
Systems Pharmacology in Small Molecular Drug Discovery


Abstract
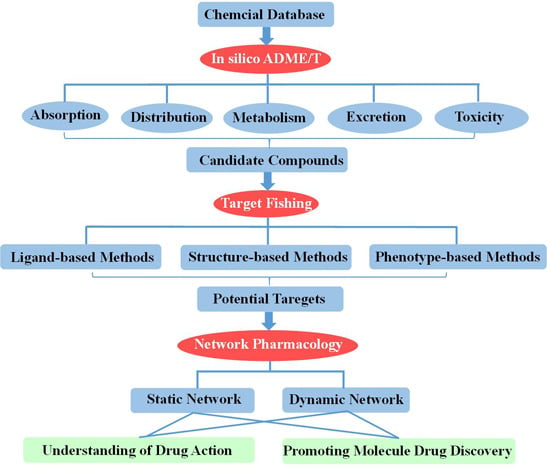
:
1. Introduction
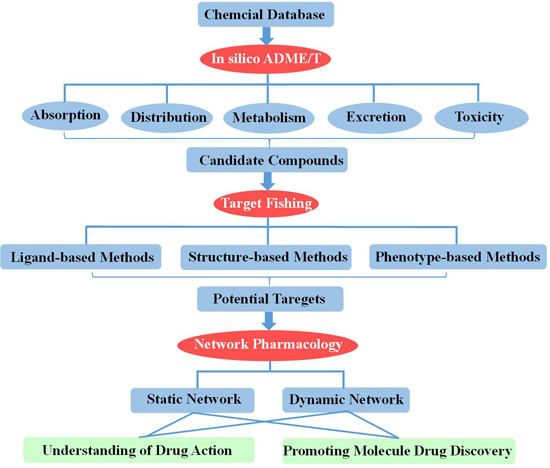
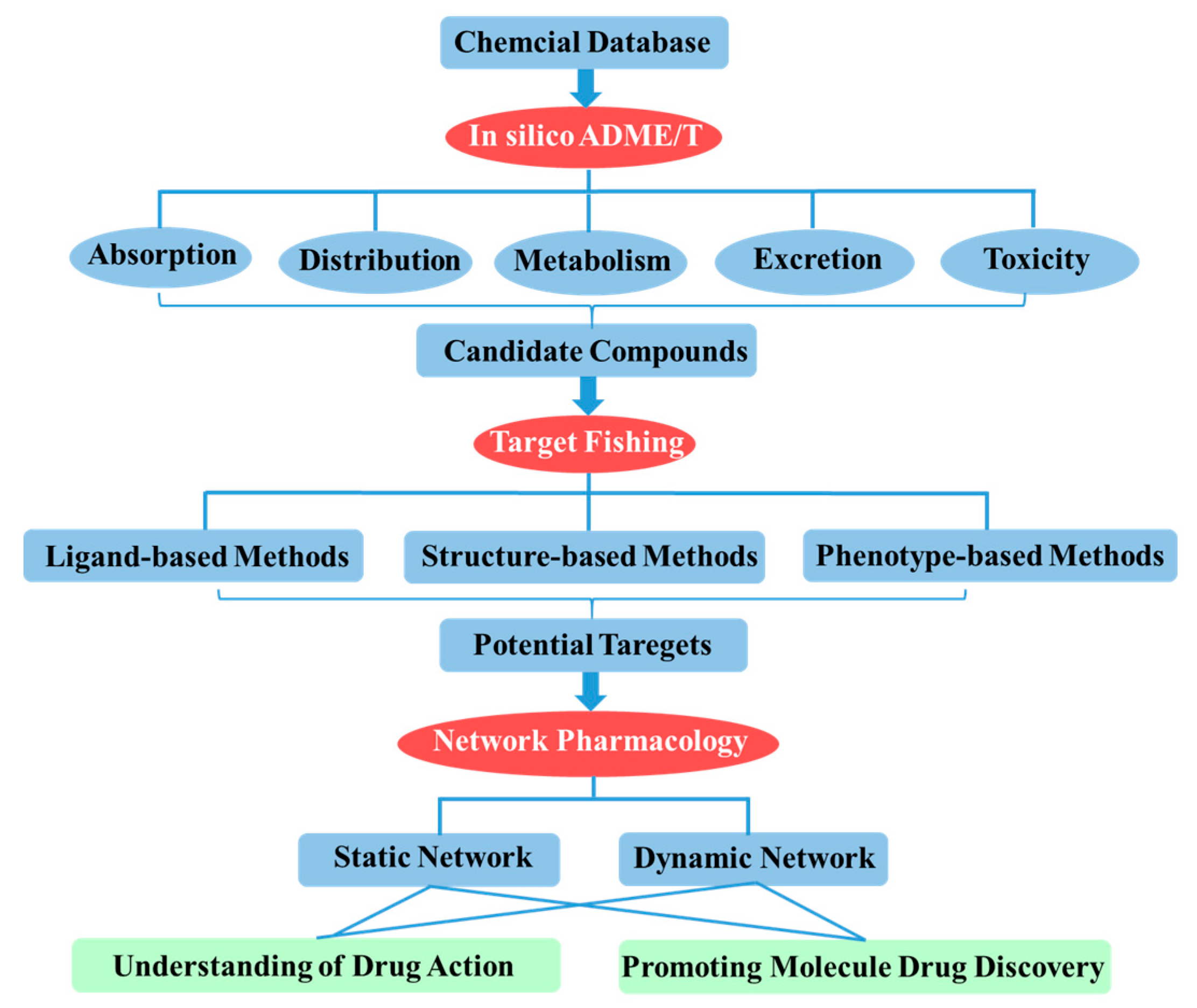
2. In Silico ADME/T (Absorption, Distribution, Metabolism, Excretion and Toxicity) Assessment for Drug Discovery
2.1. Absorption
2.2. Distribution
2.3. Metabolism
2.4. Excretion
2.5. Toxicity
3. Target Fishing
3.1. Ligand-Based Methods for Protein Target Prediction
3.2. Structure-Based Methods for Protein Target Prediction
3.3. Phenotype-Based Methods for Protein Target Prediction
4. Network-Based Drug Discovery
4.1. Static Network
4.2. Dynamic Network
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Li, P.; Fu, Y.; Wang, Y. Network based approach to drug discovery: A mini review. Mini Rev. Med. Chem. 2015, 15, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Sorger, P.K.; Allerheiligen, S.R.; Abernethy, D.R.; Altman, R.B.; Brouwer, K.L.; Califano, A.; D’Argenio, D.Z.; Iyengar, R.; Jusko, W.J.; Lalonde, R. Quantitative and Systems Pharmacology in the Post-Genomic Era: New Approaches to Discovering Drugs and Understanding Therapeutic Mechanisms; QSP workshop group: Bethesda, MD, USA, 2011; pp. 1–48. [Google Scholar]
- Chung, T.D.; Terry, D.B.; Smith, L.H. In Vitro and in Vivo Assessment of ADME and PK Properties During Lead Selection and Lead Optimization–Guidelines, Benchmarks and Rules of Thum; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2015. [Google Scholar]
- Wang, Y.; Xing, J.; Xu, Y.; Zhou, N.; Peng, J.; Xiong, Z.; Liu, X.; Luo, X.; Luo, C.; Chen, K. In silico ADME/T modelling for rational drug design. Q. Rev. Biophys. 2015, 48, 488–515. [Google Scholar] [CrossRef] [PubMed]
- Al-Awadhi, F.H.; Salvador, L.A.; Luesch, H. Screening strategies for drug discovery and target identification. Mar. Biomed. Beach Bedside 2015, 135–166. [Google Scholar]
- Caldwell, G.W. Compound optimization in early-and late-phase drug discovery: Acceptable pharmacokinetic properties utilizing combined physicochemical, in vitro and in vivo screens. Curr. Opin. Drug Discov. Dev. 2000, 3, 30–41. [Google Scholar] [CrossRef]
- Cox, P.B.; Gregg, R.J.; Vasudevan, A. Abbott Physicochemical Tiering (APT)—A unified approach to HTS triage. Bioorg. Med. Chem. 2012, 20, 4564–4573. [Google Scholar] [CrossRef] [PubMed]
- Stoner, C.L.; Troutman, M.; Gao, H.; Johnson, K.; Stankovic, C.; Brodfuehrer, J.; Gifford, E.; Chang, M. Moving in silico screening into practice: A minimalist approach to guide permeability screening!! Lett. Drug Des. Discov. 2006, 3, 575–581. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Zhang, W.; Wang, W.; Xu, X. Recent advances in computational prediction of drug absorption and permeability in drug discovery. Curr. Med. Chem. 2006, 13, 2653–2667. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Cheng, F.; Xu, Y.; Li, W.; Tang, Y. Estimation of ADME properties with substructure pattern recognition. J. Chem. Inf. Model. 2010, 50, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, W.; Huang, C.; Li, Y.; Yu, H.; Wang, Y.; Duan, J.; Ling, Y. A novel chemometric method for the prediction of human oral bioavailability. Int. J. Mol. Sci. 2012, 13, 6964–6982. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.L.; Jorgensen, W.L.; Whitehead, L. Automated ligand-and structure-based protocol for in silico prediction of human serum albumin binding. J. Chem. Inf. Model. 2013, 53, 907–922. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, T.S.; Kirshner, D.A.; Lau, E.Y.; Wong, S.E.; Nilmeier, J.P.; Lightstone, F.C. A method to predict blood-brain barrier permeability of drug-like compounds using molecular dynamics simulations. Biophys. J. 2014, 107, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Hochman, J.; Tang, C.; Prueksaritanont, T. Drug–drug interactions related to altered absorption and plasma protein binding: Theoretical and regulatory considerations, and an industry perspective. J. Pharm. Sci. 2015, 104, 916–929. [Google Scholar] [CrossRef] [PubMed]
