
Pharmacological Modulators of Endoplasmic Reticulum Stress in Metabolic Diseases

Abstract
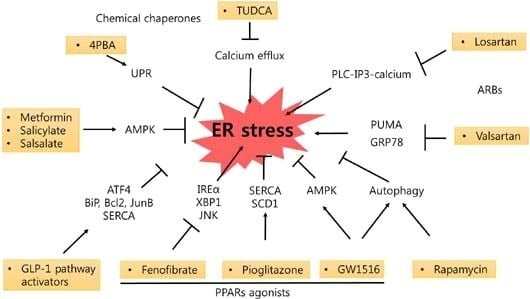
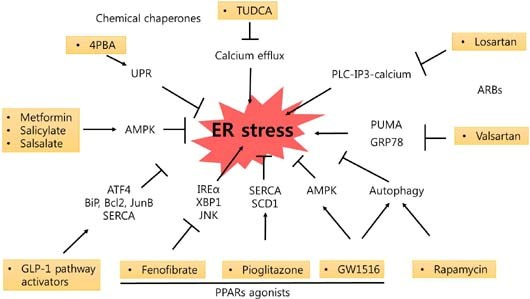
:
1. Introduction
2. Cellular Aspects of ER-Stress and Metabolic Diseases
2.1. Diabetes Mellitus
2.2. Cardiovascular Diseases (CVD)
2.3. Non-Alcoholic Fatty Liver Disease (NAFLD)
3. Effects of Pharmacologic Modulators on ER Stress-Induced Cellular Damage
3.1. Rapamycin
3.2. Chemical Chaperones
3.3. AMPK Activators
3.4. Glucagon-Like Peptide-1 (GLP-1) Receptor Agonists and Dipeptidyl Peptidase IV (DPP-IV) Inhibitors
3.5. Peroxisome Proliferator-Activated Receptor (PPAR) Agonists
3.6. Angiotensin II Type 1 Receptor Blockers (ARBs)
4. Conclusions

{kind=link}
| Category | Drug | Mediator | Effect on Disorders | Reference |
|---|---|---|---|---|
| mTOR inhibitors | Rapamycin | Autophagy ↑ | NAFLD | [6] |
| Hepatic ischemia | [7] | |||
| Insulin resistance (hepatocyte, skeletal myocytes) | [8,13] | |||
| Diabetes | [10] | |||
| IRE1/JNK ↓ | Apoptosis (renal cell) | [11,12] | ||
| Chemical chaperones | 4-PBA | GRP78 ↓, CHOP ↓ | Adipogenesis | [16] |
| TUDCA | Calcium efflux ↓ | Apoptosis (hepatocyte) | [17] | |
| eIF2α ↓, CHOP ↓ | Steatohepatitis | [19] | ||
| AMPK activators | Metformin | AMPK ↑ | Renal fibrosis | [26] |
| AMPK ↑, PPARδ ↑ | Vascular dysfunction | [27] | ||
| eIF2α ↓, JNK ↓, IRS-1 ↓ | Apoptosis (hepatocyte, endothelium) | [24,25,31] | ||
| Salicylate/Salsalate | AMPK ↑ | Apoptosis (endothelium) | [31] | |
| Insulin resistance (hepatocyte) | [32] | |||
| AICAR | AMPK ↑ | EDR (aortae) | [22] | |
| Cardiac hypoxic injury | [23] | |||
| GLP-1 receptor agonists and DPP-4 inhibitors | Exenatide | PKA ↑, ATF4 ↑, BiP ↑, Bcl2 ↑, JunB ↑, SERCA ↑, Autophagy ↑ | Apoptosis (β-cell), NAFLD | [38,39,46,47] |
| Vildagliptin | C/EBPβ ↓ | β-cell loss | [49] | |
| Gemigliptin | Akt/PERK/CHOP ↓, IRE1α/JNK-p38 ↓ | Apoptosis (cardiomyocyte) | [51] | |
| PPARs agonists | Fenofibrate | IRE1α/XBP1/JNK ↓, AMPK ↑, eNOS ↑ | NAFLD, EDV | [52,53,54] |
| Pioglitazone | SERCA ↑, SCD1 ↑ | β-cell dysfunction, Apoptosis (macrophage) | [55,59] | |
| GW1516 | AMPK ↑, ERK1/2 ↓ | Insulin resistance (skeletal myocytes) | [60] | |
| Autophagy ↑ | Cardiac hypertrophy | [62] | ||
| ARBs | Valsartan | PUMA ↓, GRP78 ↓ | Apoptosis (cardiomyocyte, renal cell) | [63,64] |
| Losartan | PLC-IP3-calcium ↓ | β-cell dysfunction | [65] | |
| Olmesartan | GRP78 ↓, CHOP ↓ | Autoimmune myocarditis | [66] | |
| Telmisartan | GRP78 ↓, CHOP ↓ | Cardiac hypertrophy | [67] |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schroder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Varadarajan, S.; Bampton, E.T.; Smalley, J.L.; Tanaka, K.; Caves, R.E.; Butterworth, M.; Wei, J.; Pellecchia, M.; Mitcheson, J.; Gant, T.W.; et al. A novel cellular stress response characterised by a rapid reorganisation of membranes of the endoplasmic reticulum. Cell Death Differ. 2012, 19, 1896–1907. [Google Scholar] [CrossRef] [PubMed]
- Lipson, K.L.; Fonseca, S.G.; Ishigaki, S.; Nguyen, L.X.; Foss, E.; Bortell, R.; Rossini, A.A.; Urano, F. Regulation of insulin biosynthesis in pancreatic β cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab. 2006, 4, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Delepine, M.; Nicolino, M.; Barrett, T.; Golamaully, M.; Lathrop, G.M.; Julier, C. EIF2AK3, encoding translation initiation factor 2-α kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat. Genet. 2000, 25, 406–409. [Google Scholar] [PubMed]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef]
