
Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos

Abstract
:
1. Introduction
2. Formulation Approach: Covalent Conjugation or Physical Complexation
3. Peptide and Protein Cargos Successfully Delivered by Cell-Penetrating Peptides

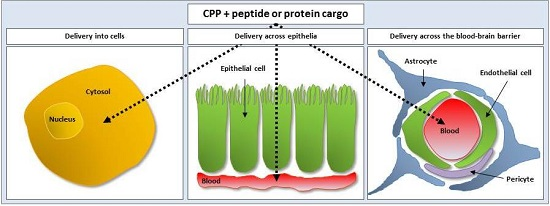
3.1. Cell-Penetrating Peptides as Vectors for Intracellular Peptide and Protein Delivery
3.2. Cell-Penetrating Peptides as Vectors for Delivery of Peptides and Proteins Across Epithelia and the Blood-Brain Barrier

{kind=link}
{kind=link}
{kind=link}
| Cell-Penetrating Peptide | Peptide or Protein Pargo | Formulation Approach | Assay | Ref. |
|---|---|---|---|---|
| Delivery into cells | ||||
| Tat | β-galactosidase | Covalent conjugation | Tissue distribution of β-galactosidase in mice following IP administration. | [29] |
| Tat | All-d-ri a-p53 | Covalent conjugation | Survival of peritoneal carcinomatosis mouse model following IP administration. | [30] |
| All-d-R11 | p53~ri-HA-2 b | Covalent conjugation | Survival of animals in a peritoneal carcinomatosis mouse model following IP administration. | [31] |
| Tat | P15 | Covalent conjugation | Apoptosis in various tumor cell lines and regression of tumor size upon intratumoral injections to mice. | [32] |
| FGF4 c-derived peptide [48] | SOCS3 d | Covalent conjugation | Uptake into mouse macrophage cells and suppression of the production of inflammatory cytokines in mice following IP administration. | [33] |
| R9 | c-Myc, Sox2, Oct4, Klf4 | Covalent conjugation | Induction of fibroblasts from human newborn into pluripotent stem cells. | [34] |
| Pep-1 | Various peptides and proteins | Physical complexation | Uptake of cargo peptide or protein in cells of various cell culture models. | [11] |
| Delivery across epithelia and the blood-brain barrier | ||||
| Tat | Insulin | Physical complexation | Insulin permeation across Caco-2 monolayers. | [21] |
| All-d-R8 | Insulin, GLP-1 e, gastrin | Physical complexation | Cargo plasma concentration following intestinal loop administration to rats. | [35] |
| Penetratin | Insulin, GLP-1, exendin-4 | Physical complexation | Cargo plasma concentration following nasal or intestinal loop administration to rats. | [36] |
| All-d-penetratin | Insulin | Physical complexation | Blood glucose level following administration by oral gavage to rats. | [43] |
| Shuffle | Insulin | Physical complexation | Insulin plasma concentration following nasal administration to rats. | [44] |
| PenetraMax | Insulin | Physical complexation | Insulin plasma concentration following intestinal loop administration to rats. | [45] |
| Tat | Bcl-xl | Covalent conjugation | Brain distribution of Bcl-xl and reduction of cerebral infarction. | [38] |
| Tat | NR2B9c f | Covalent conjugation | Brain concentration of NR2B9c in rats and reduction of cerebral infarction in mice following IP administration. | [46] |
| Tat | GDNF g | Covalent conjugation | Brain concentration of GDNF and reduction of cerebral infarction following intravenous administration to mice. | [47] |
4. Sequence-Specific Properties of Cell-Penetrating Peptides Influencing Their Membrane Permeation
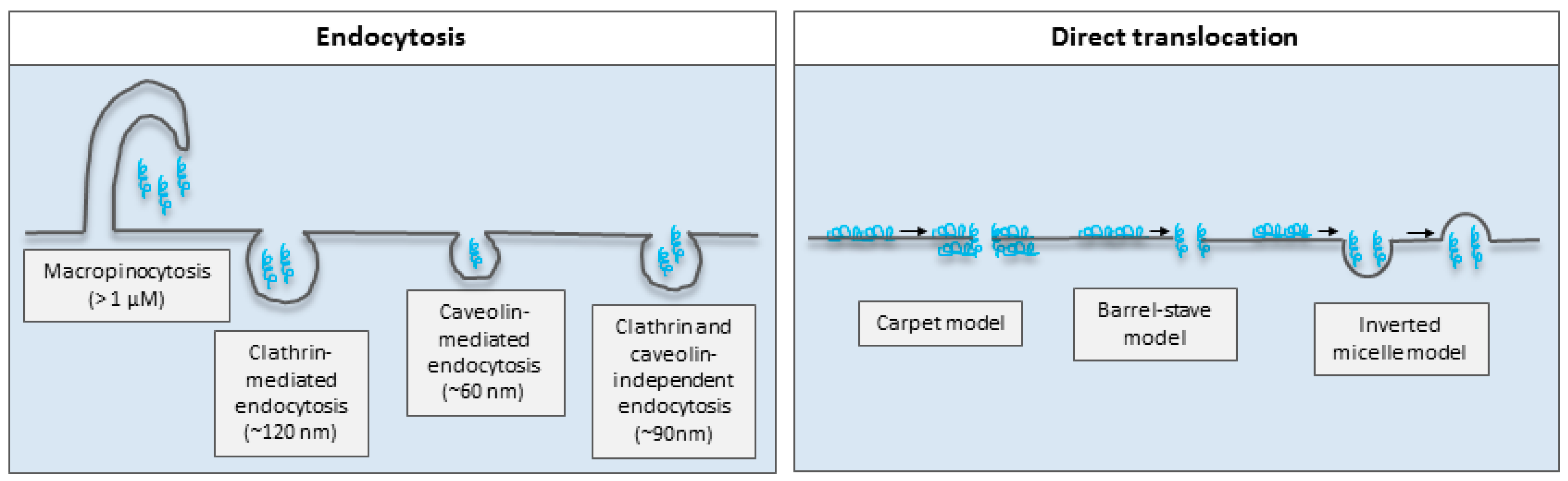
5. Mechanisms of Membrane Permeation of Cell-Penetrating Peptides

6. Challenges for the Use of Cell-Penetrating Peptides as Delivery Vectors
6.1. Stability of Cell-Penetrating Peptides
6.2. Detection of Cell-Penetrating Peptides and Their Cargo Peptide or Protein Drugs
7. Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Frankel, A.; Pabo, C. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 1189–1193. [Google Scholar] [CrossRef]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [PubMed]