- Zsila, F.; Bikadi, Z.; Malik, D.; Hari, P.; Pechan, I.; Berces, A.; Hazai, E. Evaluation of drug–human serum albumin binding interactions with support vector machine aided online automated docking. Bioinformatics 2011, 27, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Bujak, R.; Struck-Lewicka, W.; Kaliszan, M.; Kaliszan, R.; Markuszewski, M.J. Blood–brain barrier permeability mechanisms in view of quantitative structure–activity relationships (QSAR). J. Pharm. Biomed. Anal. 2015, 108, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B.; Goodacre, R. Metabolomics and systems pharmacology: Why and how to model the human metabolic network for drug discovery. Drug Discov. Today 2014, 19, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, J.; Williamson, M.J.; Tyzack, J.D.; Tan, L.; Bond, P.J.; Bender, A.; Glen, R.C. Computational prediction of metabolism: Sites, products, SAR, P450 enzyme dynamics, and mechanisms. J. Chem. Inf. Model. 2012, 52, 617–648. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y. Handbook of Essential Pharmacokinetics, Pharmacodynamics and Drug Metabolism for Industrial Scientists; Springer Science & Business Media: Berlin, Germany, 2001. [Google Scholar]
- Button, W.G.; Judson, P.N.; Long, A.; Vessey, J.D. Using absolute and relative reasoning in the prediction of the potential metabolism of xenobiotics. J. Chem. Inf. Comput. Sci. 2003, 43, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, K.L. QSAR in Environmental Toxicology-II; Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Klopman, G.; Tu, M.; Talafous, J. META. 3. A genetic algorithm for metabolic transform priorities optimization. J. Chem. Inf. Comput. Sci. 1997, 37, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Moroy, G.; Martiny, V.Y.; Vayer, P.; Villoutreix, B.O.; Miteva, M.A. Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov. Today 2012, 17, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Boobis, A.; Gundert-Remy, U.; Kremers, P.; Macheras, P.; Pelkonen, O. In silico prediction of ADME and pharmacokinetics: Report of an expert meeting organised by COST B15. Eur. J. Pharm. Sci. 2002, 17, 183–193. [Google Scholar] [CrossRef]
- Paine, S.W.; Barton, P.; Bird, J.; Denton, R.; Menochet, K.; Smith, A.; Tomkinson, N.P.; Chohan, K.K. A rapid computational filter for predicting the rate of human renal clearance. J. Mol. Graph. Model. 2010, 29, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.-W.; Fagerholm, U.; Norinder, U. In silico categorization of in vivo intrinsic clearance using machine learning. Mol. Pharm. 2013, 10, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Maltarollo, V.G.; Gertrudes, J.C.; Oliveira, P.R.; Honorio, K.M. Applying machine learning techniques for ADME-Tox prediction: A review. Expert Opin. Drug Metab. Toxicol. 2015, 11, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Colmenarejo, G. In silico ADME prediction: Data sets and models. Curr. Comput. Aided Drug Des. 2005, 1, 365–376. [Google Scholar] [CrossRef]
- Helma, C. In silico predictive toxicology: The state-of-the-art and strategies to predict human health effects. Curr. Opin. Drug Discov. Dev. 2005, 8, 27–31. [Google Scholar]
- Greene, N.; Judson, P.; Langowski, J.; Marchant, C. Knowledge-based expert systems for toxicity and metabolism prediction: DEREK, StAR and METEOR. SAR QSAR Environ. Res. 1999, 10, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, D.; Earnshaw, C. Computer prediction of possible toxic action from chemical structure; the DEREK system. Hum. Exp. Toxicol. 1991, 10, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Klopman, G. MULTICASE 1. A hierarchical computer automated structure evaluation program. Quant. Struct. Act. Relatsh. 1992, 11, 176–184. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, Q.-Y.; Xiang, M.-L.; Ma, C.-Y.; Huang, Q.; Yang, S.-Y. In silico prediction of mitochondrial toxicity by using GA-CG-SVM approach. Toxicol. Vitro 2009, 23, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Myshkin, E.; Brennan, R.; Khasanova, T.; Sitnik, T.; Serebriyskaya, T.; Litvinova, E.; Guryanov, A.; Nikolsky, Y.; Nikolskaya, T.; Bureeva, S. Prediction of organ toxicity endpoints by QSAR modeling based on precise chemical-histopathology annotations. Chem. Biol. Drug Des. 2012, 80, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Sakharkar, K.R.; Sakharkar, M.K.; Chandra, R. Drug discovery: Diseases, drugs and targets. Post Genom. Approaches Drug Vaccine Dev. 2015, 5, 1. [Google Scholar]
