
Isolation and Characterization of a Novel Dicistrovirus Associated with Moralities of the Great Freshwater Prawn, Macrobrachium rosenbergii

Abstract
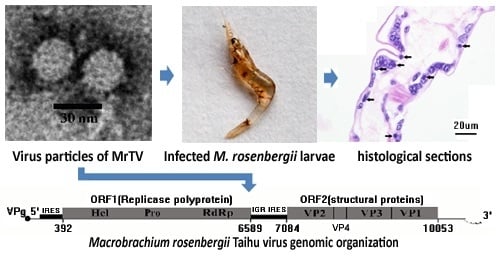
:
1. Introduction
2. Results
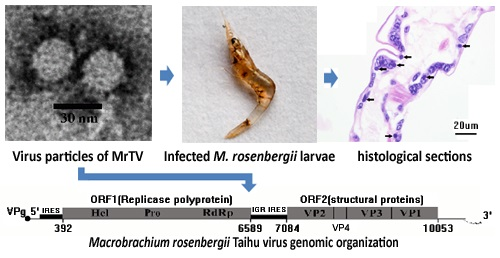
2.1. Isolation of an Unknown Virus
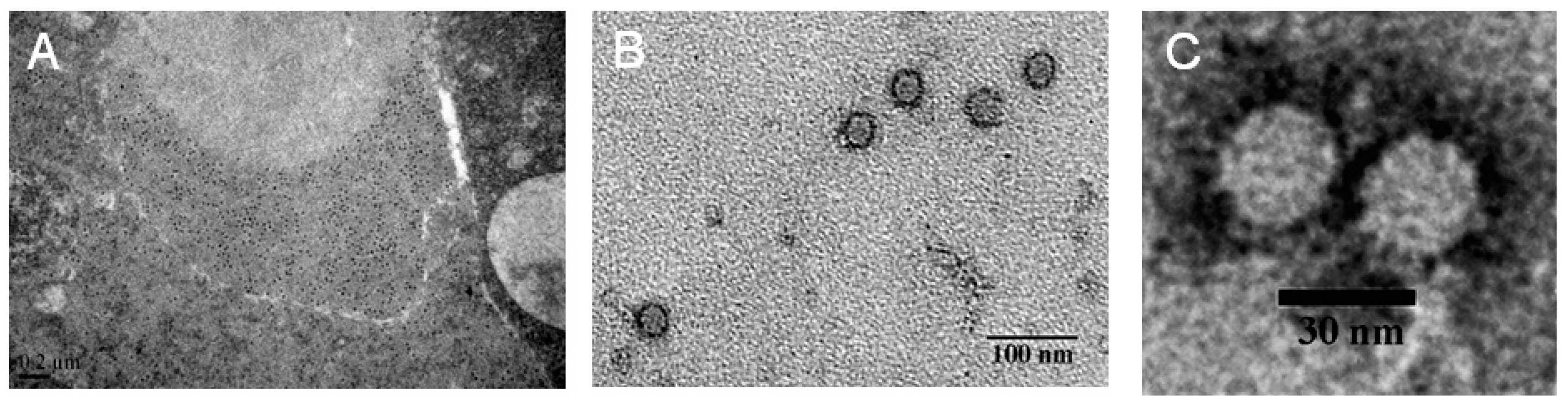
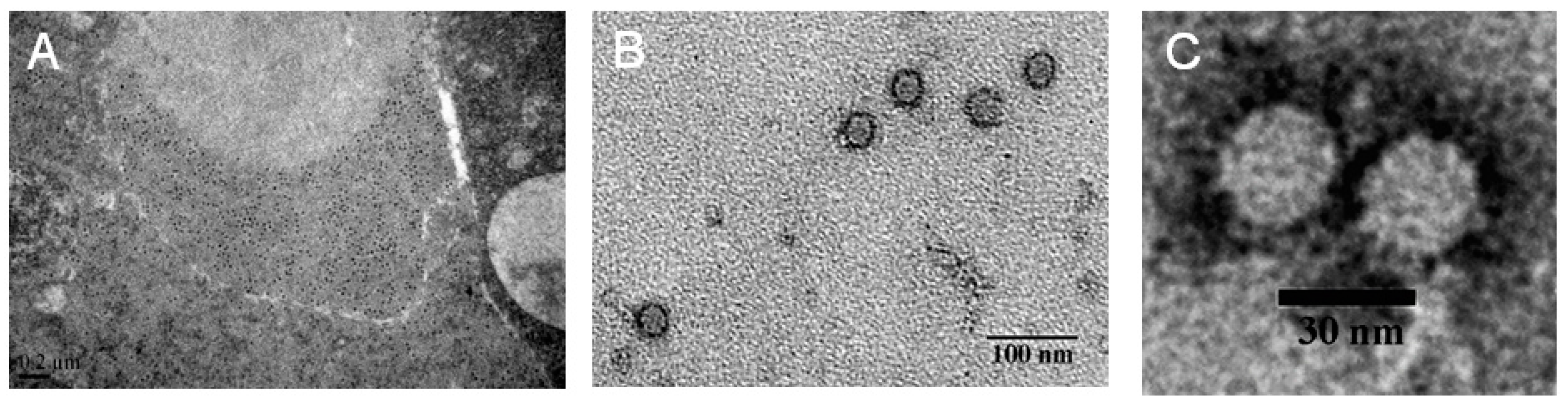
2.2. The Purified Virus Is an Unknown RNA Virus
2.3. Genome Characteristics of MrTV
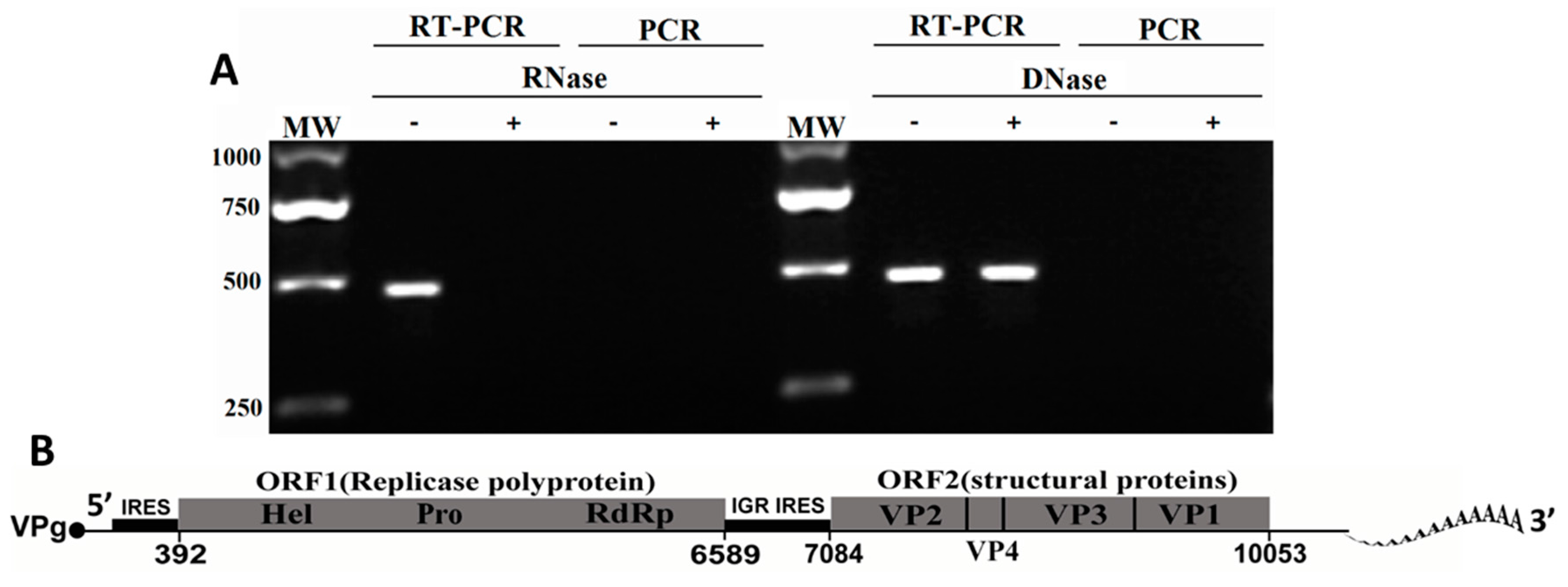
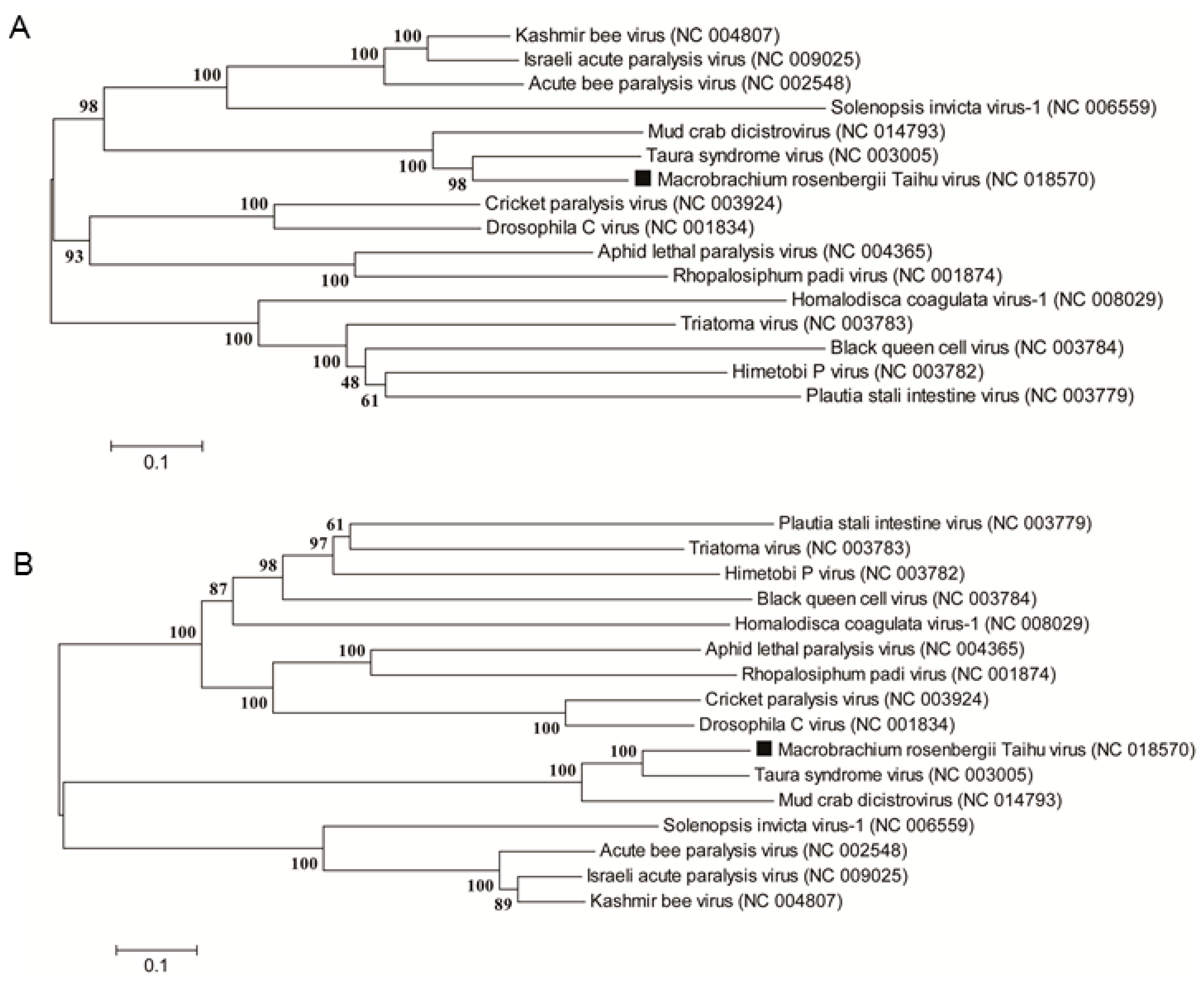
2.4. MrTV in Relation to Other Dicistroviruses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Acronym | Genome (%) | RdRp (%) | ORF2 (%) |
|---|---|---|---|---|
| Genus: Cripavirus | ||||
| Cricket paralysis virus 1 | CrPV | 32.7 | 45.2 | 18.3 |
| Aphid lethal paralysis virus | ALPV | 30.6 | 29.4 | 17.7 |
| Black queen cell virus | BQCV | 35.9 | 32.0 | 18.0 |
| Drosophila C virus | DCV | 34.0 | 41.0 | 17.3 |
| Plautia stali intestine virus | PSIV | 34.0 | 35.3 | 16.2 |