- Farrera-Sinfreu, J.; Giralt, E.; Castel, S.; Albericio, F.; Royo, M. Cell-penetrating cis-ƴ-amino-l-proline-derived peptides. J. Am. Chem. Soc. 2005, 127, 9459–9468. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, K.; El Andaloussi, S.; Zaghloul, E.M.; Lehto, T.; Lindberg, S.; Moreno, P.M.D.; Viola, J.R.; Magdy, T.; Abdo, R.; Guterstam, P.; et al. PepFect 14, a novel cell-penetrating peptide for oligonucleotide delivery in solution and as solid formulation. Nucleic Acids Res. 2011, 39, 5284–5298. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Liu, T.; Liu, Y.Y.; Briesewitz, R.; Barrios, A.M.; Jhiang, S.M.; Pei, D. Efficient delivery of cyclic peptides into mammalian cells with short sequence motifs. ACS Chem. Biol. 2013, 82, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Angeles-Boza, A.M.; Erazo-Oliveras, A.; Lee, Y.J.; Pellois, J.P. Generation of endosomolytic reagents by branching of cell-penetrating peptides: Tools for the delivery of bioactive compounds to live cells in cis or trans. Bioconjug. Chem. 2010, 21, 2164–2167. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Dougherty, P.G.; Pei, D. Monitoring the cytosolic entry of cell-penetrating peptides using a pH-sensitive fluorophore. Chem. Commun. 2015, 51, 2162–2165. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, M.; Rosenthal-Aizman, K.; Saar, K.; Eiríksdóttir, E.; Jiang, Y.; Sassian, M.; Östlund, P.; Hällbrink, M.; Langel, Ü. Overcoming methotrexate resistance in breast cancer tumour cells by the use of a new cell-penetrating peptide. Biochem. Pharmacol. 2006, 71, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Tang, J.; Fu, L.; Ran, R.; Liu, Y.; Yuan, M.; He, Q. A pH-responsive α-helical cell penetrating peptide-mediated liposomal delivery system. Biomaterials 2013, 34, 7980–7993. [Google Scholar] [CrossRef] [PubMed]
- Margus, H.; Padari, K.; Pooga, M. Cell-penetrating peptides as versatile vehicles for oligonucleotide delivery. Mol. Ther. 2012, 20, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Depollier, J.; Mery, J.; Heitz, F.; Divita, G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat. Biotechnol. 2001, 19, 1173–1176. [Google Scholar] [CrossRef] [PubMed]
- Khafagy, E.S.; Morishita, M. Oral biodrug delivery using cell-penetrating peptide. Adv. Drug Deliv. Rev. 2012, 64, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.L.; Ma, J.L.; Wang, T.; Yang, T.B.; Liu, C.B. Cell-penetrating peptide-mediated therapeutic molecule delivery into the central nervous system. Curr. Neuropharmacol. 2013, 11, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Taverner, A.; Dondi, R.; Almansour, K.; Laurent, F.; Owens, S.E.; Eggleston, I.M.; Fotaki, N.; Mrsny, R.J. Enhanced paracellular transport of insulin can be achieved via transient induction of myosin light chain phosphorylation. J. Control. Release 2015, 210, 189–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolotarevsky, Y.; Hecht, G.; Koutsouris, A.; Gonzalez, D.E.; Quan, C.; Tom, J. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology 2002, 123, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Meloni, B.P.; Craig, A.J.; Milech, N.; Hopkins, R.M.; Watt, P.M.; Knuckey, N.W. The neuroprotective efficacy of cell-penetrating peptides TAT, penetratin, Arg-9, and Pep-1 in glutamic acid, kainic acid, and in vitro ischemia injury models using primary cortical neuronal cultures. Cell. Mol. Neurobiol. 2014, 34, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Suhorutsenko, J.; Eriste, E.; Copolovici, D.M.; Langel, Ü. Human Protein 53-derived Cell-penetrating peptides. Int. J. Pept. Res. Ther. 2012, 18, 291–297. [Google Scholar] [CrossRef]