- Tersey, S.A.; Nishiki, Y.; Templin, A.T.; Cabrera, S.M.; Stull, N.D.; Colvin, S.C.; Evans-Molina, C.; Rickus, J.L.; Maier, B.; Mirmira, R.G. Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 2012, 61, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Marhfour, I.; Lopez, X.M.; Lefkaditis, D.; Salmon, I.; Allagnat, F.; Richardson, S.J.; Morgan, N.G.; Eizirik, D.L. Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia 2012, 55, 2417–2420. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zeng, H.; Zhang, Y.; Jungries, R.; Chung, P.; Plesken, H.; Sabatini, D.D.; Ron, D. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol. Cell 2001, 7, 1153–1163. [Google Scholar] [CrossRef]
- Laybutt, D.R.; Preston, A.M.; Akerfeldt, M.C.; Kench, J.G.; Busch, A.K.; Biankin, A.V.; Biden, T.J. Endoplasmic reticulum stress contributes to β cell apoptosis in type 2 diabetes. Diabetologia 2007, 50, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Koizumi, A.; Takeda, K.; Gotoh, T.; Akira, S.; Araki, E.; Mori, M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Investig. 2002, 109, 525–532. [Google Scholar] [CrossRef]
- Thameem, F.; Farook, V.S.; Bogardus, C.; Prochazka, M. Association of amino acid variants in the activating transcription factor 6 gene (ATF6) on 1q21-q23 with type 2 diabetes in Pima Indians. Diabetes 2006, 55, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.J.; Chung, H.S.; Yoon, C.S.; Ko, J.H.; Jun, H.J.; Kim, T.K.; Lee, S.H.; Ko, K.S.; Rhee, B.D.; Kim, M.K.; et al. The Effects of glyburide on apoptosis and endoplasmic reticulum stress in INS-1 cells in a glucolipotoxic condition. Diabetes Metab. J. 2011, 35, 480–488. [Google Scholar] [CrossRef]
- Minamino, T.; Komuro, I.; Kitakaze, M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ. Res. 2010, 107, 1071–1082. [Google Scholar] [CrossRef]
- Cnop, M.; Ladriere, L.; Hekerman, P.; Ortis, F.; Cardozo, A.K.; Dogusan, Z.; Flamez, D.; Boyce, M.; Yuan, J.; Eizirik, D.L. Selective inhibition of eukaryotic translation initiation factor 2α dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic β-cell dysfunction and apoptosis. J. Biol. Chem. 2007, 282, 3989–3997. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lhotak, S.; Hilditch, B.A.; Austin, R.C. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation 2005, 111, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Seimon, T.A.; Nadolski, M.J.; Liao, X.; Magallon, J.; Nguyen, M.; Feric, N.T.; Koschinsky, M.L.; Harkewicz, R.; Witztum, J.L.; Tsimikas, S.; et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010, 12, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Herzig, S.; Kulkarni, R.N.; Montminy, M. TRB3: A tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 2003, 300, 1574–1577. [Google Scholar] [CrossRef] [PubMed]
- Passos, E.; Ascensao, A.; Martins, M.J.; Magalhaes, J. Endoplasmic reticulum stress response in non-alcoholic steatohepatitis: The possible role of physical exercise. Metabolism 2015, 64, 780–792. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Mirshahi, F.; Cheung, O.; Natarajan, R.; Maher, J.W.; Kellum, J.M.; Sanyal, A.J. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Q.; Xu, C.F.; Yu, C.H.; Chen, W.X.; Li, Y.M. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Harding, H.P.; Zhang, Y.; Oyadomari, M.; Ron, D. Dephosphorylation of translation initiation factor 2α enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 2008, 7, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, S.; Malhotra, J.; Hassler, J.R.; Back, S.H.; Wang, G.; Chang, L.; Xu, W.; Miao, H.; Leonardi, R.; et al. The unfolded protein response transducer IRE1α prevents ER stress-induced hepatic steatosis. EMBO J. 2011, 30, 1357–1375. [Google Scholar] [CrossRef] [PubMed]