| Rhopalosiphum padi virus | RhPV | 30.3 | 32.4 | 19.1 |
| Triatoma virus | TrV | 35.1 | 34.3 | 19.4 |
| Homalodisca coagulata virus-1 | HoCV-1 | 34.6 | 37.0 | 17.7 |
| Himetobi P virus | HPV | 31.8 | 37.2 | 16.8 |
| Genus: Aparavirus | ||||
| Acute bee paralysis virus 1 | ABPV | 34.5 | 44.6 | 18.9 |
| Taura syndrome virus | TSV | 58.7 | 86.7 | 71.7 |
| Kashmir bee virus | KBV | 34.2 | 44.2 | 18.5 |
| Solenopsis invicta virus-1 | SINV-1 | 29.1 | 43.8 | 17.5 |
| Israeli acute paralysis virus | IAPV | 34.5 | 45.7 | 19.1 |
| Unclassed genus | ||||
| Mud crab dicistroviruses | MCDV | 55.1 | 79.6 | 60.4 |

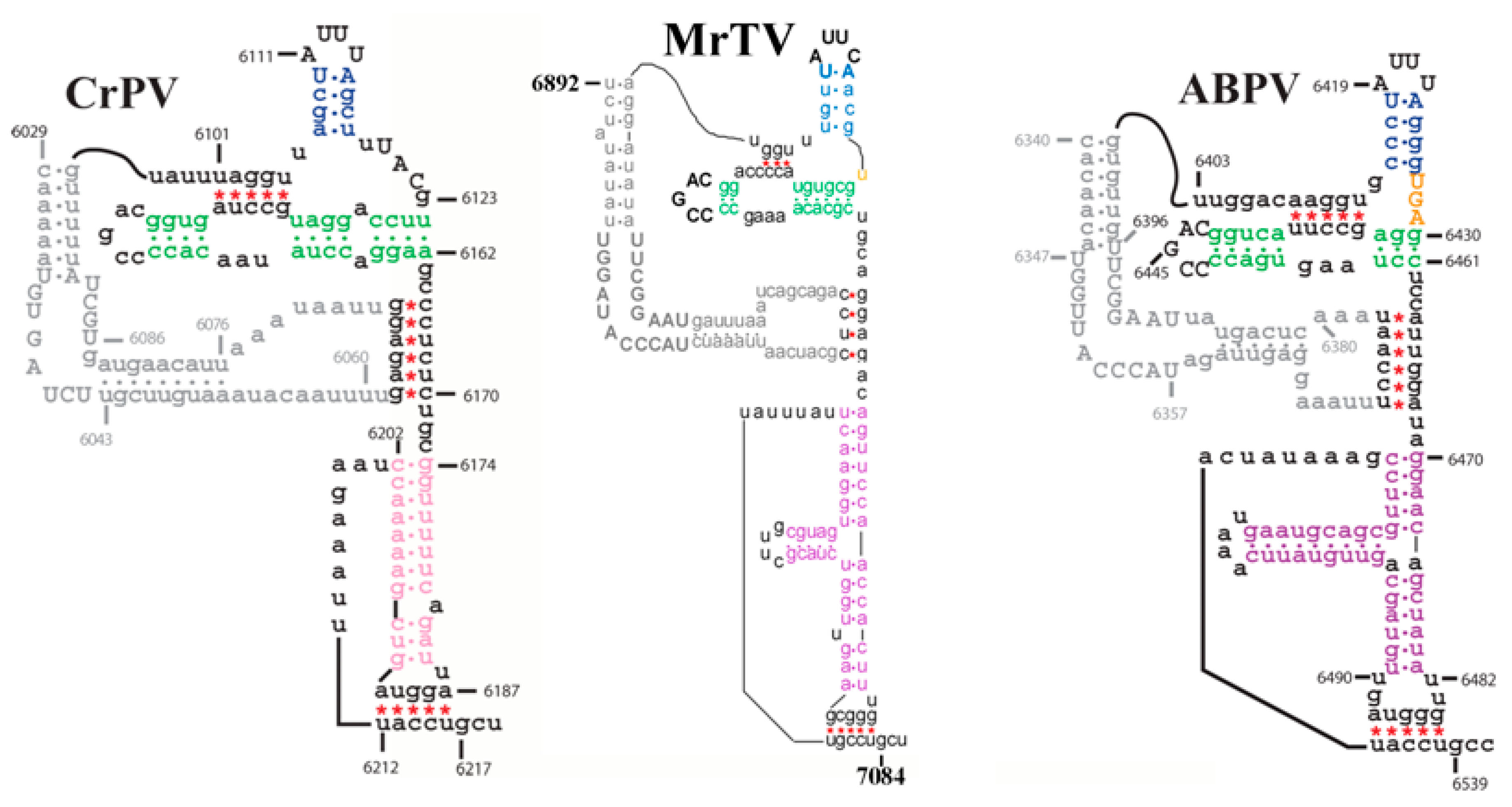
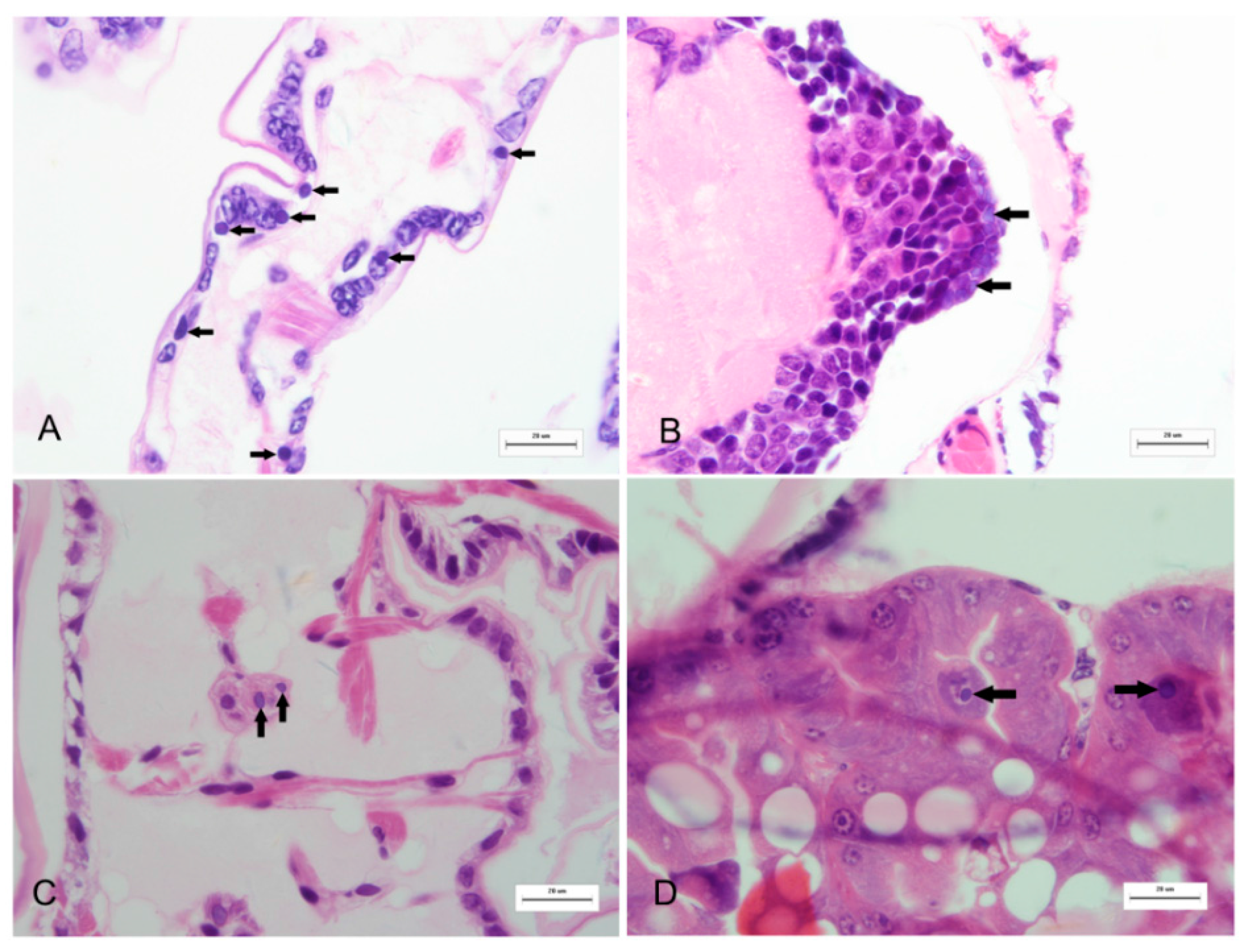
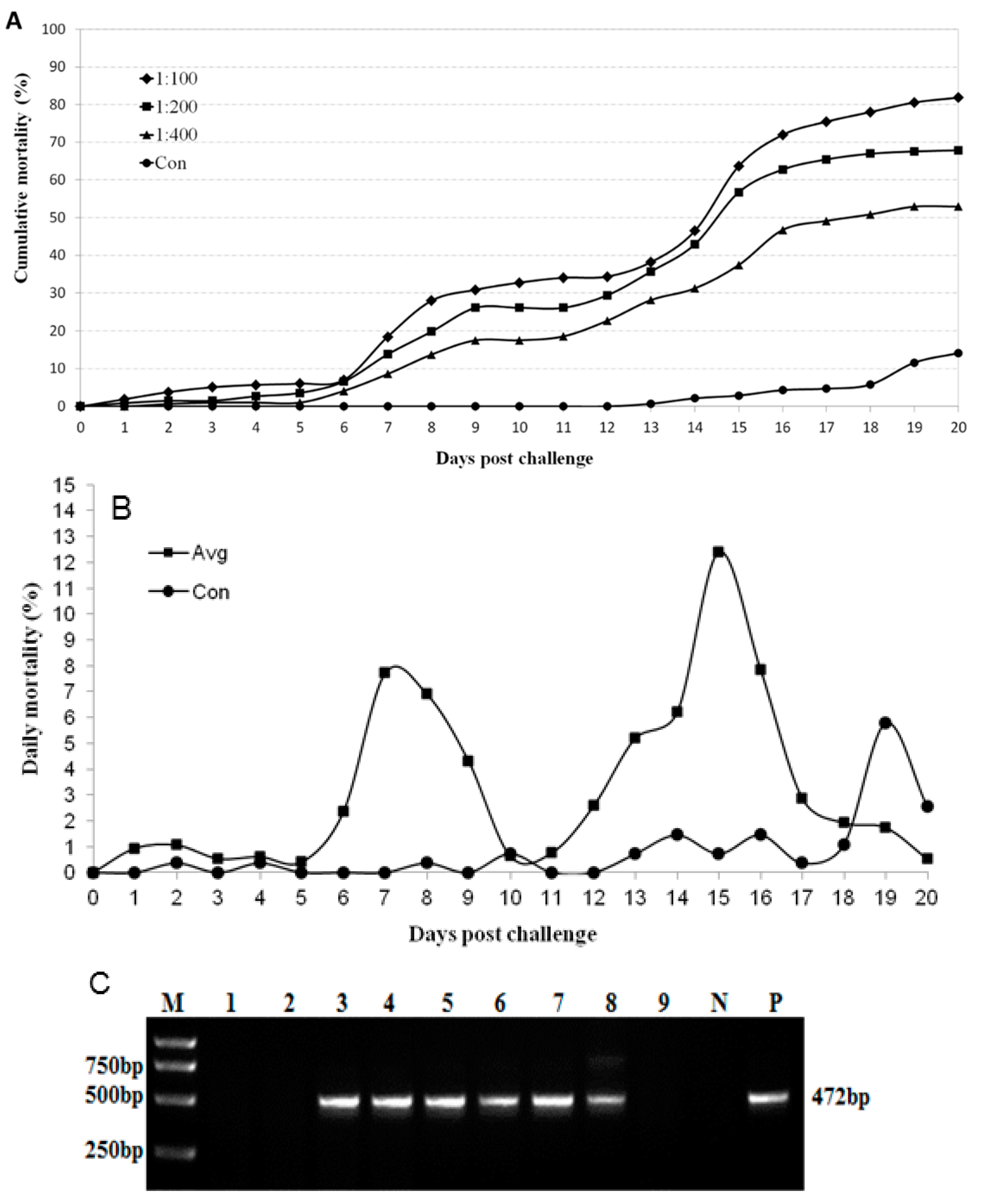
2.5. MrTV Is the Causative Agent of the Disease
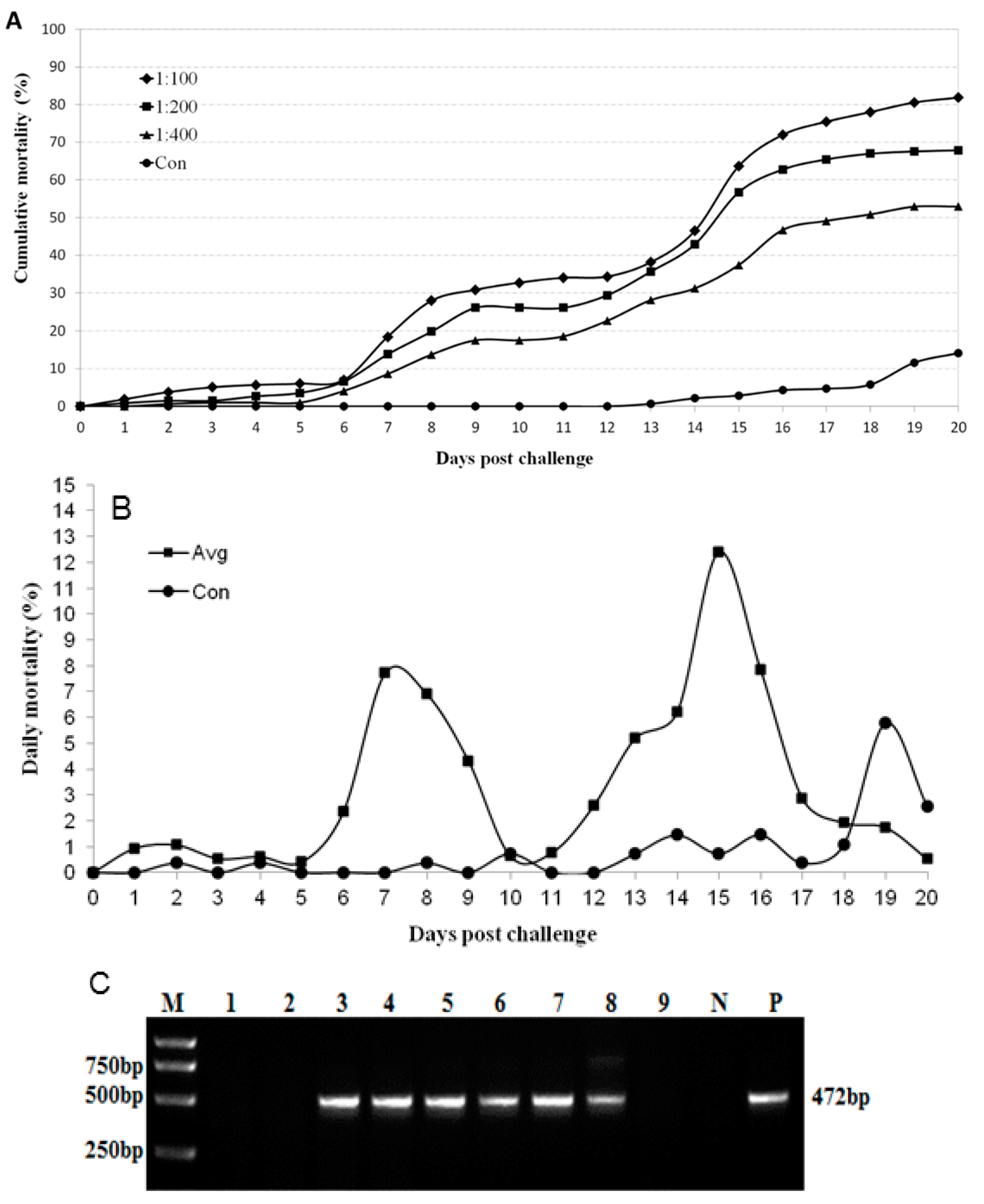
3. Discussion
4. Materials and Methods
4.1. Larvae of M. rosenbergii
4.2. Virus Isolation, Purification, and Examination by Electron Microscopy
4.3. Extraction of Viral Genome or Total RNA
4.4. Random PCR Procedure