- Virès, E.; Granier, C.; Prevot, P.; Lebleu, B. Structure-activity relationship study of the plasma membrane translocating potential of a short peptide from HIV-1 Tat protein. Lett. Pept. Sci. 1997, 4, 429–436. [Google Scholar] [CrossRef]
- Herce, H.D.; Deng, W.; Helma, J.; Leonhardt, H.; Cardoso, M.C. Visualization and targeted disruption of protein interactions in living cells. Nat. Commun. 2013, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vivès, E.; Bernard, L. The Tat-derived cell-penetrating peptide. In Cell-Penetrating Peptides: Processes and Applications; Humana Press: Boca Raton, FL, USA, 2002; pp. 3–21. [Google Scholar]
- Liang, J.F.; Yang, V.C. Insulin-cell penetrating peptide hybrids with improved intestinal absorption efficiency. Biochem. Biophys. Res. Commun. 2005, 335, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Caldinelli, L.; Albani, D.; Pollegioni, L. One single method to produce native and Tat-fused recombinant human α-synuclein in Escherichia coli. BMC Biotechnol. 2013, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, M.; de Groot, A.M.; Berthelsen, J.; Franzyk, H.; Sijts, A.; Nielsen, H.M. Conjugation of cell-penetrating peptides to parathyroid hormone affects its structure, potency, and transepithelial permeation. Bioconjug. Chem. 2015, 26, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Mie, M.; Takahashi, F.; Funabashi, H.; Yanagida, Y.; Aizawa, M.; Kobatake, E. Intracellular delivery of antibodies using TAT fusion protein A. Biochem. Biophys. Res. Commun. 2003, 310, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Hiraoka, M.; Kizaka-Kondoh, S. Antitumor effect of TAT-oxygen-dependent degradation-caspase-3 fusion protein specifically stabilized and activated in hypoxic tumor cells. Cancer Res. 2002, 62, 2013–2018. [Google Scholar] [PubMed]
- Kristensen, M.; Franzyk, H.; Klausen, M.T.; Iversen, A.; Bahnsen, J.S.; Skyggebjerg, R.B.; Foderà, V.; Nielsen, H.M. Penetratin-mediated transepithelial insulin permeation: Importance of cationic residues and pH for complexation and permeation. AAPS J. 2015, 17, 1200–1209. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Aoyama, Y.; Khafagy, E.S.; Henmi, M.; Takeda-Morishita, M. Effect of different intestinal conditions on the intermolecular interaction between insulin and cell-penetrating peptide penetratin and on its contribution to stimulation of permeation through intestinal epithelium. Eur. J. Pharm. Biopharm. 2015, 94, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, H.; Vocero-Akbani, A.M.; Snyder, E.L.; Ho, A.; Latham, D.G.; Lissy, N.A.; Becker-Hapak, M.; Ezhevsky, S.A.; Dowdy, S.F. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat. Med. 1998, 4, 1449–1452. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, S.R. In Vivo Protein Transduction: Delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef] [PubMed]
- Snyder, E.L.; Meade, B.R.; Saenz, C.C.; Dowdy, S.F. Treatment of terminal peritoneal carcinomatosis by a transducible p53-activating peptide. PLoS Biol. 2004, 2, 186–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, D.; Takayama, K.; Inoue, M.; Watanabe, T.; Kumon, H.; Futaki, S.; Kumon, H.; Futaki, S.; Matsui, H.; Tomizava, H. Cell-penetrating d-Isomer peptides of p53 C-terminus: Long-term inhibitory effect on the growth of bladder cancer. Urology 2010, 75, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Perea, S.E.; Reyes, O.; Puchades, Y.; Mendoza, O.; Vispo, N.S.; Torrens, I.; Santos, A.; Silva, R.; Acevedo, B.; López, E.; et al. Antitumor effect of a novel proapoptotic peptide that impairs the phosphorylation by the protein kinase 2 (casein kinase 2). Cancer Res. 2004, 64, 7127–7129. [Google Scholar] [CrossRef] [PubMed]