- Schneider, H.-J. Limitations and extensions of the lock-and-key principle: Differences between gas state, solution and solid state structures. Int. J. Mol. Sci. 2015, 16, 6694–6717. [Google Scholar] [CrossRef] [PubMed]
- Sams-Dodd, F. Target-based drug discovery: Is something wrong? Drug Discov. Today 2005, 10, 139–147. [Google Scholar] [CrossRef]
- Szuromi, P.; Vinson, V.; Marshall, E. Rethinking drug discovery. Science 2004, 303, 1795. [Google Scholar] [CrossRef]
- Food and Drug Administration. Innovation or Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products; Food and Drug Administration: Washington, DC, USA, 2004. [Google Scholar]
- Ghasemi, J.; Abdolmaleki, A. Computer aided drug design for multi-target drug design: SAR/QSAR, molecular docking and pharmacophore methods. Curr. Drug Targets 2016. [Google Scholar] [CrossRef]
- Yan, X.; Liao, C.; Liu, Z.; Hagler, A.; Gu, Q.; Xu, J. Chemical Structure Similarity Search for Ligand-Based Virtual Screening: Methods and Computational Resources. Curr. Drug Targets 2015. [Google Scholar] [CrossRef]
- Xie, L.; Xie, L.; Kinnings, S.L.; Bourne, P.E. Novel computational approaches to polypharmacology as a means to define responses to individual drugs. Annu. Rev. Pharm. Toxicol. 2012, 52, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Martin, Y.C.; Kofron, J.L.; Traphagen, L.M. Do structurally similar molecules have similar biological activity? J. Med. Chem. 2002, 45, 4350–4358. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, Y.; Kotera, M.; Kanehisa, M.; Goto, S. Drug-target interaction prediction from chemical, genomic and pharmacological data in an integrated framework. Bioinformatics 2010, 26, i246–i254. [Google Scholar] [CrossRef] [PubMed]
- Nidhi; Glick, M.; Davies, J.W.; Jenkins, J.L. Prediction of biological targets for compounds using multiple-category Bayesian models trained on chemogenomics databases. J. Chem. Inf. Model. 2006, 46, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Setola, V.; Irwin, J.J.; Laggner, C.; Abbas, A.I.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B. Predicting new molecular targets for known drugs. Nature 2009, 462, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Koutsoukas, A.; Simms, B.; Kirchmair, J.; Bond, P.J.; Whitmore, A.V.; Zimmer, S.; Young, M.P.; Jenkins, J.L.; Glick, M.; Glen, R.C. From in silico target prediction to multi-target drug design: Current databases, methods and applications. J. Proteom. 2011, 74, 2554–2574. [Google Scholar] [CrossRef] [PubMed]
- DeGraw, A.J.; Keiser, M.J.; Ochocki, J.D.; Shoichet, B.K.; Distefano, M.D. Prediction and evaluation of protein farnesyltransferase inhibition by commercial drugs. J. Med. Chem. 2010, 53, 2464–2471. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, J.; Luo, F.; Deng, Z.; Hu, Q.-N. Predicting target-ligand interactions using protein ligand-binding site and ligand substructures. BMC Syst. Biol. 2015, 9, S2. [Google Scholar] [CrossRef] [PubMed]
- Khamis, M.A.; Gomaa, W.; Ahmed, W.F. Machine learning in computational docking. Artif. Intell. Med. 2015, 63, 135–152. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, N.M.; Gesto, D.; Oliveira, E.F.; Santos-Martins, D.; Brás, N.F.; Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Receptor-based virtual screening protocol for drug discovery. Arch. Biochem. Biophys. 2015, 582, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wang, B.; Meroueh, S.O. Structure-based computational approaches for small-molecule modulation of protein-protein interactions. Protein Protein Interact. Methods Appl. 2015, 1278, 77–92. [Google Scholar]