- Usui, M.; Yamaguchi, S.; Tanji, Y.; Tominaga, R.; Ishigaki, Y.; Fukumoto, M.; Katagiri, H.; Mori, K.; Oka, Y.; Ishihara, H. Atf6α-null mice are glucose intolerant due to pancreatic β-cell failure on a high-fat diet but partially resistant to diet-induced insulin resistance. Metabolism 2012, 61, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.T.; Yoshimori, T. A current perspective of autophagosome biogenesis. Cell Res. 2014, 24, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wang, Z.; Tao, L.; Wang, Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 2010, 6, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Klionsky, D.J. Endoplasmic reticulum stress: A new pathway to induce autophagy. Autophagy 2007, 3, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillon, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Hua, X.; Li, D.; Zhang, J.; Xia, Q. Rapamycin attenuates mouse liver ischemia and reperfusion injury by inhibiting endoplasmic reticulum stress. Transplant. Proc. 2015, 47, 1646–1652. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, R.Q.; Zeng, X.Y.; Zhou, X.; Li, S.; Jo, E.; Molero, J.C.; Ye, J.M. Restoration of autophagy alleviates hepatic ER stress and impaired insulin signalling transduction in high fructose-fed male mice. Endocrinology 2015, 156, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Gu, L.; Wang, Y.; Fan, N.; Ma, Y.; Peng, Y. Rapamycin improves palmitate-induced ER stress/NF κB pathways associated with stimulating autophagy in adipocytes. Mediat. Inflamm. 2015, 2015, 272313. [Google Scholar]
- Bachar-Wikstrom, E.; Wikstrom, J.D.; Kaiser, N.; Cerasi, E.; Leibowitz, G. Improvement of ER stress-induced diabetes by stimulating autophagy. Autophagy 2013, 9, 626–628. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Nakajima, S.; Saito, Y.; Takahashi, S.; Katoh, R.; Kitamura, M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell Death Differ. 2012, 19, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Liu, Y.; Zhang, L.; Huang, S.; Ding, H.F.; Dong, Z. mTOR contributes to ER stress and associated apoptosis in renal tubular cells. Am. J. Physiol. Ren. Physiol. 2015, 308, F267–F274. [Google Scholar] [CrossRef]
- Hwang, S.L.; Li, X.; Lee, J.Y.; Chang, H.W. Improved insulin sensitivity by rapamycin is associated with reduction of mTOR and S6K1 activities in L6 myotubes. Biochem. Biophys. Res. Commun. 2012, 418, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Engin, F.; Hotamisligil, G.S. Restoring endoplasmic reticulum function by chemical chaperones: An emerging therapeutic approach for metabolic diseases. Diabetes Obes. Metab. 2010, 12, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Welch, W.J.; Brown, C.R. Influence of molecular and chemical chaperones on protein folding. Cell Stress Chaperones 1996, 1, 109–115. [Google Scholar] [CrossRef]