- Jo, D.; Liu, D.; Yao, S.; Collins, R.D.; Hawiger, J. Intracellular protein therapy with SOCS3 inhibits inflammation and apoptosis. Nat. Med. 2005, 11, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, C.H.; Moon, J.I.; Chung, Y.G.; Chang, M.Y.; Han, B.S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, A.; et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Morishita, M.; Eda, Y.; Ida, N.; Nishio, R.; Takayama, K. Usefulness of cell-penetrating peptides to improve intestinal insulin absorption. J. Control. Release 2008, 132, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Khafagy, E.S.; Morishita, M.; Kamei, N.; Eda, Y.; Ikeno, Y.; Takayama, K. Efficiency of cell-penetrating peptides on the nasal and intestinal absorption of therapeutic peptides and proteins. Int. J. Pharm. 2009, 381, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.; Wang, J.; Kim, K. Conjugation with cationic cell-penetrating peptide increases pulmonary absorption of insulin. Mol. Pharm. 2009, 6, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Pei, W.; Ge, H.; Liang, Q.; Luo, Y.; Sharp, F.R.; Lu, A.; Ran, R.; Graham, S.H.; Chen, J. In Vivo Delivery of a Bcl-xL fusion protein containing the TAT protein transduction domain protects against ischemic brain injury and neuronal apoptosis. J. Neurosci. 2002, 22, 5423–5431. [Google Scholar] [PubMed]
- Whitehead, K.; Karr, N.; Mitragotri, S. Safe and effective permeation enhancers for oral drug delivery. Pharm. Res. 2008, 25, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Van Der Walle, C.F.; Schmidt, E. Modulation of the intestinal tight junctions using bacterial enterotoxins. Pept. Protein Deliv. 2011, 195–219. [Google Scholar] [CrossRef]
- Kamei, N.; Morishita, M.; Takayama, K. Importance of intermolecular interaction on the improvement of intestinal therapeutic peptide/protein absorption using cell-penetrating peptides. J. Control. Release 2009, 136, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Morishita, M.; Kanayama, Y.; Hasegawa, K.; Nishimura, M.; Hayashinaka, E.; Wada, Y.; Watanabe, Y.; Takayama, K. Molecular imaging analysis of intestinal insulin absorption boosted by cell-penetrating peptides by using positron emission tomography. J. Control. Release 2010, 146, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, E.J.B.; Yoshida, S.; Kamei, N.; Iwamae, R.; Khafagy, E.S.; Olsen, J.; Rahbek, U.L.; Pedersen, B.L.; Takayama, K.; Morishita, T.M. In vivo proof of concept of oral insulin delivery based on a co-administration strategy with the cell-penetrating peptide penetratin. J. Control. Release 2014, 189, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Khafagy, E.S.; Morishita, M.; Ida, N.; Nishio, R.; Isowa, K.; Takayama, K. Structural requirements of penetratin absorption enhancement efficiency for insulin delivery. J. Control. Release 2010, 143, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Kikuchi, S.; Morishita, T.M.; Terasawa, Y.; Yasuda, A.; Yamamoto, S.; Ida, N.; Nishio, R.; Takayama, K. Determination of the optimal cell-penetrating peptide sequence for intestinal insulin delivery based on molecular orbital analysis with self-organizing maps. J. Pharm. Sci. 2013, 102, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.; Liu, Y.; Li, L. Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science 2002, 298, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Kilic, E.; Kilic, U.; Hermann, D.M. TAT-GDNF in neurodegeneration and ischemic stroke. CNS Drug Rev. 2005, 11, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Hawiger, J. Noninvasive intracellular delivery of functional peptides and proteins. Curr. Opin. Chem. Biol. 1999, 3, 89–94. [Google Scholar] [CrossRef]