- Kellenberger, E.; Rodrigo, J.; Muller, P.; Rognan, D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins Struct. Funct. Bioinform. 2004, 57, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Tabei, Y.; Pauwels, E.; Stoven, V.; Takemoto, K.; Yamanishi, Y. Identification of chemogenomic features from drug–target interaction networks using interpretable classifiers. Bioinformatics 2012, 28, i487–i494. [Google Scholar] [CrossRef] [PubMed]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y. Combining label-free cell phenotypic profiling with computational approaches for novel drug discovery. Expert Opin.Drug Discov. 2015, 10, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J. The Connectivity Map: A new tool for biomedical research. Nat. Rev. Cancer 2007, 7, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Fliri, A.F.; Loging, W.T.; Volkmann, R.A. Drug effects viewed from a signal transduction network perspective. J. Med. Chem. 2009, 52, 8038–8046. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Young, D.W.; Jenkins, J.L.; Serrano, M.; Mikhailov, D.; Clemons, P.A.; Davies, J.W. Chemogenomic data analysis: Prediction of small-molecule targets and the advent of biological fingerprints. Comb. Chem. High Throughput Screen. 2007, 10, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Iskar, M.; Campillos, M.; Kuhn, M.; Jensen, L.J.; Van Noort, V.; Bork, P. Drug-induced regulation of target expression. PLoS Comput. Biol. 2010, 6, e1000925. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Agarwal, P. Human disease-drug network based on genomic expression profiles. PLoS ONE 2009, 4, e6536. [Google Scholar] [CrossRef] [PubMed]
- Suthram, S.; Dudley, J.T.; Chiang, A.P.; Chen, R.; Hastie, T.J.; Butte, A.J. Network-based elucidation of human disease similarities reveals common functional modules enriched for pluripotent drug targets. PLoS Comput. Biol. 2010, 6, e1000662. [Google Scholar] [CrossRef] [PubMed]
- Campillos, M.; Kuhn, M.; Gavin, A.-C.; Jensen, L.J.; Bork, P. Drug target identification using side-effect similarity. Science 2008, 321, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Young, D.W.; Bender, A.; Hoyt, J.; McWhinnie, E.; Chirn, G.-W.; Tao, C.Y.; Tallarico, J.A.; Labow, M.; Jenkins, J.L.; Mitchison, T.J. Integrating high-content screening and ligand-target prediction to identify mechanism of action. Nat. Chem. Biol. 2008, 4, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Bai, H.; Ouyang, Q.; Lai, L.; Tang, C. Finding multiple target optimal intervention in disease-related molecular network. Mol. Syst. Biol. 2008, 4, 228. [Google Scholar] [CrossRef] [PubMed]
- Berg, E.L. Systems biology in drug discovery and development. Drug Discov. Today 2014, 19, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Westerhoff, H.V. Network-based pharmacology through systems biology. Drug Discov. Today Technol. 2015, 15, 15. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Zhou, W.; Wan, J.-B.; Zhang, Q.; Ni, J.; Hu, Y. An integrative thrombosis network: Visualization and topological analysis. Evid. Based Complement. Altern. Med. 2015, 501, 265303. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Xing, X.; Xue, J.; Hou, T.; Liu, F. Topological properties in metabolic functional module of transcriptional regulatory networks. J. Comput. Theor. Nanosci. 2015, 12, 2776–2785. [Google Scholar] [CrossRef]
- Wu, S.; Chen, B.; Xiong, D. Topological properties of stock index futures based on network approach. In LISS 2013; Springer: Berlin, Germany, 2015; pp. 135–140. [Google Scholar]
- Lee, J.-S.; Pfeffer, J. Estimating Centrality Statistics for Complete and Sampled Networks: Some Approaches and Complications. In Proceedings of the 2015 48th Hawaii International Conference on System Sciences (HICSS), Kauai, Hawaii, HI, USA, 5–8 January 2015; pp. 1686–1695.
