
Canine Models for Copper Homeostasis Disorders

Abstract
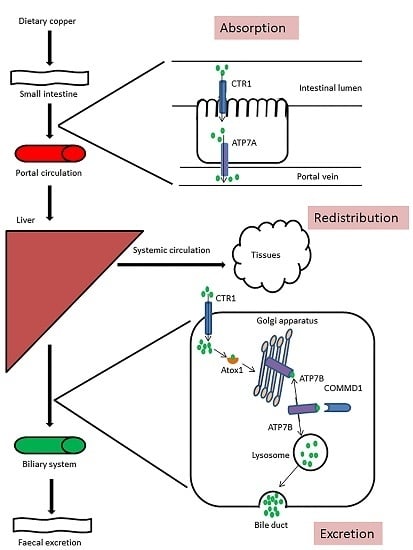
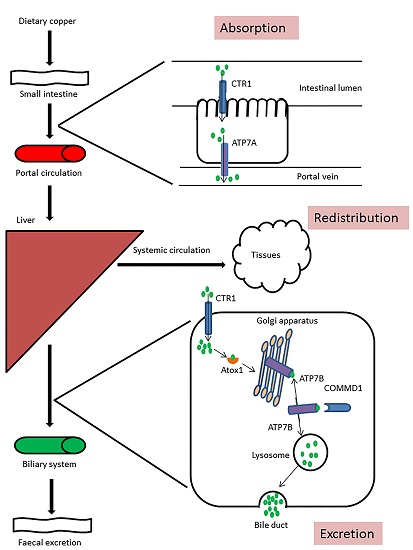
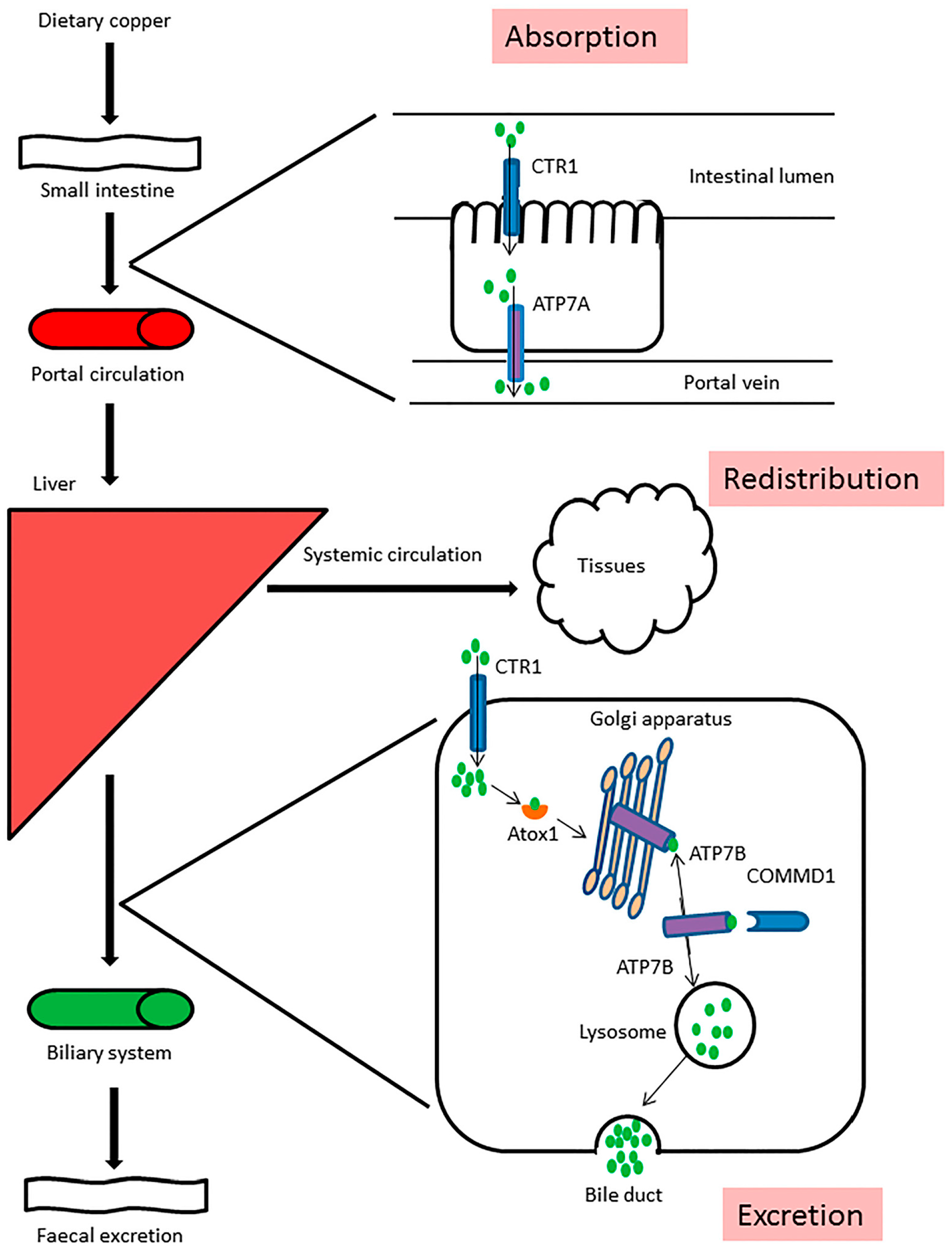
:
{kind=link}
{kind=link}
1. Introduction
2. Copper Homeostasis
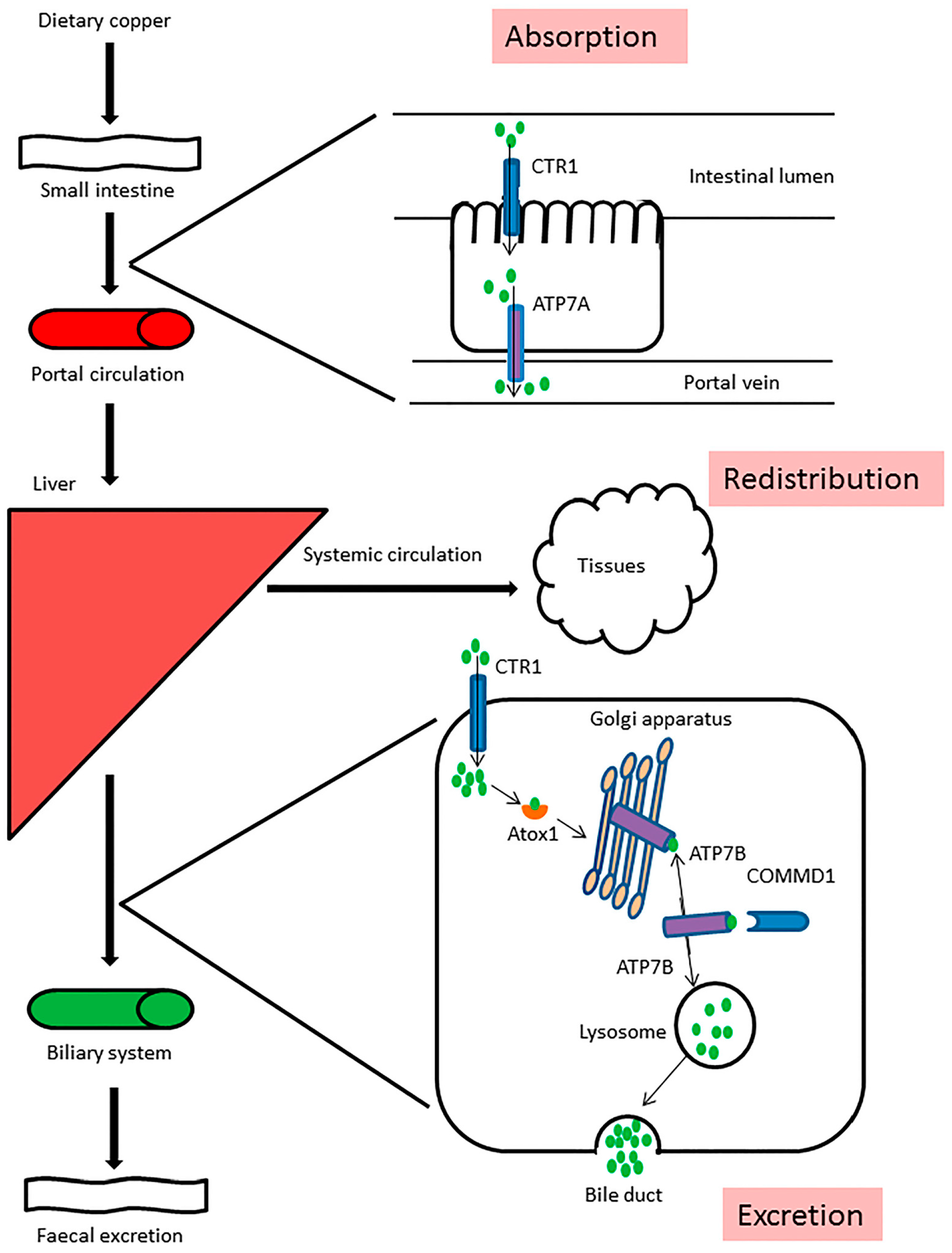
3. Copper Metabolism Disorders in Humans
4. Copper Metabolism Disorders in Dogs
5. The Power of Gene-Mapping Studies in Dog for Identification of Genes Involved in Copper Metabolism
6. Canine Models for Development of New Chelation and Dietary Treatments
6.1. Chelation Therapy
6.2. Dietary Strategy
7. Organoids and Transplantation Studies
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Osredkar, J.; Sustar, N. Copper and zinc, biological role and significance of Copper/Zinc imbalance. J. Clin. Toxicol. 2011. [Google Scholar] [CrossRef]
- Opazo, C.M.; Greenough, M.A.; Bush, A.I. Copper: From neurotransmission to neuroproteostasis. Front. Aging Neurosci. 2014, 6, 143. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.Y.; Rennert, O.M. The role of copper in iron metabolism. Ann. Clin. Lab. Sci. 1980, 10, 338–344. [Google Scholar] [PubMed]
- Horn, D.; Barrientos, A. Mitochondrial copper metabolism and delivery to cytochrome C oxidase. IUBMB Life 2008, 60, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.; Bartoli, G.; Ragnelli, A.M.; Cittadini, A.; Palozza, P.; Aimola, P.; Zarivi, O.; Bonfigli, A. Copper deficiency and pigmentation in the Rat: Morphofunctional aspects. J. Submicrosc. Cytol. Pathol. 1992, 24, 273–279. [Google Scholar] [PubMed]
- Bousquet-Moore, D.; Prohaska, J.R.; Nillni, E.A.; Czyzyk, T.; Wetsel, W.C.; Mains, R.E.; Eipper, B.A. Interactions of peptide amidation and copper: Novel biomarkers and mechanisms of neural dysfunction. Neurobiol. Dis. 2010, 37, 130–140. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, B.L. Roles for iron and copper in connective tissue biosynthesis. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1981, 294, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Kaler, S.G. ATP7A-related copper transport diseases-emerging concepts and future trends. Nat. Rev. Neurol. 2011, 7, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Menkes, J.H.; Alter, M.; Steigleder, G.K.; Weakley, D.R.; Sung, J.H. A Sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degeneration. Pediatrics 1962, 29, 764–779. [Google Scholar] [PubMed]
- Vulpe, C.; Levinson, B.; Whitney, S.; Packman, S.; Gitschier, J. Isolation of a Candidate gene for menkes disease and evidence that it encodes a copper-transporting ATPase. Nat. Genet. 1993, 3, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Bull, P.C.; Thomas, G.R.; Rommens, J.M.; Forbes, J.R.; Cox, D.W. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the menkes gene. Nat. Genet. 1993, 5, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E.; Petrukhin, K.; Chernov, I.; Pellequer, J.L.; Wasco, W.; Ross, B.; Romano, D.M.; Parano, E.; Pavone, L.; Brzustowicz, L.M. The Wilson disease gene is a copper transporting ATPase with homology to the menkes disease gene. Nat. Genet. 1993, 5, 344–350. [Google Scholar] [CrossRef]