- Poon, G.M.K.; Gariépy, J. Cell-surface proteoglycans as molecular portals for cationic peptide and polymer entry into cells. Biochem. Soc. Trans. 2007, 35, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.; Seelig, J. Binding and clustering of glycosaminoglycans: A common property of mono- and multivalent cell-penetrating compounds. Biophys. J. 2008, 94, 2142–2149. [Google Scholar] [CrossRef] [PubMed]
- Letoha, T.; Keller-Pintér, A.; Kusz, E.; Kolozsi, C.; Bozsó, Z.; Tóth, G.; Vizler, C.; Oláh, Z.; Szilák, L. Cell-penetrating peptide exploited syndecans. Biochim. Biophys. Acta 2010, 1798, 2258–2265. [Google Scholar] [CrossRef] [PubMed]
- Alves, I.D.; Bechara, C.; Walrant, A.; Zaltsman, Y.; Jiao, C.Y.; Sagan, S. Relationships between membrane binding, affinity and cell internalization efficacy of a cell-penetrating peptide: Penetratin as a case study. PLoS ONE 2011, 6, e24096. [Google Scholar] [CrossRef] [PubMed]
- Bechara, C.; Pallerla, M.; Zaltsman, Y.; Burlina, F.; Alves, I.D.; Lequin, O.; Sagan, S. Tryptophan within basic peptide sequences triggers glycosaminoglycan-dependent endocytosis. Biochemistry 2013, 27, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rothbard, J.B.; Jessop, T.C.; Lewis, R.S.; Murray, B.A.; Wender, P.A. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J. Am. Chem. Soc. 2004, 126, 9506–9507. [Google Scholar] [CrossRef] [PubMed]
- Ryser, H.J.; Hancock, R. Histones and basic polyamino acids stimulate the uptake of albumin by tumor cells in culture. Science 1965, 150, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Tünnemann, G.; Ter-Avetisyan, G.; Martin, G.M.; Stoockl, M.; Hermann, A.C.C. Live-cell analysis of cell penetration ability and toxicity of oligo-arginines. J. Pept. Sci. 2008, 14, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar] [CrossRef] [PubMed]
- Jiao, C.Y.; Delaroche, D.; Burlina, F.; Alves, I.D.; Chassaing, G.; Sagan, S. Translocation and endocytosis for cell-penetrating peptide internalization. J. Biol. Chem. 2009, 284, 33957–33965. [Google Scholar] [CrossRef] [PubMed]
- Ram, N.; Aroui, S.; Jaumain, E.; Bichraoui, H.; Mabrouk, K.; Ronjat, M.; Lortat-Jacob, H.; de Waard, M. Direct peptide interaction with surface glycosaminoglycans contributes to the cell penetration of maurocalcine. J. Biol. Chem. 2008, 283, 24274–24284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rydberg, H.A.; Matson, M.; Amand, H.L.; Esbjörner, E.K.; Nordén, B. Effects of tryptophan content and backbone spacing on the uptake efficiency of cell-penetrating peptides. Biochemistry 2012, 51, 5531–5539. [Google Scholar] [CrossRef] [PubMed]
- Magzoub, M.; Eriksson, L.E.G.; Gräslund, A. Comparison of the interaction, positioning, structure induction and membrane perturbation of cell-penetrating peptides and non-translocating variants with phospholipid vesicles. Biophys. Chem. 2003, 103, 271–288. [Google Scholar] [CrossRef]
- Caesar, C.E.B.; Esbjo, E.K.; Lincoln, P.; Norde, B. Membrane interactions of cell-penetrating peptides probed by tryptophan fluorescence and dichroism techniques: Correlations of structure to cellular uptake. Biochemistry 2006, 45, 7682–7692. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, H.A.; Carlsson, N.; Nordén, B. Membrane interaction and secondary structure of de novo designed arginine-and tryptophan peptides with dual function. Biochem. Biophys. Res. Commun. 2012, 427, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Fittipaldi, A.; Ferrari, A.; Zoppé, M.; Arcangeli, C.; Pellegrini, V.; Beltram, F.; Giazza, M. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J. Biol. Chem. 2003, 278, 34141–34149. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Niwa, M.; Takeuchi, T.; Sonomura, K.; Kawabata, N.; Koike, Y.; Takehashi, M.; Tanaka, S.; Ueda, K.; Simpson, J.D.; et al. Cellular uptake of arginine-rich peptides: Roles for macropinocytosis and actin rearrangement. Mol. Ther. 2004, 10, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Pujals, S.; Fernández-Carneado, J.; López-Iglesias, C.; Kogan, M.J.; Giralt, E. Mechanistic aspects of CPP-mediated intracellular drug delivery: Relevance of CPP self-assembly. Biochim. Biophys. Acta 2006, 758, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Lundin, P.; Johansson, H.; Guterstam, P.; Holm, T.; Hansen, M.; Langel, U.; Andaloussi, S.E.L. Distinct uptake routes of cell-penetrating peptide conjugates. Bioconjug. Chem. 2008, 19, 2535–2542. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.T.; Sayers, E.J. Cell entry of cell penetrating peptides: Tales of tails wagging dogs. J. Control. Release 2012, 161, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Tréhin, R.; Krauss, U.; Beck-Sickinger, A.G.; Merkle, H.P.; Nielsen, H.M. Cellular uptake but low permeation of human calcitonin-derived cell penetrating peptides and Tat(47-57) through well-differentiated epithelial models. Pharm. Res. 2004, 21, 1248–1256. [Google Scholar] [CrossRef]
- Foerg, C.; Ziegler, U.; Fernandez-Carneado, J.; Giralt, E.; Merkle, H.P. Differentiation restricted endocytosis of cell penetrating peptides in MDCK cells corresponds with activities of Rho-GTPases. Pharm. Res. 2007, 24, 628–642. [Google Scholar] [CrossRef] [PubMed]
- Bárány-Wallje, E.; Gaur, J.; Lundberg, P.; Langel, U.; Gräslund, A. Differential membrane perturbation caused by the cell penetrating peptide Tp10 depending on attached cargo. FEBS Lett. 2007, 1, 2389–2393. [Google Scholar] [CrossRef] [PubMed]
- Tünnemann, G.; Martin, R.M.; Haupt, S.; Patsch, C.; Edenhofer, F.; Cardoso, M.C. Cargo-dependent mode of uptake and bioavailability of TAT-containing proteins and peptides in living cells. FASEB J. 2006, 20, 1775–1784. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Waizenegger, T.; Köhler, K.; Brock, R. A quantitative validation of fluorophore-labelled cell-permeable peptide conjugates: Fluorophore and cargo dependence of import. Biochim. Biophys. Acta 2002, 1564, 365–374. [Google Scholar] [CrossRef]
- Hirose, H.; Takeuchi, T.; Osakada, H.; Pujals, S.; Katayama, S.; Nakase, I.; Kobayashi, S.; Haraguchi, T.; Futaki, S. Transient focal membrane deformation induced by arginine-rich peptides leads to their direct penetration into cells. Mol. Ther. 2012, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [PubMed]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nature 2003, 422, 37–44. [Google Scholar] [CrossRef]
- Plapied, L.; Duhem, N.; des Rieux, A.; Préat, V. Fate of polymeric nanocarriers for oral drug delivery. Curr. Opin. Colloid Interface Sci. 2011, 16, 228–237. [Google Scholar] [CrossRef]
- Futaki, S.; Nakase, I.; Tadokoro, A.; Takeuchi, T.; Jones, A.T. Arginine-rich peptides and their internalization mechanisms. Biochem. Soc. Trans. 2007, 35, 784–787. [Google Scholar] [CrossRef] [PubMed]
- Khalil, L.A.; Kogure, K.; Futaki, S.; Harashima, H. High density of octaarginine stimulates macropinocytosis leading to efficient intracellular trafficking for gene expression. J. Biol. Chem. 2006, 281, 3544–3551. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Melikov, K.; Brooks, H.; Prevot, P.; Lebleu, B.; Chernomordik, L.V. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J. Biol. Chem. 2005, 280, 15300–15306. [Google Scholar] [CrossRef] [PubMed]