- Basseri, S.; Lhotak, S.; Sharma, A.M.; Austin, R.C. The chemical chaperone 4-phenylbutyrate inhibits adipogenesis by modulating the unfolded protein response. J. Lipid Res. 2009, 50, 2486–2501. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Khaoustov, V.I.; Chung, C.C.; Sohn, J.; Krishnan, B.; Lewis, D.E.; Yoffe, B. Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress-induced caspase-12 activation. Hepatology 2002, 36, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.J.; Yoon, J.H.; Kwak, M.S.; Jang, E.S.; Lee, J.H.; Yu, S.J.; Kim, Y.J.; Kim, C.Y.; Lee, H.S. Tauroursodeoxycholic acid attenuates progression of steatohepatitis in mice fed a methionine-choline-deficient diet. Dig. Dis. Sci. 2014, 59, 1461–1474. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Kemp, B.E. AMPK in health and disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhang, M.; Wang, S.; Liang, B.; Zhao, Z.; Liu, C.; Wu, M.; Choi, H.C.; Lyons, T.J.; Zou, M.H. Activation of AMP-activated protein kinase inhibits oxidized LDL-triggered endoplasmic reticulum stress in vivo. Diabetes 2010, 59, 1386–1396. [Google Scholar] [CrossRef] [PubMed]
- Terai, K.; Hiramoto, Y.; Masaki, M.; Sugiyama, S.; Kuroda, T.; Hori, M.; Kawase, I.; Hirota, H. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol. Cell. Biol. 2005, 25, 9554–9575. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Jeong, S.K.; Kim, H.R.; Chae, S.W.; Chae, H.J. Metformin regulates palmitate-induced apoptosis and ER stress response in HepG2 liver cells. Immunopharmacol. Immunotoxicol. 2010, 32, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Simon-Szabo, L.; Kokas, M.; Mandl, J.; Keri, G.; Csala, M. Metformin attenuates palmitate-induced endoplasmic reticulum stress, serine phosphorylation of IRS-1 and apoptosis in rat insulinoma cells. PLoS ONE 2014, 9, e97868. [Google Scholar]
- Kim, H.; Moon, S.Y.; Kim, J.S.; Baek, C.H.; Kim, M.; Min, J.Y.; Lee, S.K. Activation of AMP-activated protein kinase inhibits ER stress and renal fibrosis. Am. J. Physiol. Ren. Physiol. 2015, 308, F226–F236. [Google Scholar] [CrossRef] [PubMed]
- Cheang, W.S.; Tian, X.Y.; Wong, W.T.; Lau, C.W.; Lee, S.S.; Chen, Z.Y.; Yao, X.; Wang, N.; Huang, Y. Metformin protects endothelial function in diet-induced obese mice by inhibition of endoplasmic reticulum stress through 5' adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor delta pathway. Arterioscler. Thromb. Vasc Biol. 2014, 34, 830–836. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Kundu, B.K.; Wu, H.C.; Hashmi, S.S.; Guthrie, P.; Locke, L.W.; Roy, R.J.; Matherne, G.P.; Berr, S.S.; Terwelp, M.; et al. Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart. J. Am. Heart Assoc. 2013, 2, e004796. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Alhusaini, S.; McGee, K.; Schisano, B.; Harte, A.; McTernan, P.; Kumar, S.; Tripathi, G. Lipopolysaccharide, high glucose and saturated fatty acids induce endoplasmic reticulum stress in cultured primary human adipocytes: Salicylate alleviates this stress. Biochem. Biophys. Res. Commun. 2010, 397, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.; Wen, X.; Ma, X.N.; Chen, W.; Huang, F.; Kou, J.; Qi, L.W.; Liu, B.; Liu, K. Pharmacological activation of AMPK prevents Drp1-mediated mitochondrial fission and alleviates endoplasmic reticulum stress-associated endothelial dysfunction. J. Mol. Cell. Cardiol. 2015, 86, 62–74. [Google Scholar] [CrossRef]
- Jung, T.W.; Lee, S.Y.; Hong, H.C.; Choi, H.Y.; Yoo, H.J.; Baik, S.H.; Choi, K.M. AMPK activator-mediated inhibition of endoplasmic reticulum stress ameliorates carrageenan-induced insulin resistance through the suppression of selenoprotein P in HepG2 hepatocytes. Mol. Cell. Endocrinol. 2014, 382, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, T.J.; Habener, J.F. The glucagon-like peptides. Endocr. Rev. 1999, 20, 876–913. [Google Scholar] [CrossRef] [PubMed]