- Zhu, C.; Wu, C.; Jegga, A.G. Network biology methods for drug repositioning. Post Genom. Approaches Drug Vaccine Dev. 2015, 5, 115. [Google Scholar]
- Kondofersky, I.; Fuchs, C.; Theis, F.J. Identifying latent dynamic components in biological systems. IET Syst. Biol. 2015, 9, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Mohsenizadeh, D.N.; Hua, J.; Bittner, M.; Dougherty, E.R. Dynamical modeling of uncertain interaction-based genomic networks. BMC Bioinform. 2015, 16, S3. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lan, Y. Hierarchical feedback modules and reaction hubs in cell signaling networks. PLoS ONE 2015, 10, e0125886. [Google Scholar] [CrossRef] [PubMed]
- Pfaffelhuber, P.; Popovic, L. How spatial heterogeneity shapes multiscale biochemical reaction network dynamics. J. R. Soc. Interface 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Draghi, J.; Whitlock, M. Robustness to noise in gene expression evolves despite epistatic constraints in a model of gene networks. Evolution 2015, 69, 2345–2358. [Google Scholar] [CrossRef] [PubMed]
- Iooss, B.; Lemaître, P. A review on global sensitivity analysis methods. In Uncertainty Management in Simulation-Optimization of Complex Systems. Springer US 2015, 101–122. [Google Scholar] [CrossRef]
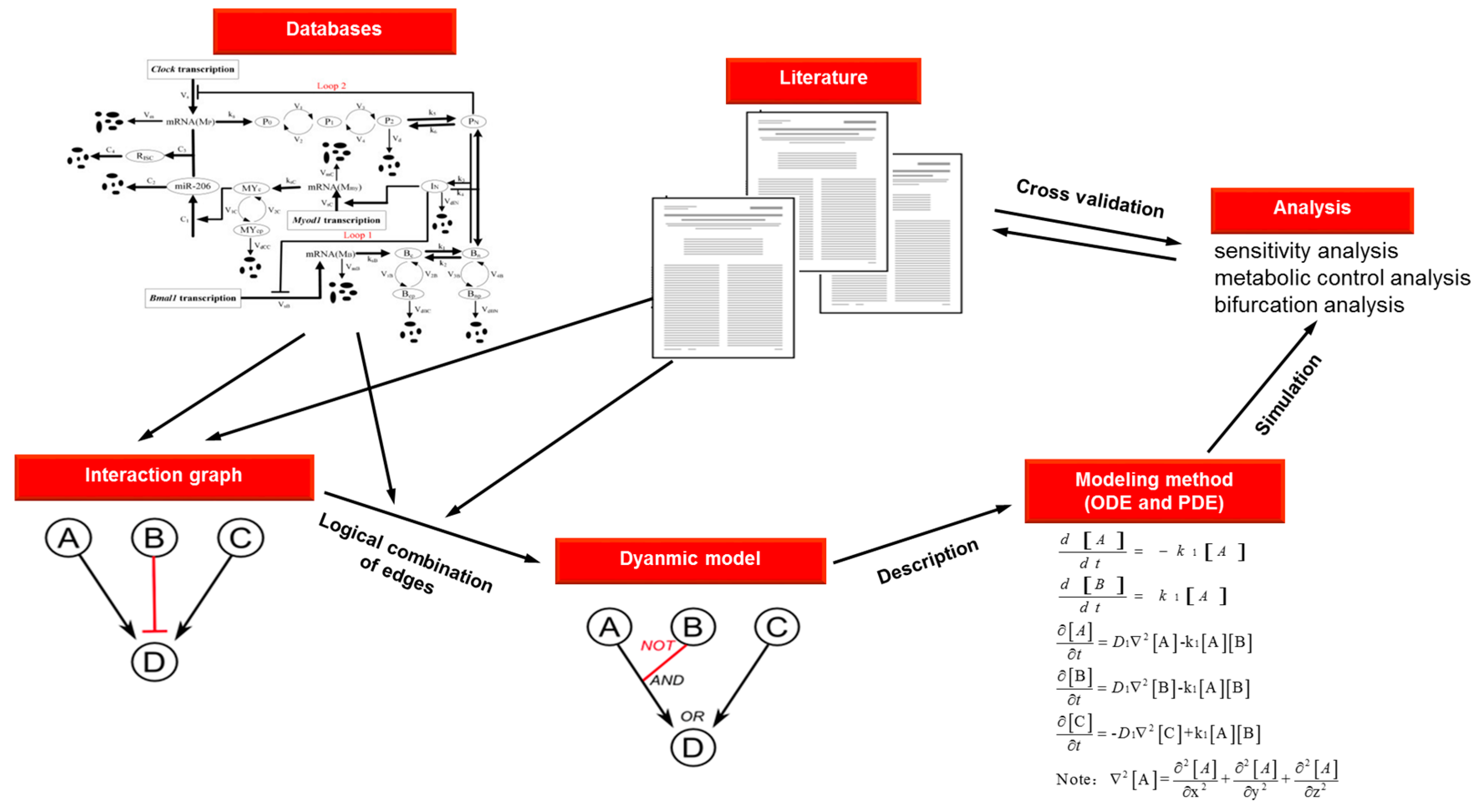