- Roberts, E.A.; Schilsky, M.L. American association for study of liver diseases (AASLD). Diagnosis and treatment of Wilson disease: An update. Hepatology 2008, 47, 2089–2111. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, P. Phenotype-genotype correlations in patients with Wilson’s disease. Ann. N. Y. Acad. Sci. 2014, 1315, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, N.; Yamaguchi, Y.; Aoki, T. Treatment and management of Wilson’s disease. Pediatr. Int. 1999, 41, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Sternlieb, I. Wilson’s disease: Indications for liver transplants. Hepatology 1984, 4, 15S–17S. [Google Scholar] [CrossRef] [PubMed]
- Tanner, M.S. Role of Copper in Indian childhood cirrhosis. Am. J. Clin. Nutr. 1998, 67, 1074S–1081S. [Google Scholar] [PubMed]
- Muller, T.; Feichtinger, H.; Berger, H.; Muller, W. Endemic tyrolean infantile cirrhosis: An ecogenetic disorder. Lancet 1996, 347, 877–880. [Google Scholar] [CrossRef]
- Scheinberg, I.H.; Sternlieb, I. Wilson disease and idiopathic copper toxicosis. Am. J. Clin. Nutr. 1996, 63, 842S–845S. [Google Scholar] [PubMed]
- Fieten, H.; Leegwater, P.A.; Watson, A.L.; Rothuizen, J. Canine models of copper toxicosis for understanding mammalian copper metabolism. Mamm. Genome 2012, 23, 62–75. [Google Scholar] [CrossRef] [PubMed]
- van De Sluis, B.; Rothuizen, J.; Pearson, P.L.; van Oost, B.A.; Wijmenga, C. Identification of a new copper metabolism gene by positional cloning in a purebred dog population. Hum. Mol. Genet. 2002, 11, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Fieten, H.; Gill, Y.; Martin, A.J.; Concilli, M.; Dirksen, K.; van Steenbeek, F.G.; Spee, B.; van den Ingh, T.S.; Martens, E.C.; Festa, P.; et al. The Menkes and Wilson disease genes counteract in copper toxicosis in labrador retrievers: A new canine model for copper-metabolism disorders. Dis. Model. Mech. 2016, 9, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Shearin, A.L.; Ostrander, E.A. Leading the way: Canine models of genomics and disease. Dis. Model. Mech. 2010, 3, 27–34. [Google Scholar] [CrossRef]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J., 3rd; Zody, M.C.; et al. Genome Sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Kasai, N.; Osanai, T.; Miyoshi, I.; Kamimura, E.; Yoshida, M.C.; Dempo, K. Clinico-pathological studies of LEC rats with hereditary hepatitis and hepatoma in the acute phase of hepatitis. Lab. Anim. Sci. 1990, 40, 502–505. [Google Scholar] [PubMed]
- Theophilos, M.B.; Cox, D.W.; Mercer, J.F. The toxic milk mouse is a murine model of wilson disease. Hum. Mol. Genet. 1996, 5, 1619–1624. [Google Scholar] [CrossRef] [PubMed]
- Huster, D.; Finegold, M.J.; Morgan, C.T.; Burkhead, J.L.; Nixon, R.; Vanderwerf, S.M.; Gilliam, C.T.; Lutsenko, S. Consequences of copper accumulation in the livers of the ATP7B−/− (Wilson disease gene) knockout mice. Am. J. Pathol. 2006, 168, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, S.; Weisman, G.A.; Gitlin, J.D.; Petris, M.J. Conditional knockout of the Menkes disease copper transporter demonstrates its critical role in embryogenesis. PLoS ONE 2012, 7, e43039. [Google Scholar] [CrossRef] [PubMed]
- Mason, K.E. A conspectus of research on copper metabolism and requirements of man. J. Nutr. 1979, 109, 1979–2066. [Google Scholar] [PubMed]
- Moriya, M.; Ho, Y.H.; Grana, A.; Nguyen, L.; Alvarez, A.; Jamil, R.; Ackland, M.L.; Michalczyk, A.; Hamer, P.; Ramos, D.; et al. Copper is taken up efficiently from albumin and α2-macroglobulin by cultured human cells by more than one mechanism. Am. J. Physiol. Cell Physiol. 2008, 295, C708–C721. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, P.V.; Klomp, L.W. New Developments in the regulation of intestinal copper absorption. Nutr. Rev. 2009, 67, 658–672. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Gitschier, J. hCTR1: A human gene for copper uptake identified by complementation in yeast. Proc. Natl. Acad. Sci. USA. 1997, 94, 7481–7486. [Google Scholar] [CrossRef] [PubMed]