- Farilla, L.; Bulotta, A.; Hirshberg, B.; Li Calzi, S.; Khoury, N.; Noushmehr, H.; Bertolotto, C.; di Mario, U.; Harlan, D.M.; Perfetti, R. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology 2003, 144, 5149–5158. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.Y.; Oh, T.J.; Cho, Y.M. Glucagon-like peptide-1 increases mitochondrial biogenesis and function in INS-1 Rat Insulinoma Cells. Endocrinol. Metab. (Seoul) 2015, 30, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Buteau, J.; Foisy, S.; Joly, E.; Prentki, M. Glucagon-like peptide 1 induces pancreatic β-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes 2003, 52, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Hui, H.; Wright, C.; Perfetti, R. Glucagon-like peptide 1 induces differentiation of islet duodenal homeobox-1-positive pancreatic ductal cells into insulin-secreting cells. Diabetes 2001, 50, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Yusta, B.; Baggio, L.L.; Estall, J.L.; Koehler, J.A.; Holland, D.P.; Li, H.; Pipeleers, D.; Ling, Z.; Drucker, D.J. GLP-1 receptor activation improves β cell function and survival following induction of endoplasmic reticulum stress. Cell Metab. 2006, 4, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Cunha, D.A.; Ladriere, L.; Ortis, F.; Igoillo-Esteve, M.; Gurzov, E.N.; Lupi, R.; Marchetti, P.; Eizirik, D.L.; Cnop, M. Glucagon-like peptide-1 agonists protect pancreatic β-cells from lipotoxic endoplasmic reticulum stress through upregulation of BiP and JunB. Diabetes 2009, 58, 2851–2862. [Google Scholar] [CrossRef] [PubMed]
- Yamane, S.; Hamamoto, Y.; Harashima, S.; Harada, N.; Hamasaki, A.; Toyoda, K.; Fujita, K.; Joo, E.; Seino, Y.; Inagaki, N. GLP-1 receptor agonist attenuates endoplasmic reticulum stress-mediated β-cell damage in Akita mice. J. Diabetes Investig. 2011, 2, 104–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, Y.S.; Lee, Y.J.; Kang, Y.; Han, J.; Lim, O.K.; Jun, H.S. Exendin-4 inhibits glucolipotoxic ER stress in pancreatic beta cells via regulation of SREBP1c and C/EBPβ transcription factors. J. Endocrinol. 2013, 216, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Y.; Jiang, S.; Yu, H.; An, W. Enhanced endoplasmic reticulum SERCA activity by overexpression of hepatic stimulator substance gene prevents hepatic cells from ER stress-induced apoptosis. Am. J. Physiol. Cell Physiol. 2014, 306, C279–C290. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younce, C.W.; Burmeister, M.A.; Ayala, J.E. Exendin-4 attenuates high glucose-induced cardiomyocyte apoptosis via inhibition of endoplasmic reticulum stress and activation of SERCA2a. Am. J. Physiol. Cell Physiol. 2013, 304, C508–C518. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hong, S.W.; Park, S.E.; Rhee, E.J.; Park, C.Y.; Oh, K.W.; Park, S.W.; Lee, W.Y. Exendin-4 attenuates endoplasmic reticulum stress through a SIRT1-dependent mechanism. Cell Stress Chaperones 2014, 19, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Mells, J.E.; Fu, P.P.; Saxena, N.K.; Anania, F.A. GLP-1 analogs reduce hepatocyte steatosis and improve survival by enhancing the unfolded protein response and promoting macroautophagy. PLoS ONE 2011, 6, e25269. [Google Scholar] [CrossRef] [PubMed]
- Barnett, A. DPP-4 inhibitors and their potential role in the management of type 2 diabetes. Int. J. Clin. Pract. 2006, 60, 1454–1470. [Google Scholar] [CrossRef]