- Zhou, W.; Li, Y.; Wang, X.; Wu, L.; Wang, Y. MiR-206-mediated dynamic mechanism of the mammalian circadian clock. BMC Syst. Biol. 2011, 5, 141. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M.; Snader, K.M. Natural products as sources of new drugs over the period 1981–2002. J. Nat. Prod. 2003, 66, 1022–1037. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, J.R. Drugs, leads, and drug-likeness: An analysis of some recently launched drugs. Bioorg. Med. Chem. Lett. 2002, 12, 1647–1650. [Google Scholar] [CrossRef]
- Brown, D.; Superti-Furga, G. Rediscovering the sweet spot in drug discovery. Drug Discov. Today 2003, 8, 1067–1077. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Description | Advantages | Disadvantages |
|---|---|---|---|
| Ligand-based | Based on the similarity of known ligands | Applicable when the structure of the receptor site is unknown | Not applicable when no ligands for a given protein exist |
| Structure-based | Based on binding of ligands to active sites of the target protein | Rich information on various target proteins | Not applicable to proteins whose 3D structures are unknown |
| Phenotype-based | Based on the desired biological phenotypic information | Applicable to the genome-scale computation | Possibly ignore valuable computation from other types of data sources |
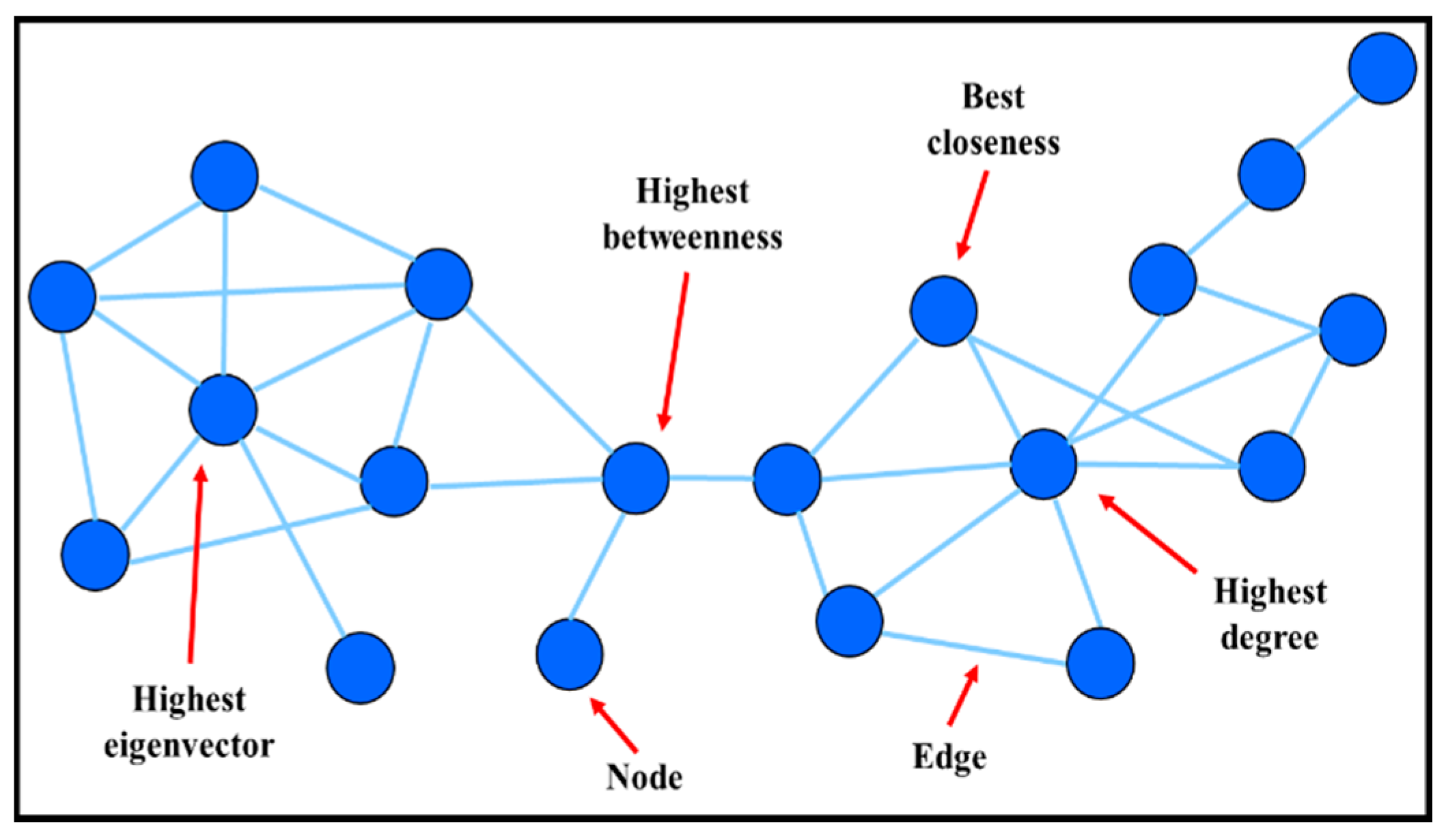