- Coyle, P.; Philcox, J.C.; Carey, L.C.; Rofe, A.M. Metallothionein: The multipurpose protein. Cell. Mol. Life Sci. 2002, 59, 627–647. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.H.; Ciriolo, M.R.; Peisach, J. The role of glutathione in copper metabolism and toxicity. J. Biol. Chem. 1989, 264, 5598–5605. [Google Scholar] [PubMed]
- Rosenzweig, A.C. Copper delivery by metallochaperone proteins. Acc. Chem. Res. 2001, 34, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.J.; Kunst, C.; Culotta, V.C. Copper activation of superoxide dismutase 1 (SOD1) in vivo. Role for protein-protein interactions with the copper chaperone for SOD1. J. Biol. Chem. 2000, 275, 33771–33776. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.; Glerum, D.M.; Tzagoloff, A. Isolation of a cDNA encoding the human homolog of COX17, a yeast gene essential for mitochondrial copper recruitment. Hum. Genet. 1997, 99, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Klomp, L.W.; Lin, S.J.; Yuan, D.S.; Klausner, R.D.; Culotta, V.C.; Gitlin, J.D. Identification and functional expression of HAH1, a novel human gene involved in copper homeostasis. J. Biol. Chem. 1997, 272, 9221–9226. [Google Scholar] [PubMed]
- Wang, Y.; Hodgkinson, V.; Zhu, S.; Weisman, G.A.; Petris, M.J. Advances in the Understanding of Mammalian Copper Transporters. Adv. Nutr. 2011, 2, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.E.; Turski, M.L.; Nose, Y.; Casad, M.; Rockman, H.A.; Thiele, D.J. Cardiac copper deficiency activates a systemic signaling mechanism that communicates with the copper acquisition and storage organs. Cell Metab. 2010, 11, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Yanagimoto, C.; Harada, M.; Kumemura, H.; Abe, M.; Koga, H.; Sakata, M.; Kawaguchi, T.; Terada, K.; Hanada, S.; Taniguchi, E.; et al. Copper incorporation into ceruloplasmin is regulated by Niemann-Pick C1 protein. Hepatol. Res. 2011, 41, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Cater, M.A.; La Fontaine, S.; Shield, K.; Deal, Y.; Mercer, J.F. ATP7B mediates vesicular sequestration of copper: Insight into biliary copper excretion. Gastroenterology 2006, 130, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Nyasae, L.; Bustos, R.; Braiterman, L.; Eipper, B.; Hubbard, A. Dynamics of endogenous ATP7A (Menkes Protein) in intestinal epithelial cells: Copper-dependent redistribution between two intracellular sites. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1181–G1194. [Google Scholar] [CrossRef] [PubMed]
- Polishchuk, E.V.; Concilli, M.; Iacobacci, S.; Chesi, G.; Pastore, N.; Piccolo, P.; Paladino, S.; Baldantoni, D.; van IJzendoorn, S.C.; Chan, J.; et al. Wilson disease protein ATP7B utilizes lysosomal exocytosis to maintain copper homeostasis. Dev. Cell 2014, 29, 686–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips-Krawczak, C.A.; Singla, A.; Starokadomskyy, P.; Deng, Z.; Osborne, D.G.; Li, H.; Dick, C.J.; Gomez, T.S.; Koenecke, M.; Zhang, J.S.; et al. COMMD1 is linked to the WASH complex and regulates endosomal trafficking of the copper transporter ATP7A. Mol. Biol. Cell 2015, 26, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonk, W.I.; de Bie, P.; Wichers, C.G.; van den Berghe, P.V.; van der Plaats, R.; Berger, R.; Wijmenga, C.; Klomp, L.W.; van de Sluis, B. The copper-transporting capacity of ATP7A mutants associated with menkes disease is ameliorated by COMMD1 as a result of improved protein expression. Cell. Mol. Life Sci. 2012, 69, 149–163. [Google Scholar] [CrossRef] [PubMed]