4.5. Full Genome Sequencing
4.6. Phylogenetic Analysis
4.7. Experimental Infection
4.8. RT-PCR Assay
4.9. Nucleotide Sequence Accession Numbers
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Anderson, I.G.; Law, A.T.; Shariff, M.; Nash, G. A Parvo-like Virus in the Giant Freshwater Prawn, Macrobrachium rosenbergii. J. Invertebr. Pathol. 1990, 55, 447–449. [Google Scholar] [CrossRef]
- Qian, D.; Shi, Z.; Zhang, S.; Cao, Z.; Liu, W.; Li, L.; Xie, Y.; Cambournac, I.; Bonami, J.R. Extra small virus-like particles (XSV) and nodavirus associated with whitish muscle disease in the giant freshwater prawn, Macrobrachium rosenbergii. J. Fish Dis. 2003, 26, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Arcier, J.M.; Herman, F.; Lightner, D.V.; Redman, R.; Mari, J.; Bonami, J.R. Viral disease associated with mortalities in hatchery-reared postlarvae of the giant freshwater prawn Macrobrachium rosenbergii. Dis. Aquat. Org. 1999, 38, 177–181. [Google Scholar] [CrossRef]
- Hsieh, C.Y.; Chuang, P.C.; Chen, L.C.; Tu, C.; Chien, M.S.; Huang, K.C.; Kao, H.F.; Tung, M.C.; Tsai, S.S. Infectious hypodermal and haematopoietic necrosis virus (IHHNV) infections in giant freshwater prawn, Macrobrachium rosenbergii. Aquaculture 2006, 258, 73–79. [Google Scholar] [CrossRef]
- Tung, C.W.; Wang, C.S.; Chen, S.N. Histological and electron microscopic study on Macrobrachium muscle virus (MMV) infection in the giant freshwater prawn, Macrobrachium rosenbergii (de Man), cultured in Taiwan. J. Fish Dis. 1999, 22, 319–323. [Google Scholar] [CrossRef]
- Lightner, D.V.; Redman, R.M.; Poulos, B.T.; Mari, J.L.; Bonami, J.R.; Shariff, M. Distinction of HPV-type viruses in Penaeus chinensis and Macrobrachium rosenbergii using a DNA probe. Asian Fish. Sci. 1994, 7, 267–272. [Google Scholar]
- Zhang, Q.; Liu, Q.; Liu, S.; Yang, H.; Liu, S.; Zhu, L.; Yang, B.; Jin, J.; Ding, L.; Wang, X.; et al. A new nodavirus is associated with covert mortality disease of shrimp. J. Gen. Virol. 2014, 95, 2700–2709. [Google Scholar] [CrossRef] [PubMed]
