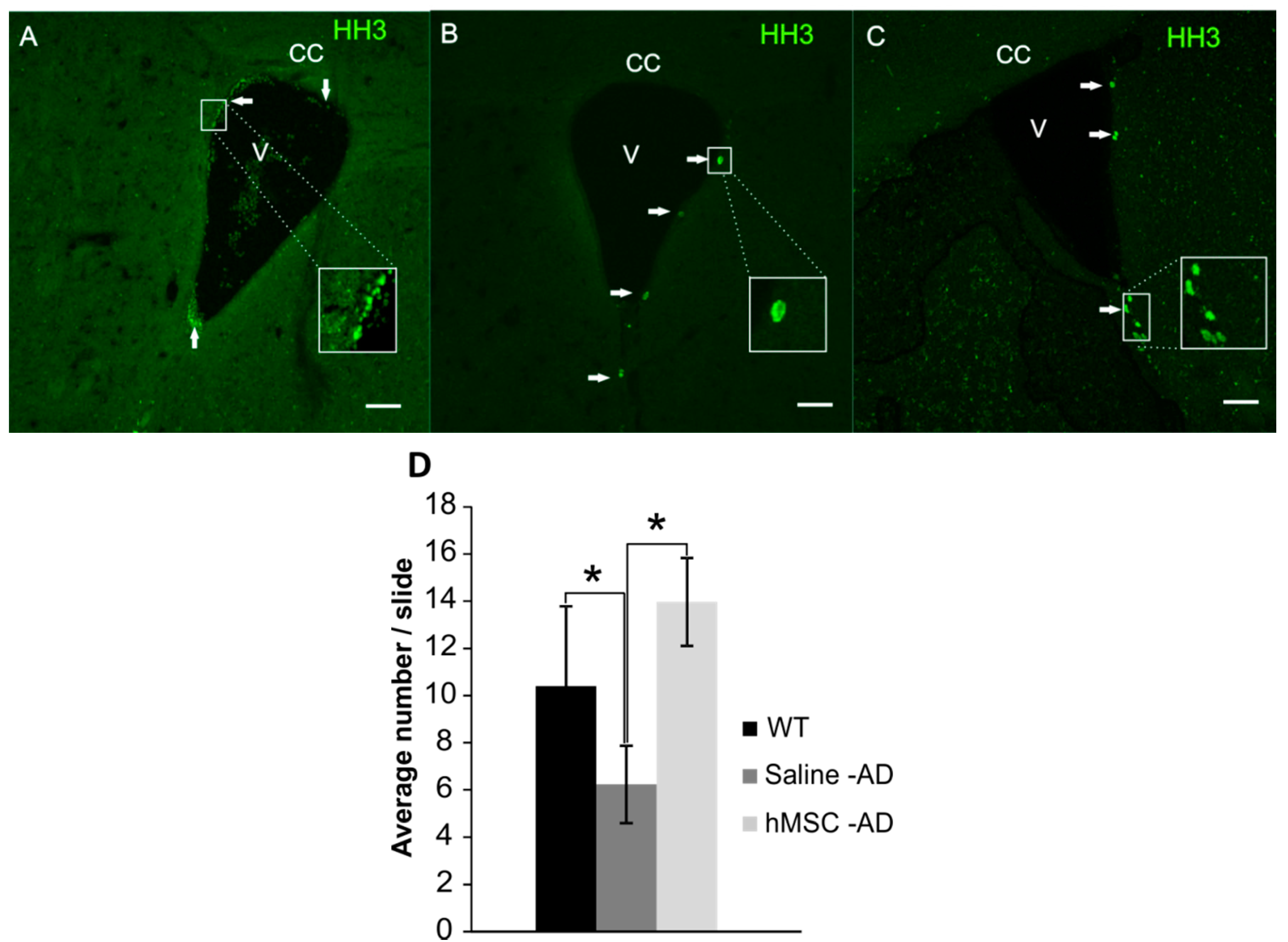
3. Discussion
4. Material and Methods
4.1. Animals and Cell Transplantation
4.2. Human MSC Cultures
4.3. Transplantation
4.4. Behavioral Evaluation
4.4.1. Spatial Reference Memory
4.4.2. Working Memory
4.5. Western Blotting
4.6. Immunohistochemical Staining and Microscopy
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blurton-Jones, M.; Laferla, F.M. Pathways by which Aβ facilitates tau pathology. Curr. Alzheimer Res. 2006, 3, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Nistico, R.; Pignatelli, M.; Piccinin, S.; Mercuri, N.B.; Collingridge, G. Targeting synaptic dysfunction in Alzheimer’s disease therapy. Mol. Neurobiol. 2012, 46, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Pozueta, J.; Lefort, R.; Shelanski, M.L. Synaptic changes in Alzheimer’s disease and its models. Neuroscience 2013, 251, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Pratico, D.; Trojanowski, J.Q. Inflammatory hypotheses: Novel mechanisms of Alzheimer’s neurodegeneration and new therapeutic targets? Neurobiol. Aging 2000, 21, 441–445; discussion 451–443. [Google Scholar] [CrossRef]
- Dantuma, E.; Merchant, S.; Sugaya, K. Stem cells for the treatment of neurodegenerative diseases. Stem Cell Res. Ther. 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Lee, J.K.; Lee, H.; Carter, J.E.; Chang, J.W.; Oh, W.; Yang, Y.S.; Suh, J.G.; Lee, B.H.; Jin, H.K.; et al. Human umbilical cord blood-derived mesenchymal stem cells improve neuropathology and cognitive impairment in an Alzheimer’s disease mouse model through modulation of neuroinflammation. Neurobiol. Aging 2012, 33, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Jin, H.K.; Bae, J.S. Bone marrow-derived mesenchymal stem cells reduce brain amyloid-β deposition and accelerate the activation of microglia in an acutely induced Alzheimer’s disease mouse model. Neurosci. Lett. 2009, 450, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.S.; Jin, H.K.; Lee, J.K.; Richardson, J.C.; Carter, J.E. Bone marrow-derived mesenchymal stem cells contribute to the reduction of amyloid-β deposits and the improvement of synaptic transmission in a mouse model of pre-dementia Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Jin, H.K.; Bae, J.S. Bone marrow-derived mesenchymal stem cells attenuate amyloid β-induced memory impairment and apoptosis by inhibiting neuronal cell death. Curr. Alzheimer Res. 2010, 7, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.Y.; Park, H.J.; Kim, H.N.; Oh, S.H.; Bae, J.S.; Ha, H.J.; Lee, P.H. Mesenchymal stem cells enhance autophagy and increase β-amyloid clearance in Alzheimer disease models. Autophagy 2014, 10, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, W.V.; Hou, H.; Town, T.; Zhu, Y.; Giunta, B.; Sanberg, C.D.; Zeng, J.; Luo, D.; Ehrhart, J.; Mori, T.; et al. Peripherally administered human umbilical cord blood cells reduce parenchymal and vascular β-amyloid deposits in Azheimer mice. Stem Cells Dev. 2008, 17, 423–439. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Sun, D.; Tang, X.; Cai, Y.; Yin, Z.Q.; Xu, H. Stem-cell challenges in the treatment of Alzheimer’s disease: A long way from bench to bedside. Med. Res. Rev. 2014, 34, 957–978. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.E.; Farag, A.; Althani, A.; Afifi, N.; Ahmed, A.E.; Lashen, S.; Rezk, S.; Pallini, R.; Casalbore, P.; Cenciarelli, C. Human olfactory bulb neural stem cells expressing hNGF restore cognitive deficit in Alzheimer’s disease rat model. J. Cell. Physiol. 2015, 230, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; Cho, T.; Wang, Y.T.; McLarnon, J.G. Neural progenitor cells attenuate inflammatory reactivity and neuronal loss in an animal model of inflamed AD brain. J. Neuroinflamm. 2009, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Blurton-Jones, M. Concise review: Can stem cells be used to treat or model Alzheimer’s disease? Stem Cells 2012, 30, 2612–2618. [Google Scholar] [CrossRef] [PubMed]
- Blurton-Jones, M.; Kitazawa, M.; Martinez-Coria, H.; Castello, N.A.; Muller, F.J.; Loring, J.F.; Yamasaki, T.R.; Poon, W.W.; Green, K.N.; LaFerla, F.M. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc. Natl. Acad. Sci. USA 2009, 106, 13594–13599. [Google Scholar] [CrossRef] [PubMed]
- Blurton-Jones, M.; Spencer, B.; Michael, S.; Castello, N.A.; Agazaryan, A.A.; Davis, J.L.; Muller, F.J.; Loring, J.F.; Masliah, E.; LaFerla, F.M. Neural stem cells genetically-modified to express neprilysin reduce pathology in alzheimer transgenic models. Stem Cell Res. Ther. 2014, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Sasaki, A.; Yoshimoto, R.; Kawahara, Y.; Manabe, T.; Kataoka, K.; Asashima, M.; Yuge, L. Neural stem cells improve learning and memory in rats with Alzheimer’s disease. Pathobiology 2008, 75, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, G.M.; Wang, P.J.; Zhang, Q.; Sha, S.H. Effects of neural stem cells on synaptic proteins and memory in a mouse model of Alzheimer’s disease. J. Neurosci. Res. 2014, 92, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, P.J.; Sha, H.Y.; Ni, J.; Li, M.H.; Gu, G.J. Neural stem cell transplants improve cognitive function without altering amyloid pathology in an APP/PS1 double transgenic model of Alzheimer’s disease. Mol. Neurobiol. 2014, 50, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Forostyak, S.; Jendelova, P.; Kapcalova, M.; Arboleda, D.; Sykova, E. Mesenchymal stromal cells prolong the lifespan in a rat model of amyotrophic lateral sclerosis. Cytotherapy 2011, 13, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Forostyak, S.; Jendelova, P.; Sykova, E. The role of mesenchymal stromal cells in spinal cord injury, regenerative medicine and possible clinical applications. Biochimie 2013, 95, 2257–2270. [Google Scholar] [CrossRef] [PubMed]