- Shimizu, S.; Hosooka, T.; Matsuda, T.; Asahara, S.; Koyanagi-Kimura, M.; Kanno, A.; Bartolome, A.; Etoh, H.; Fuchita, M.; Teruyama, K.; et al. DPP4 inhibitor vildagliptin preserves β-cell mass through amelioration of endoplasmic reticulum stress in C/EBPB transgenic mice. J. Mol. Endocrinol. 2012, 49, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.A.; Choi, Y.K.; Jung, G.S.; Seo, H.Y.; Kim, H.S.; Jang, B.K.; Kim, J.G.; Lee, I.K.; Kim, M.K.; Park, K.G. Sitagliptin attenuates methionine/choline-deficient diet-induced steatohepatitis. Diabetes Res. Clin. Pract. 2014, 105, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.J.; Jung, T.W.; Ryu, J.Y.; Hong, H.C.; Choi, H.Y.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Choi, K.M.; Choi, D.S.; et al. Dipeptidyl petidase-IV inhibitor (gemigliptin) inhibits tunicamycin-induced endoplasmic reticulum stress, apoptosis and inflammation in H9c2 cardiomyocytes. Mol. Cell. Endocrinol. 2014, 392, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Lu, Y.; Shen, X.; Bao, Y.; Cheng, J.; Chen, L.; Li, B.; Zhang, Q. Fenofibrate treatment attenuated chronic endoplasmic reticulum stress in the liver of nonalcoholic fatty liver disease mice. Pharmacology 2015, 95, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.M.; Qadri, I.; Janssen, R.C.; Friedman, J.E. Fenofibrate and PBA prevent fatty acid-induced loss of adiponectin receptor and pAMPK in human hepatoma cells and in hepatitis C virus-induced steatosis. J. Lipid Res. 2009, 50, 2193–2202. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Cheng, J.; Chen, L.; Li, C.; Chen, G.; Gui, L.; Shen, B.; Zhang, Q. Endoplasmic reticulum stress involved in high-fat diet and palmitic acid-induced vascular damages and fenofibrate intervention. Biochem. Biophys. Res. Commun. 2015, 458, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kono, T.; Ahn, G.; Moss, D.R.; Gann, L.; Zarain-Herzberg, A.; Nishiki, Y.; Fueger, P.T.; Ogihara, T.; Evans-Molina, C. PPAR-γ activation restores pancreatic islet SERCA2 levels and prevents β-cell dysfunction under conditions of hyperglycemic and cytokine stress. Mol. Endocrinol. 2012, 26, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Yoshiuchi, K.; Kaneto, H.; Matsuoka, T.A.; Kasami, R.; Kohno, K.; Iwawaki, T.; Nakatani, Y.; Yamasaki, Y.; Shimomura, I.; Matsuhisa, M. Pioglitazone reduces ER stress in the liver: Direct monitoring of in vivo ER stress using ER stress-activated indicator transgenic mice. Endocr. J. 2009, 56, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Peter, A.; Weigert, C.; Staiger, H.; Machicao, F.; Schick, F.; Machann, J.; Stefan, N.; Thamer, C.; Haring, H.U.; Schleicher, E. Individual stearoyl-coa desaturase 1 expression modulates endoplasmic reticulum stress and inflammation in human myotubes and is associated with skeletal muscle lipid storage and insulin sensitivity in vivo. Diabetes 2009, 58, 1757–1765. [Google Scholar] [CrossRef]
- Thorn, K.; Hovsepyan, M.; Bergsten, P. Reduced levels of SCD1 accentuate palmitate-induced stress in insulin-producing β-cells. Lipids Health Dis. 2010, 9, 108. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, J.; Ichiki, T.; Takahara, Y.; Kojima, H.; Sankoda, C.; Kitamoto, S.; Tokunou, T.; Sunagawa, K. PPARγ agonists attenuate palmitate-induced ER stress through up-regulation of SCD-1 in macrophages. PLoS ONE 2015, 10, e0128546. [Google Scholar] [CrossRef]