- Bonning, B.C. The Dicistroviridae: An emerging family of invertebrate viruses. Virol. Sin. 2009, 24, 415–427. [Google Scholar] [CrossRef]
- Christian, P.D.; Carstens, E.B.; Domier, L.; Johnson, J.; Johnson, K.N.; Nakashima, N.; Scotti, P.D.; van der Wilk, F. Dicistroviridae in virus taxonomy. In Virus Taxonomy: The Eighth Report of the International Committee on Taxonomy Viruses; Fauquet, C.M., Mayo, M.A., Maniloff, J., Desselberger, U., Ball, L.A., Eds.; Academic Press: San Diego, CA, USA, 2005; pp. 678–683. [Google Scholar]
- De Miranda, J.R.; Drebot, M.; Tyler, S.; Shen, M.; Cameron, C.E.; Stoltz, D.B.; Camazine, S.M. Complete nucleotide sequence of Kashmir bee virus and comparison with acute bee paralysis virus. J. Gen. Virol. 2004, 85, 2263–2270. [Google Scholar] [CrossRef] [PubMed]
- Maori, E.; Lavi, S.; Mozes-Koch, R.; Gantman, Y.; Peretz, Y.; Edelbaum, O.; Tanne, E.; Sela, I. Isolation and characterization of Israeli acute paralysis virus, a dicistrovirus affecting honeybees in Israel: Evidence for diversity due to intra- and interspecies recombination. J. Gen. Virol. 2007, 88, 3428–3438. [Google Scholar] [CrossRef] [PubMed]