- De Bie, P.; van de Sluis, B.; Burstein, E.; van de Berghe, P.V.; Muller, P.; Berger, R.; Gitlin, J.D.; Wijmenga, C.; Klomp, L.W. Distinct Wilson’s disease mutations in ATP7B are associated with enhanced binding to COMMD1 and reduced stability of ATP7B. Gastroenterology 2007, 133, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Miyayama, T.; Hiraoka, D.; Kawaji, F.; Nakamura, E.; Suzuki, N.; Ogra, Y. Roles of COMM-domain-containing 1 in stability and recruitment of the copper-transporting ATPase in a mouse hepatoma cell line. Biochem. J. 2010, 429, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.S.; Holmes, C.S.; Kaler, S.G. Relative efficiencies of plasma catechol levels and ratios for neonatal diagnosis of menkes disease. Neurochem. Res. 2009, 34, 1464–1468. [Google Scholar] [CrossRef]
- Kaler, S.G.; Holmes, C.S.; Goldstein, D.S.; Tang, J.; Godwin, S.C.; Donsante, A.; Liew, C.J.; Sato, S.; Patronas, N. Neonatal diagnosis and treatment of menkes disease. N. Engl. J. Med. 2008, 358, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Abdel Ghaffar, T.Y.; Elsayed, S.M.; Elnaghy, S.; Shadeed, A.; Elsobky, E.S.; Schmidt, H. Phenotypic and genetic characterization of a cohort of pediatric Wilson disease patients. BMC Pediatr. 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, P.; Czlonkowska, A.; Merle, U.; Ferenc, S.; Gromadzka, G.; Yurdaydin, C.; Vogel, W.; Bruha, R.; Schmidt, H.T.; Stremmel, W. Late-onset Wilson’s disease. Gastroenterology 2007, 132, 1294–1298. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Ray, K. Wilson’s disease: An Update. Nat. Clin. Pract. Neurol. 2006, 2, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Ala, A.; Walker, A.P.; Ashkan, K.; Dooley, J.S.; Schilsky, M.L. Wilson’s disease. Lancet 2007, 369, 397–408. [Google Scholar] [CrossRef]
- Wilson Disease Mutation Database. Available online: http://www.Wilsondisease.Med.Ualberta.Ca/Database.Asp (accessed on 7 October 2009).
- Czlonkowska, A.; Gromadzka, G.; Chabik, G. Monozygotic female twins discordant for phenotype of Wilson’s disease. Mov. Disord. 2009, 24, 1066–1069. [Google Scholar] [CrossRef] [PubMed]
- Gromadzka, G.; Rudnicka, M.; Chabik, G.; Przybylkowski, A.; Czlonkowska, A. Genetic variability in the methylenetetrahydrofolate reductase gene (MTHFR) affects clinical expression of Wilson’s disease. J. Hepatol. 2011, 55, 913–919. [Google Scholar] [CrossRef]
- Schiefermeier, M.; Kollegger, H.; Madl, C.; Polli, C.; Oder, W.; Kuhn, H.; Berr, F.; Ferenci, P. The impact of apolipoprotein E genotypes on age at onset of symptoms and phenotypic expression in Wilson’s disease. Brain 2000, 123 Pt 3, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Merle, U.; Stremmel, W.; Gessner, R. Influence of homozygosity for methionine at codon 129 of the human prion gene on the onset of neurological and hepatic symptoms in Wilson disease. Arch. Neurol. 2006, 63, 982–985. [Google Scholar] [CrossRef]