- Salvado, L.; Barroso, E.; Gomez-Foix, A.M.; Palomer, X.; Michalik, L.; Wahli, W.; Vazquez-Carrera, M. PPARβ/δ prevents endoplasmic reticulum stress-associated inflammation and insulin resistance in skeletal muscle cells through an AMPK-dependent mechanism. Diabetologia 2014, 57, 2126–2135. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Guardia, D.; Palomer, X.; Coll, T.; Serrano, L.; Rodriguez-Calvo, R.; Davidson, M.M.; Merlos, M.; El Kochairi, I.; Michalik, L.; Wahli, W.; et al. PPARβ/δ activation blocks lipid-induced inflammatory pathways in mouse heart and human cardiac cells. Biochim. Biophys. Acta 2011, 1811, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Palomer, X.; Capdevila-Busquets, E.; Botteri, G.; Salvado, L.; Barroso, E.; Davidson, M.M.; Michalik, L.; Wahli, W.; Vazquez-Carrera, M. PPARβ/δ attenuates palmitate-induced endoplasmic reticulum stress and induces autophagic markers in human cardiac cells. Int. J. Cardiol. 2014, 174, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Dong, Z.; Geng, J.; Sun, Y.; Liu, G.; Kang, W.; Zhang, Y.; Ge, Z. Valsartan protects against ER stress-induced myocardial apoptosis via CHOP/Puma signaling pathway in streptozotocin-induced diabetic rats. Eur. J. Pharm. Sci. 2011, 42, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Peng, P.A.; Wang, L.; Ma, Q.; Xin, Y.; Zhang, O.; Han, H.Y.; Liu, X.L.; Ji, Q.W.; Zhou, Y.J.; Zhao, Y.X. Valsartan protects HK-2 cells from contrast media-induced apoptosis by inhibiting endoplasmic reticulum stress. Cell Biol. Int. 2015, 39, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Madec, A.M.; Cassel, R.; Dubois, S.; Ducreux, S.; Vial, G.; Chauvin, M.A.; Mesnier, A.; Chikh, K.; Bosco, D.; Rieusset, J.; et al. Losartan, an angiotensin II type 1 receptor blocker, protects human islets from glucotoxicity through the phospholipase C pathway. FASEB. J. 2013, 27, 5122–5130. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, V.; Watanabe, K.; Veeraveedu, P.T.; Gurusamy, N.; Ma, M.; Thandavarayan, R.A.; Lakshmanan, A.P.; Yamaguchi, K.; Suzuki, K.; Kodama, M. Olmesartan, an AT1 antagonist, attenuates oxidative stress, endoplasmic reticulum stress and cardiac inflammatory mediators in rats with heart failure induced by experimental autoimmune myocarditis. Int. J. Biol. Sci. 2011, 7, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.S.; Shangguan, H.J.; Shang, Z.; Yang, L.; Meng, X.M.; Qiao, S.B. Endoplasmic reticulum stress caused by left ventricular hypertrophy in rats: Effects of telmisartan. Am. J. Med. Sci. 2011, 342, 318–323. [Google Scholar] [CrossRef]
- Chan, J.Y.; Cooney, G.J.; Biden, T.J.; Laybutt, D.R. Differential regulation of adaptive and apoptotic unfolded protein response signalling by cytokine-induced nitric oxide production in mouse pancreatic beta cells. Diabetologia 2011, 54, 1766–1776. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, T.W.; Choi, K.M. Pharmacological Modulators of Endoplasmic Reticulum Stress in Metabolic Diseases. Int. J. Mol. Sci. 2016, 17, 192. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17020192
Jung TW, Choi KM. Pharmacological Modulators of Endoplasmic Reticulum Stress in Metabolic Diseases. International Journal of Molecular Sciences. 2016; 17(2):192. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17020192
Chicago/Turabian StyleJung, Tae Woo, and Kyung Mook Choi. 2016. "Pharmacological Modulators of Endoplasmic Reticulum Stress in Metabolic Diseases" International Journal of Molecular Sciences 17, no. 2: 192. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17020192