- Amand, H.L.; Fant, K.; Nordén, B.; Esbjörner, E.K. Stimulated endocytosis in penetratin uptake: Effect of arginine and lysine. Biochem. Biophys. Res. 2008, 371, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Fretz, M.M.; Penning, N.A.; Al-Taei, S.; Futaki, S.; Takeuchi, T.; Nakase, I.; Storm, G.; Jones, A.T. Temperature-, concentration- and cholesterol-dependent translocation of l- and d-octa-arginine across the plasma and nuclear membrane of CD34+ leukaemia cells. Biochem. J. 2007, 403, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Duchardt, F.; Fotin-Mleczek, M.; Schwarz, H.; Fischer, R.; Brock, R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 2007, 8, 848–866. [Google Scholar] [CrossRef] [PubMed]
- Al Soraj, M.; He, L.; Peynshaert, K.; Cousaert, J.; Vercauteren, D.; Braeckmans, K.; de Smedt, S.C.; Jones, A.T. siRNA and pharmacological inhibition of endocytic pathways to characterize the differential role of macropinocytosis and the actin cytoskeleton on cellular uptake of dextran and cationic cell penetrating peptides octaarginine (R8) and HIV-Tat. J. Control. Release 2012, 161, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Larochelle, J.R.; Jiang, B.; Lian, W.; Hard, R.L.; Selner, N.G.; Leuchapanichul, R.; Barrios, A.M.; Pei, D. Early endosomal escape of a cyclic cell-penetrating peptide allows effective cytosolic cargo delivery. Biochemistry 2014, 53, 4034–4046. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, C.R.; Hajnóczky, G.; Maksymowych, A.B.; Simpson, L.L. Visualization of binding and transcytosis of botulinum toxin by human intestinal epithelial cells. J. Pharmacol. Exp. Ther. 2005, 315, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Lukyanenko, V.; Malyukova, I.; Hubbard, A.; Delannoy, M.; Boedeker, E.; Zhu, C.; Cebotaru, L.; Kovbasnjuk, O. Enterohemorrhagic Escherichia coli infection stimulates Shiga toxin 1 macropinocytosis and transcytosis across intestinal epithelial cells. Am. J. Physiol. Cell. Physiol. 2011, 301, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, M.E.; Hällbrink, M.M.; Elmquist, A.M.; Langel, U. Passage of cell-penetrating peptides across a human epithelial cell layer in vitro. Biochem. J. 2004, 377, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.H.A.; Atcliffe, B.W.; Howlett, G.J.; Sawyer, W.H. Conformation and orientation of penetratin in phospholipid membranes. J. Pept. Sci. 2006, 12, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Derossi, D.; Calvet, S.; Trembleau, A.; Brunissen, A.; Chassaing, G.; Prochiantz, A. Cell internalization of the third helix of the Antennapedia homeodomain is receptor-independent. J. Biol. Chem. 1996, 271, 18188–18193. [Google Scholar] [CrossRef] [PubMed]
- Binder, H.; Lindblom, G. Charge-dependent translocation of the Trojan peptide penetratin across lipid membranes. Biophys. J. 2003, 85, 982–995. [Google Scholar] [CrossRef]
- Sakai, N.; Takeuchi, T.; Futaki, S.; Matile, S. Direct observation of anion-mediated translocation of fluorescent oligoarginine carriers into and across bulk liquid and anionic bilayer membranes. Chembiochem 2005, 6, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Joanne, P.; Galanth, C.; Goasdoué, N.; Nicolas, P.; Sagan, S.; Lavielle, S.; Chassaing, G.; Amri, C.E.; Alved, I.D. Lipid reorganization induced by membrane-active peptides probed using differential scanning calorimetry. Biochim. Biophys. Acta 2009, 1788, 1772–1781. [Google Scholar] [CrossRef] [PubMed]
- Alves, I.D.; Goasdoué, N.; Correia, I.; Aubry, S.; Galanth, C.; Sagan, S.; Lavielle, S.; Chassaing, G. Membrane interaction and perturbation mechanisms induced by two cationic cell penetrating peptides with distinct charge distribution. Biochim. Biophys. Acta 2008, 1780, 948–959. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, M.; Foged, C.; Berthelsen, J.; Nielsen, H.M. Peptide-enhanced oral delivery of therapeutic peptides and proteins. J. Drug Del. Sci. Tech. 2013, 23, 365–373. [Google Scholar] [CrossRef]