- Litwin, T.; Gromadzka, G.; Czlonkowska, A. Apolipoprotein E gene (APOE) genotype in Wilson’s disease: impact on clinical presentation. Parkinsonism Relat. Disord. 2012, 18, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Gromadzka, G.; Czlonkowska, A. Influence of IL-1RN intron 2 variable number of tandem repeats (VNTR) polymorphism on the age at onset of neuropsychiatric symptoms in Wilson’s disease. Int. J. Neurosci. 2011, 121, 8–15. [Google Scholar] [CrossRef]
- Weiss, K.H.; Runz, H.; Noe, B.; Gotthardt, D.N.; Merle, U.; Ferenci, P.; Stremmel, W.; Fullekrug, J. Genetic analysis of BIRC4/XIAP as a Putative modifier gene of wilson disease. J. Inherit. Metab. Dis. 2010, 33 (Suppl. 3), S233–S240. [Google Scholar] [CrossRef] [PubMed]
- Schilsky, M.L. Treatment of Wilson’s disease: What are the relative roles of penicillamine, trientine, and zinc supplementation? Curr. Gastroenterol. Rep. 2001, 3, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Czlonkowska, A.; Litwin, T.; Karlinski, M.; Dziezyc, K.; Chabik, G.; Czerska, M. d-Penicillamine vs. zinc sulfate as first-line therapy for Wilson’s disease. Eur. J. Neurol. 2014, 21, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Boga, S.; Jain, D.; Schilsky, M.L. Trientine induced colitis during therapy for Wilson disease: A case report and review of the literature. BMC Pharmacol. Toxicol. 2015, 16. 30–015–0031-z. [Google Scholar] [CrossRef] [PubMed]
- Dahlman, T.; Hartvig, P.; Lofholm, M.; Nordlinder, H.; Loof, L.; Westermark, K. Long-term treatment of Wilson’s disease with Triethylene tetramine dihydrochloride (Trientine). QJM 1995, 88, 609–616. [Google Scholar] [PubMed]
- Brewer, G.J.; Terry, C.A.; Aisen, A.M.; Hill, G.M. Worsening of neurologic syndrome in patients with Wilson’s disease with initial Penicillamine therapy. Arch. Neurol. 1987, 44, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Turkay, A.; Yuzbaziyan-Gurkan, V. Development of neurologic symptoms in a patient with asymptomatic Wilson’s disease treated with penicillamine. Arch. Neurol. 1994, 51, 304–305. [Google Scholar] [CrossRef] [PubMed]
- Ranucci, G.; di Dato, F.; Spagnuolo, M.I.; Vajro, P.; Iorio, R. Zinc Monotherapy is effective in Wilson’s disease patients with mild liver disease diagnosed in childhood: a retrospective study. Orphanet J. Rare Dis. 2014, 9. [Google Scholar] [CrossRef]
- Marin, C.; Robles, R.; Parrilla, G.; Ramirez, P.; Bueno, F.S.; Parrilla, P. Liver Transplantation in Wilson’s disease: Are its indications established? Transplant. Proc. 2007, 39, 2300–2301. [Google Scholar] [CrossRef] [PubMed]
- Tamura, S.; Sugawara, Y.; Kishi, Y.; Akamatsu, N.; Kaneko, J.; Makuuchi, M. Living-related liver transplantation for Wilson’s disease. Clin. Transplant. 2005, 19, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Emre, S.; Atillasoy, E.O.; Ozdemir, S.; Schilsky, M.; Rathna Varma, C.V.; Thung, S.N.; Sternlieb, I.; Guy, S.R.; Sheiner, P.A.; Schwartz, M.E.; et al. Orthotopic liver transplantation for Wilson’s disease: A single-center experience. Transplantation 2001, 72, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Bax, R.T.; Hassler, A.; Luck, W.; Hefter, H.; Krageloh-Mann, I.; Neuhaus, P.; Emmrich, P. Cerebral manifestation of Wilson’s disease successfully treated with liver transplantation. Neurology 1998, 51, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Podgaetz, E.; Chan, C. Liver transplant team. Liver transplantation for Wilson s disease: Our experience with review of the literature. Ann. Hepatol. 2003, 2, 131–134. [Google Scholar] [PubMed]