- Laroni, A.; Novi, G.; Kerlero de Rosbo, N.; Uccelli, A. Towards clinical application of mesenchymal stem cells for treatment of neurological diseases of the central nervous system. J. Neuroimmune Pharmacol. 2013, 8, 1062–1076. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Amemori, T.; Ermakova, I.V.; Buresova, O.; Zigova, T.; Racekova, E.; Bures, J. Brain transplants enhance rather than reduce the impairment of spatial memory and olfaction in bulbectomized rats. Behav. Neurosci. 1989, 103, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Sterniczuk, R.; Antle, M.C.; Laferla, F.M.; Dyck, R.H. Characterization of the 3× Tg-AD mouse model of Alzheimer’s disease: Part 2. Behavioral and cognitive changes. Brain Res. 2010, 1348, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Clemente, A.S.; Velasco, P.T.; Wood, M.; Viola, K.L.; Klein, W.L. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 2007, 27, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Lesne, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-β protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Lesne, S.E.; Sherman, M.A.; Grant, M.; Kuskowski, M.; Schneider, J.A.; Bennett, D.A.; Ashe, K.H. Brain amyloid-β oligomers in ageing and Alzheimer’s disease. Brain 2013, 136, 1383–1398. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.N.; Hofmeister, J.J.; Jungbauer, L.; Welzel, A.T.; Yu, C.; Sherman, M.A.; Lesne, S.; LaDu, M.J.; Walsh, D.M.; Ashe, K.H.; et al. Cognitive effects of cell-derived and synthetically derived Aβ oligomers. Neurobiol. Aging 2011, 32, 1784–1794. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; Klein, W.L. The Aβ oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease. Neurobiol. Learn. Mem. 2011, 96, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Kemper, L.J.; Wang, J.; Zahs, K.R.; Ashe, K.H.; Pasinetti, G.M. Grape seed polyphenolic extract specifically decreases Aβ*56 in the brains of Tg2576 mice. J. Alzheimers Dis. 2011, 26, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Scherzer-Attali, R.; Farfara, D.; Cooper, I.; Levin, A.; Ben-Romano, T.; Trudler, D.; Vientrov, M.; Shaltiel-Karyo, R.; Shalev, D.E.; Segev-Amzaleg, N.; et al. Naphthoquinone-tyrptophan reduces neurotoxic Aβ*56 levels and improves cognition in Alzheimer’s disease animal model. Neurobiol. Dis. 2012, 46, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Zahs, K.R.; Ashe, K.H. β-amyloid oligomers in aging and Alzheimer’s disease. Front. Aging Neurosci. 2013, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Behrens, P.F.; Franz, P.; Woodman, B.; Lindenberg, K.S.; Landwehrmeyer, G.B. Impaired glutamate transport and glutamate-glutamine cycling: Downstream effects of the huntington mutation. Brain 2002, 125, 1908–1922. [Google Scholar] [CrossRef] [PubMed]
- Coulter, D.A.; Eid, T. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia 2012, 60, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Duan, W.; Li, Z.; Huang, J.; Yin, Y.; Zhang, K.; Wang, Q.; Zhang, Z.; Li, C. Decreased GLT-1 and increased SOD1 and HO-1 expression in astrocytes contribute to lumbar spinal cord vulnerability of SOD1-G93A transgenic mice. FEBS Lett. 2010, 584, 1615–1622. [Google Scholar] [CrossRef] [PubMed]