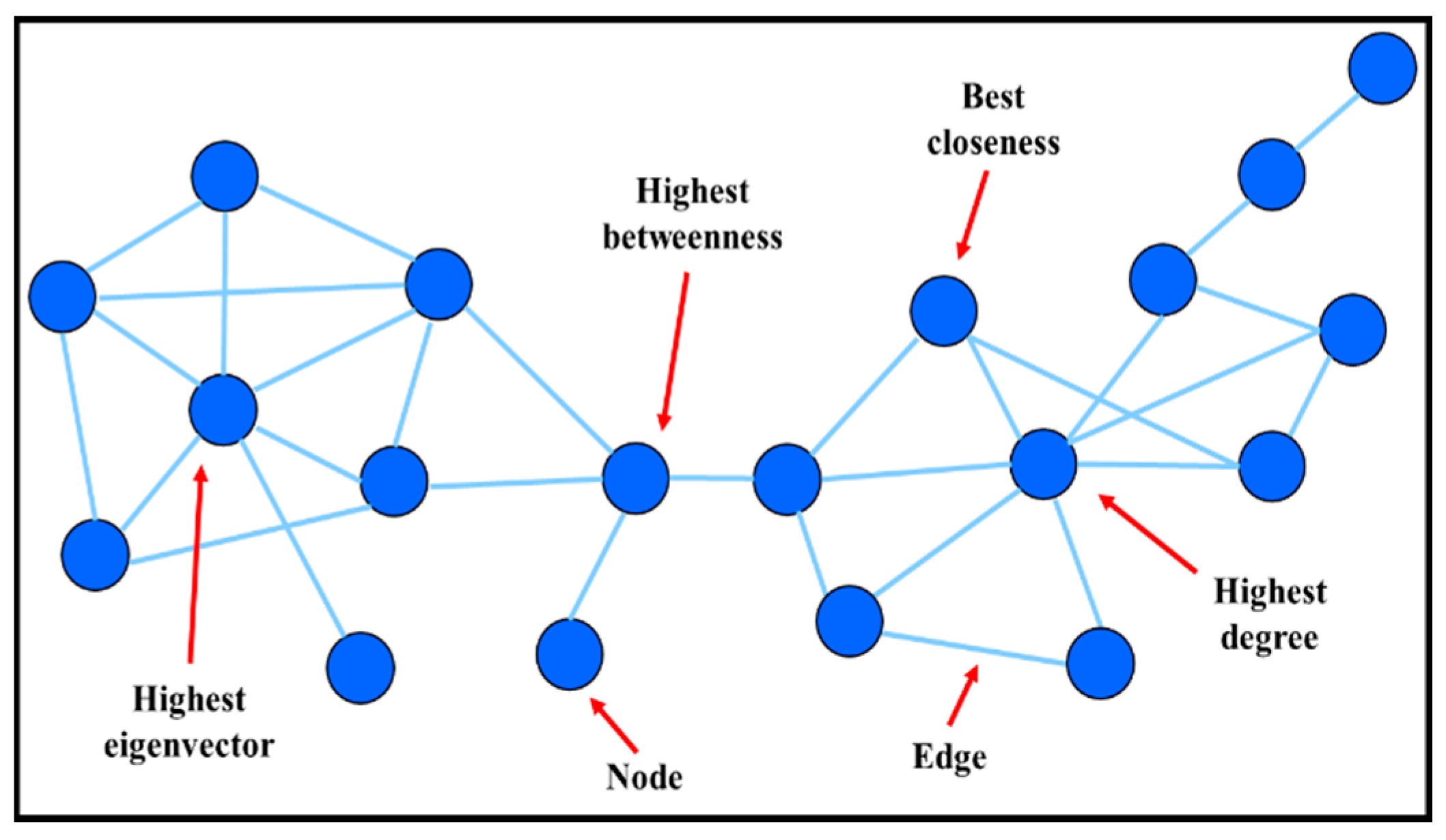
| Network Characteristics | Definition | Biological Entities and Functions |
|---|---|---|
| Node | Basic component interacting (pair-wise) with other node(s) | Small-molecular (metabolic network) |
| Genes (genetic regulatory network) | ||
| Proteins (protein-protein network) | ||
| Edge | A relationship between the nodes | Connection may be physical, regulatory, genetic interaction |
| Metabolic network: enzyme-catalyzed reactions | ||
| Genetic regulatory network: expression data | ||
| Degree | Number of links to other nodes | Associated with topological robustness of biological networks i.e., small degree nodes are more “disposable” than hubs |
| Betweenness | Number of shortest paths that pass through each node | Important for finding non-hub crucial nodes or classifying hubs according to their positions in the network |
| Closeness | Number of link to the center | Only applicable to connected networks |
| Eigenvector | Influence of a node in a network | Assigning relative scores to all nodes in the network |
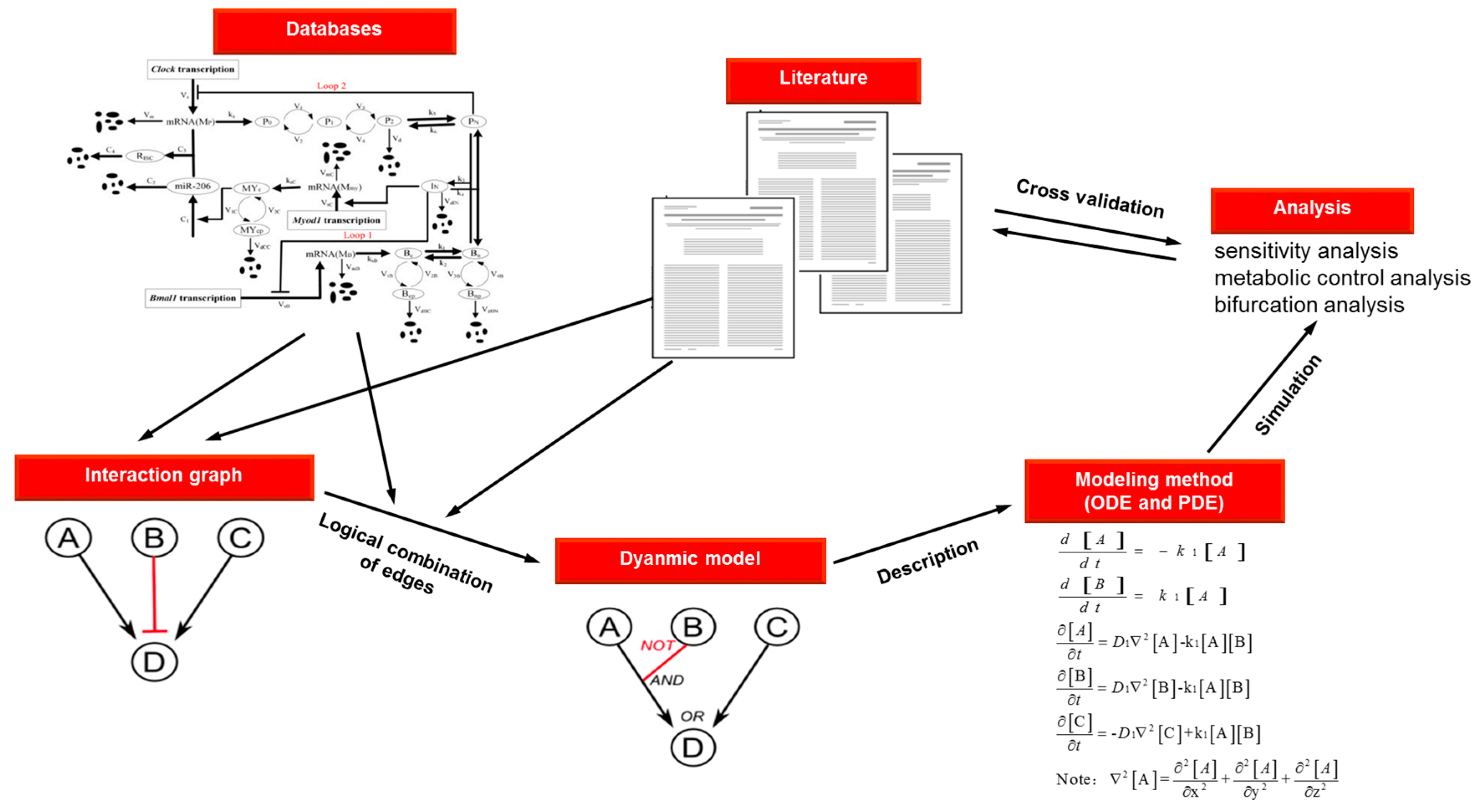
| Method | Description | Reaction | Equation | Advantages | Disadvantages |
|---|---|---|---|---|---|
| ODEs | Series of reaction-rate equations solved using numerical methods | Well understood formalism | Limited to temporal modeling | ||
| Deterministic | Assumed high concentrations and uniform mixing | ||||
| Produces graphs or tables of reagent production and consumption | Fast | Brittle | |||
| Mathematically robust | |||||
| PDEs | Expresses spatial and temporal dependence through partial derivatives | Well understood formalism | Complicated | ||
| Possible to be fast | Difficult to implement or generalize | ||||
| Bases on numerical methods | With diffusion of molecules at rate D1 | Mathematically robust | Unable to model state of discontinuous transitions | ||
| Produces numeric output of concentrations and x, y, z coordinates | Enables modeling of time- and space-dependent process | Brittle |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, W.; Wang, Y.; Lu, A.; Zhang, G. Systems Pharmacology in Small Molecular Drug Discovery. Int. J. Mol. Sci. 2016, 17, 246. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17020246
Zhou W, Wang Y, Lu A, Zhang G. Systems Pharmacology in Small Molecular Drug Discovery. International Journal of Molecular Sciences. 2016; 17(2):246. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17020246
Chicago/Turabian StyleZhou, Wei, Yonghua Wang, Aiping Lu, and Ge Zhang. 2016. "Systems Pharmacology in Small Molecular Drug Discovery" International Journal of Molecular Sciences 17, no. 2: 246. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17020246