- Nayak, N.C.; Chitale, A.R. Indian Childhood Cirrhosis (ICC) & ICC-like diseases: The changing scenario of facts vs. notions. Indian J. Med. Res. 2013, 137, 1029–1042. [Google Scholar] [PubMed]
- Li, Y.; Togashi, Y.; Sato, S.; Emoto, T.; Kang, J.H.; Takeichi, N.; Kobayashi, H.; Kojima, Y.; Une, Y.; Uchino, J. Spontaneous hepatic copper accumulation in long-evans cinnamon rats with hereditary hepatitis. A model of Wilson’s disease. J. Clin. Investig. 1991, 87, 1858–1861. [Google Scholar] [CrossRef] [PubMed]
- Haywood, S.; Muller, T.; Muller, W.; Heinz-Erian, P.; Tanner, M.S.; Ross, G. Copper-Associated Liver Disease in north ronaldsay sheep: A possible animal model for non-wilsonian hepatic copper toxicosis of infancy and childhood. J. Pathol. 2001, 195, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Puls, R. Mineral Levels in Animal Health: Diagnostic Data, 2nd ed.; Clearbrook, B.C., Ed.; Sherpa international, cop.: Dalhousie, QC, Canada, 1994. [Google Scholar]
- Fieten, H.; Dirksen, K.; van den Ingh, T.S.; Winter, E.A.; Watson, A.L.; Leegwater, P.A.; Rothuizen, J. d-Penicillamine treatment of copper-associated hepatitis in labrador retrievers. Vet. J. 2013, 196, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Mandigers, P.J.; van den Ingh, T.S.; Bode, P.; Teske, E.; Rothuizen, J. Association between Liver Copper Concentration and subclinical hepatitis in doberman pinschers. J. Vet. Intern. Med. 2004, 18, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Twedt, D.C.; Hunsaker, H.A.; Allen, K.G. Use of 2,3,2-Tetramine as a hepatic copper chelating agent for treatment of copper hepatotoxicosis in bedlington terriers. J. Am. Vet. Med. Assoc. 1988, 192, 52–56. [Google Scholar] [PubMed]
- Brewer, G.J.; Dick, R.D.; Schall, W.; Yuzbasiyan-Gurkan, V.; Mullaney, T.P.; Pace, C.; Lindgren, J.; Thomas, M.; Padgett, G. Use of zinc acetate to treat copper toxicosis in dogs. J. Am. Vet. Med. Assoc. 1992, 201, 564–568. [Google Scholar] [PubMed]
- Twedt, D.C.; Sternlieb, I.; Gilbertson, S.R. Clinical, morphologic, and chemical studies on copper toxicosis of bedlington terriers. J. Am. Vet. Med. Assoc. 1979, 175, 269–275. [Google Scholar] [PubMed]
- Hyun, C.; Filippich, L.J. Inherited canine copper toxicosis in australian bedlington terriers. J. Vet. Sci. 2004, 5, 19–28. [Google Scholar] [PubMed]
- Vonk, W.I.; Bartuzi, P.; de Bie, P.; Kloosterhuis, N.; Wichers, C.G.; Berger, R.; Haywood, S.; Klomp, L.W.; Wijmenga, C.; van de Sluis, B. Liver-specific commd1 knockout mice are susceptible to hepatic copper accumulation. PLoS ONE 2011, 6, e29183. [Google Scholar] [CrossRef] [PubMed]
- Klomp, A.E.; van de Sluis, B.; Klomp, L.W.; Wijmenga, C. The ubiquitously expressed MURR1 protein is absent in canine copper toxicosis. J. Hepatol. 2003, 39, 703–709. [Google Scholar] [CrossRef]
- Su, L.C.; Ravanshad, S.; Owen, C.A., Jr.; McCall, J.T.; Zollman, P.E.; Hardy, R.M. A Comparison of copper-loading disease in bedlington terriers and Wilson’s disease in humans. Am. J. Physiol. 1982, 243, G226–G230. [Google Scholar] [PubMed]