- Le Prince, G.; Delaere, P.; Fages, C.; Lefrancois, T.; Touret, M.; Salanon, M.; Tardy, M. Glutamine synthetase (GS) expression is reduced in senile dementia of the alzheimer type. Neurochem. Res. 1995, 20, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Minkeviciene, R.; Ihalainen, J.; Malm, T.; Matilainen, O.; Keksa-Goldsteine, V.; Goldsteins, G.; Iivonen, H.; Leguit, N.; Glennon, J.; Koistinaho, J.; et al. Age-related decrease in stimulated glutamate release and vesicular glutamate transporters in APP/PS1 transgenic and wild-type mice. J. Neurochem. 2008, 105, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.R. Neuronal expression of glutamine synthetase in Alzheimer’s disease indicates a profound impairment of metabolic interactions with astrocytes. Neurochem. Int. 2000, 36, 471–482. [Google Scholar] [CrossRef]
- Timmer, N.M.; Herbert, M.K.; Claassen, J.A.; Kuiperij, H.B.; Verbeek, M.M. Total glutamine synthetase levels in cerebrospinal fluid of Alzheimer’s disease patients are unchanged. Neurobiol. Aging 2015, 36, 1271–1273. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.; Shaw, C.A. Late appearance of glutamate transporter defects in a murine model of ALS-parkinsonism dementia complex. Neurochem. Int. 2007, 50, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Kitazawa, M.; Tseng, B.P.; LaFerla, F.M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Rohn, T.T.; Vyas, V.; Hernandez-Estrada, T.; Nichol, K.E.; Christie, L.A.; Head, E. Lack of pathology in a triple transgenic mouse model of Alzheimer’s disease after overexpression of the anti-apoptotic protein Bcl-2. J. Neurosci. 2008, 28, 3051–3059. [Google Scholar] [CrossRef] [PubMed]
- Bobkova, N.V.; Poltavtseva, R.A.; Samokhin, A.N.; Sukhikh, G.T. Therapeutic effect of mesenchymal multipotent stromal cells on memory in animals with Alzheimer-type neurodegeneration. Bull. Exp. Biol. Med. 2013, 156, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.A.; Kim, H.J.; Joo, Y.; Ha, S.; Suh, Y.H. The therapeutic effects of human adipose-derived stem cells in Alzheimer’s disease mouse models. Neurodegener. Dis. 2014, 13, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Salem, A.M.; Ahmed, H.H.; Atta, H.M.; Ghazy, M.A.; Aglan, H.A. Potential of bone marrow mesenchymal stem cells in management of Alzheimer’s disease in female rats. Cell Biol. Int. 2014, 38, 1367–1383. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.M.; Kim, H.S.; Park, K.R.; Shin, J.M.; Kang, A.R.; il Lee, K.; Song, S.; Kim, Y.B.; Han, S.B.; Chung, H.M.; et al. Placenta-derived mesenchymal stem cells improve memory dysfunction in an Aβ1–42-infused mouse model of Alzheimer’s disease. Cell Death Dis. 2013, 4, e958. [Google Scholar] [CrossRef] [PubMed]
- Eichenbaum, H.; Stewart, C.; Morris, R.G. Hippocampal representation in place learning. J. Neurosci. 1990, 10, 3531–3542. [Google Scholar] [PubMed]
- Buzsaki, G.; Moser, E.I. Memory, navigation and theta rhythm in the hippocampal-entorhinal system. Nat. Neurosci. 2013, 16, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Leutgeb, S.; Leutgeb, J.K. Spatial and memory circuits in the medial entorhinal cortex. Curr. Opin. Neurobiol. 