- Mari, J.; Poulos, B.T.; Lightner, D.V.; Bonami, J.R. Shrimp Taura syndrome virus: Genomic characterization and similarity with members of the genus Cricket paralysis-like viruses. J. Gen. Virol. 2002, 83, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Valles, S.M.; Strong, C.A.; Oi, D.H.; Porter, S.D.; Pereira, R.M.; Vander Meer, R.K.; Hashimoto, Y.; Hooper-Bùi, L.M.; Sánchez-Arroyo, H.; Davis, T.; et al. Phenology, distribution, and host specificity of Solenopsis invicta virus-1. J. Invertebr. Pathol. 2007, 96, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Overstreet, R.M.; Lightner, D.V.; Hasson, K.W.; McIlwain, S.; Lotz, J.M. Susceptibility to Taura syndrome virus of some penaeid shrimp species native to the Gulf of Mexico and the southeastern United States. J. Invertebr. Pathol. 1997, 69, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.X.; He, J.G.; Xu, H.D.; Weng, S.P. Pathogenicity and complete genome sequence analysis of the mud crab dicistrovirus-1. Virus Res. 2013, 171, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Pfingsten, J.S.; Costantino, D.A.; Kieft, J.S. Conservation and diversity among the three -dimensional folds of the Dicistroviridae intergenic region IRESes. J. Mol. Biol. 2007, 370, 856–869. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, N.; Uchiumi, T. Functional analysis of structural motifs in dicistroviruses. Virus Res. 2009, 139, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Froussard, P. A random-PCR method (rPCR) to construct whole cDNA library from low amounts of RNA. Nucleic Acids Res. 1992, 20, 2900. [Google Scholar] [CrossRef] [PubMed]
- Allander, T.; Tammi, M.T.; Eriksson, M.; Bjerkner, A.; Tiveljung-Lindell, A.; Andersson, B. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc. Natl. Acad. Sci. USA 2005, 102, 12891–12896. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, X.; Cao, Z.; Yuan, J.; Shi, Z.; Yuan, X.; Lin, L.; Xu, Y.; Yao, J.; Hao, G.; Shen, J. Isolation and Characterization of a Novel Dicistrovirus Associated with Moralities of the Great Freshwater Prawn, Macrobrachium rosenbergii. Int. J. Mol. Sci. 2016, 17, 204. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17020204
Pan X, Cao Z, Yuan J, Shi Z, Yuan X, Lin L, Xu Y, Yao J, Hao G, Shen J. Isolation and Characterization of a Novel Dicistrovirus Associated with Moralities of the Great Freshwater Prawn, Macrobrachium rosenbergii. International Journal of Molecular Sciences. 2016; 17(2):204. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17020204
Chicago/Turabian StylePan, Xiaoyi, Zheng Cao, Junfa Yuan, Zhengli Shi, Xuemei Yuan, Lingyun Lin, Yang Xu, Jiayun Yao, Guijie Hao, and Jinyu Shen. 2016. "Isolation and Characterization of a Novel Dicistrovirus Associated with Moralities of the Great Freshwater Prawn, Macrobrachium rosenbergii" International Journal of Molecular Sciences 17, no. 2: 204. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17020204