- Favier, R.P.; Spee, B.; Penning, L.C.; Rothuizen, J. Copper-induced hepatitis: The COMMD1 deficient dog as a translational animal model for human chronic hepatitis. Vet. Q. 2011, 31, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, G.; van den Ingh, T.S.; Bode, P.; Rothuizen, J. Copper-associated chronic hepatitis in labrador retrievers. J. Vet. Intern. Med. 2006, 20, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.L.; Keating, J.H.; Freeman, L.M.; Webster, C.R. Chronic hepatitis in labrador retrievers: Clinical presentation and prognostic factors. J. Vet. Intern. Med. 2007, 21, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Smedley, R.; Mullaney, T.; Rumbeiha, W. Copper-Associated hepatitis in labrador retrievers. Vet. Pathol. 2009, 46, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, G.; Heuven, H.C.; Leegwater, P.A.; Jones, P.G.; van den Ingh, T.S.; Bode, P.; Rothuizen, J. Heritabilities of copper-accumulating traits in labrador retrievers. Anim. Genet. 2008, 39, 454. [Google Scholar] [CrossRef] [PubMed]
- Haywood, S.; Rutgers, H.C.; Christian, M.K. Hepatitis and copper accumulation in skye terriers. Vet. Pathol. 1988, 25, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, L.P.; Shaw, D.; Dolan, M.; Raisbeck, M.; Crawford, S.; Dennis, G.L.; Olwin, D.B. Hereditary copper toxicosis in West Highland white terriers. Vet. Pathol. 1986, 23, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Webb, C.B.; Twedt, D.C.; Meyer, D.J. Copper-associated liver disease in dalmatians: A review of 10 dogs (1998–2001). J. Vet. Intern. Med. 2002, 16, 665–668. [Google Scholar]
- Karlsson, E.K.; Baranowska, I.; Wade, C.M.; Salmon Hillbertz, N.H.; Zody, M.C.; Anderson, N.; Biagi, T.M.; Patterson, N.; Pielberg, G.R.; Kulbokas, E.J., 3rd; et al. Efficient mapping of mendelian traits in dogs through genome-wide association. Nat. Genet. 2007, 39, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, E.K.; Lindblad-Toh, K. Leader of the pack: Gene mapping in dogs and other model organisms. Nat. Rev. Genet. 2008, 9, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.G.; Kim, L.V.; Sutter, N.B.; Carlson, S.; Lorentzen, T.D.; Malek, T.B.; Johnson, G.S.; DeFrance, H.B.; Ostrander, E.A.; Kruglyak, L. Genetic structure of the purebred domestic dog. Science 2004, 304, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Mandigers, P.J.; van den Ingh, T.S.; Bode, P.; Rothuizen, J. Improvement in Liver Pathology After 4 Months of d-penicillamine in 5 doberman pinschers with subclinical hepatitis. J. Vet. Intern. Med. 2005, 19, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Graham, D.W.; DiSpirito, A.A.; Alterman, M.A.; Galeva, N.; Larive, C.K.; Asunskis, D.; Sherwood, P.M. Methanobactin, a copper-acquisition compound from methane-oxidizing bacteria. Science 2004, 305, 1612–1615. [Google Scholar] [CrossRef] [PubMed]
- Summer, K.H.; Lichtmannegger, J.; Bandow, N.; Choi, D.W.; DiSpirito, A.A.; Michalke, B. The biogenic methanobactin is an effective chelator for copper in a rat model for wilson disease. J. Trace Elem. Med. Biol. 2011, 25, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Fieten, H.; Hooijer-Nouwens, B.D.; Biourge, V.C.; Leegwater, P.A.; Watson, A.L.; van den Ingh, T.S.; Rothuizen, J. Association of Dietary Copper and Zinc Levels with Hepatic Copper and Zinc Concentration in Labrador Retrievers. J. Vet. Intern. Med. 2012, 26, 1274–1280. [Google Scholar] [CrossRef] [PubMed]