2015, 32, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Huntley, J.D.; Howard, R.J. Working memory in early Alzheimer’s disease: A neuropsychological review. Int. J. Geriatr. Psychiatry 2010, 25, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Micotti, E.; Paladini, A.; Balducci, C.; Tolomeo, D.; Frasca, A.; Marizzoni, M.; Filibian, M.; Caroli, A.; Valbusa, G.; Dix, S.; et al. Striatum and entorhinal cortex atrophy in ad mouse models: Mri comprehensive analysis. Neurobiol. Aging 2015, 36, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.Y.; Vadhwana, B.; Verkhratsky, A.; Rodriguez, J.J. Early astrocytic atrophy in the entorhinal cortex of a triple transgenic animal model of Alzheimer’s disease. ASN Neuro 2011, 3, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.Y.; Verkhratsky, A.; Terzieva, S.; Rodriguez, J.J. Glutamine synthetase in astrocytes from entorhinal cortex of the triple transgenic animal model of Alzheimer’s disease is not affected by pathological progression. Biogerontology 2013, 14, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Kulijewicz-Nawrot, M.; Sykova, E.; Chvatal, A.; Verkhratsky, A.; Rodriguez, J.J. Astrocytes and glutamate homoeostasis in Alzheimer’s disease: A decrease in glutamine synthetase, but not in glutamate transporter-1, in the prefrontal cortex. ASN Neuro 2013, 5, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Olabarria, M.; Noristani, H.N.; Verkhratsky, A.; Rodriguez, J.J. Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer’s disease mouse model: Mechanism for deficient glutamatergic transmission? Mol. Neurodegener. 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.J.; Jones, V.C.; Tabuchi, M.; Allan, S.M.; Knight, E.M.; LaFerla, F.M.; Oddo, S.; Verkhratsky, A. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PLoS ONE 2008, 3, e2935. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.J.; Jones, V.C.; Verkhratsky, A. Impaired cell proliferation in the subventricular zone in an Alzheimer’s disease model. Neuroreport 2009, 20, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Paul, G.; Anisimov, S.V. The secretome of mesenchymal stem cells: Potential implications for neuroregeneration. Biochimie 2013, 95, 2246–2256. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Qiu, R.; Xu, Q. Mesenchymal stem cell therapy for neurodegenerative diseases. J. Nanosci. Nanotechnol. 2014, 14, 969–975. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruzicka, J.; Kulijewicz-Nawrot, M.; Rodrigez-Arellano, J.J.; Jendelova, P.; Sykova, E. Mesenchymal Stem Cells Preserve Working Memory in the 3xTg-AD Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2016, 17, 152. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17020152
Ruzicka J, Kulijewicz-Nawrot M, Rodrigez-Arellano JJ, Jendelova P, Sykova E. Mesenchymal Stem Cells Preserve Working Memory in the 3xTg-AD Mouse Model of Alzheimer’s Disease. International Journal of Molecular Sciences. 2016; 17(2):152. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17020152
Chicago/Turabian StyleRuzicka, Jiri, Magdalena Kulijewicz-Nawrot, Jose Julio Rodrigez-Arellano, Pavla Jendelova, and Eva Sykova. 2016. "Mesenchymal Stem Cells Preserve Working Memory in the 3xTg-AD Mouse Model of Alzheimer’s Disease" International Journal of Molecular Sciences 17, no. 2: 152. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17020152