- Palm, C.; Jayamanne, M.; Kjellander, M.; Hällbrink, M. Peptide degradation is a critical determinant for cell-penetrating peptide uptake. Biochim. Biophys. Acta 2007, 1768, 1769–1776. [Google Scholar] [CrossRef] [PubMed]
- Rennert, R.; Wespe, C.; Beck-Sickinger, A.G.; Neundorf, I. Developing novel hCT derived cell-penetrating peptides with improved metabolic stability. Biochim. Biophys. Acta 2006, 1758, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Foerg, C.; Weller, K.M.; Rechsteiner, H.; Nielsen, H.M.; Fernández-Carneado, J.; Brunisholz, R.; Giralt, E.; Merkle, H.P. Metabolic cleavage and translocation efficiency of selected cell penetrating peptides: A comparative study with epithelial cell cultures. AAPS J. 2008, 10, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Tréhin, R.; Nielsen, H.M.; Jahnke, H.G.; Krauss, U.; Beck-Sickinger, A.G.; Merkle, H.P. Metabolic cleavage of cell-penetrating peptides in contact with epithelial models: Human calcitonin (hCT)-derived peptides, Tat(47-57) and penetratin(43-58). Biochem. J. 2004, 382, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Yang, M.; Kasimova, M.R.; Malmsten, M.; Franzyk, H.; Jorgensen, L.; Foged, C.; Nielsen, H.M. Membrane adsorption and binding, cellular uptake and cytotoxicity of cell-penetrating peptidomimetics with α-peptide/β-peptoid backbone: Effects of hydrogen bonding and α-chirality in the β-peptoid residues. Biochim. Biophys. Acta 2012, 1818, 2660–2668. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Kasimova, M.R.; Simonsen, A.H.; Jorgensen, L.; Malmsten, M.; Franzyk, H.; Foged, C.; Nielsen, H.M. Interaction of peptidomimetics with bilayer membranes: Biophysical characterization and cellular uptake. Langmuir 2012, 28, 5167–5175. [Google Scholar] [CrossRef] [PubMed]
- Nyakatura, E.K.; Mortier, J.; Radtke, V.S.; Wieczorek, S.; Araghi, R.R.; Baldauf, C.; Wolber, G.; Koksch, B. β- and γ-Amino Acids at α-Helical Interfaces. Toward the formation of highly stable foldameric coiled coils. Med. Chem. Lett. 2014, 5, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Horne, W.S.; Gellman, S.H. Foldamers with heterogeneous backbones. Acc. Chem. Res. 2008, 41, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Hyrup Møller, L.; Bahnsen, J.S.; Nielsen, H.M.; Østergaard, J.; Stürup, S.; Gammelgaard, B. Selenium as an alternative peptide label—Comparison to fluorophore-labelled penetratin. Eur. J. Pharm. Sci. 2015, 67, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Wolfhard Semmler MS. Peptides, multimers and polymers. In Handbook of Experimental Pharmacology; Springer: Berlin, Germany, 2008; pp. 168–212. [Google Scholar]
- Anderson, C.J.; Welch, M.J. Radiometal-labeled agents (non-technetium) for diagnostic imaging. Chem. Rev. 1999, 99, 2219–2239. [Google Scholar] [CrossRef] [PubMed]
- Walther, C.; Ott, I.; Gust, R.; Neundorf, I. Specific labeling with potent radiolabels alters the uptake of cell-penetrating peptides. Biopolymers 2009, 92, 445–451. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kristensen, M.; Birch, D.; Mørck Nielsen, H. Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos. Int. J. Mol. Sci. 2016, 17, 185. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17020185
Kristensen M, Birch D, Mørck Nielsen H. Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos. International Journal of Molecular Sciences. 2016; 17(2):185. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17020185
Chicago/Turabian StyleKristensen, Mie, Ditlev Birch, and Hanne Mørck Nielsen. 2016. "Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos" International Journal of Molecular Sciences 17, no. 2: 185. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17020185