- Fieten, H.; Biourge, V.C.; Watson, A.L.; Leegwater, P.A.; van den Ingh, T.S.; Rothuizen, J. Dietary management of labrador retrievers with subclinical hepatic copper accumulation. J. Vet. Intern. Med. 2015, 29, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Fieten, H.; Biourge, V.C.; Watson, A.L.; Leegwater, P.A.; van den Ingh, T.S.; Rothuizen, J. Nutritional management of inherited copper-associated hepatitis in the labrador retriever. Vet. J. 2014, 199, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, G.; Jones, P.G.; Biourge, V.; van den Ingh, T.S.; Mesu, S.J.; Bode, P.; Rothuizen, J. Dietary management of hepatic copper accumulation in labrador retrievers. J. Vet. Intern. Med. 2009, 23, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, K.; Nakagama, H.; Tajima, R.; Ushigome, M.; Ogra, Y.; Suzuki, K.T.; Yoshikawa, K.; Nagao, M. Effects of soy protein isolate on LEC rats, a model of Wilson disease: Mechanisms underlying enhancement of liver cell damage. Biochem. Biophys. Res. Commun. 2003, 302, 271–274. [Google Scholar] [CrossRef]
- Yonezawa, K.; Nunomiya, S.; Daigo, M.; Ogra, Y.; Suzuki, K.T.; Enomoto, K.; Nakagama, H.; Yoshikawa, K.; Nagao, M. Soy protein isolate enhances hepatic copper accumulation and cell damage in LEC rats. J. Nutr. 2003, 133, 1250–1254. [Google Scholar] [PubMed]
- Schilsky, M.L.; Scheinberg, I.H.; Sternlieb, I. Liver transplantation for Wilson’s disease: Indications and outcome. Hepatology 1994, 19, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Dalgetty, D.M.; Medine, C.N.; Iredale, J.P.; Hay, D.C. Progress and future challenges in stem cell-derived liver technologies. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G241–G248. [Google Scholar] [CrossRef] [PubMed]
- Van Laecke, S.; Desideri, F.; Geerts, A.; van Vlierberghe, H.; Berrevoet, F.; Rogiers, X.; Troisi, R.; de Hemptinne, B.; Vanholder, R.; Colle, I. Hypomagnesemia and the risk of new-onset diabetes after liver transplantation. Liver Transpl. 2010, 16, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Dorrell, C.; Boj, S.F.; van Es, J.H.; Li, V.S.; van de Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J.; et al. In Vitro Expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 2013, 494, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Gehart, H.; van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.; Ellis, E.; van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Nantasanti, S.; Spee, B.; Kruitwagen, H.S.; Chen, C.; Geijsen, N.; Oosterhoff, L.A.; van Wolferen, M.E.; Pelaez, N.; Fieten, H.; Wubbolts, R.W.; et al. Disease modeling and gene therapy of copper storage disease in canine hepatic organoids. Stem Cell. Rep. 2015, 5, 895–907. [Google Scholar] [CrossRef]
- Boch, J. TALEs of Genome Targeting. Nat. Biotechnol. 2011, 29, 135–136. [Google Scholar] [CrossRef] [PubMed]
- Horvath, P.; Barrangou, R. CRISPR/Cas, the immune system of bacteria and archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Leegwater, P.A.J.; Fieten, H. Canine Models for Copper Homeostasis Disorders. Int. J. Mol. Sci. 2016, 17, 196. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17020196
Wu X, Leegwater PAJ, Fieten H. Canine Models for Copper Homeostasis Disorders. International Journal of Molecular Sciences. 2016; 17(2):196. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17020196
Chicago/Turabian StyleWu, Xiaoyan, Peter A. J. Leegwater, and Hille Fieten. 2016. "Canine Models for Copper Homeostasis Disorders" International Journal of Molecular Sciences 17, no. 2: 196. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17020196