
Pressure Combined with Ischemia/Reperfusion Injury Induces Deep Tissue Injury via Endoplasmic Reticulum Stress in a Rat Pressure Ulcer Model

 and
and
Abstract
:
1. Introduction
2. Results
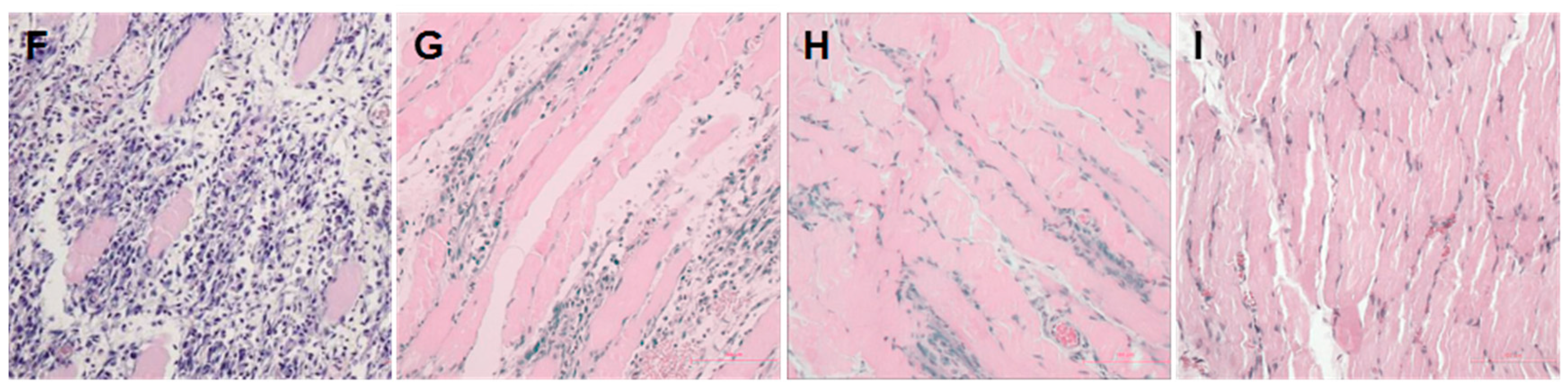
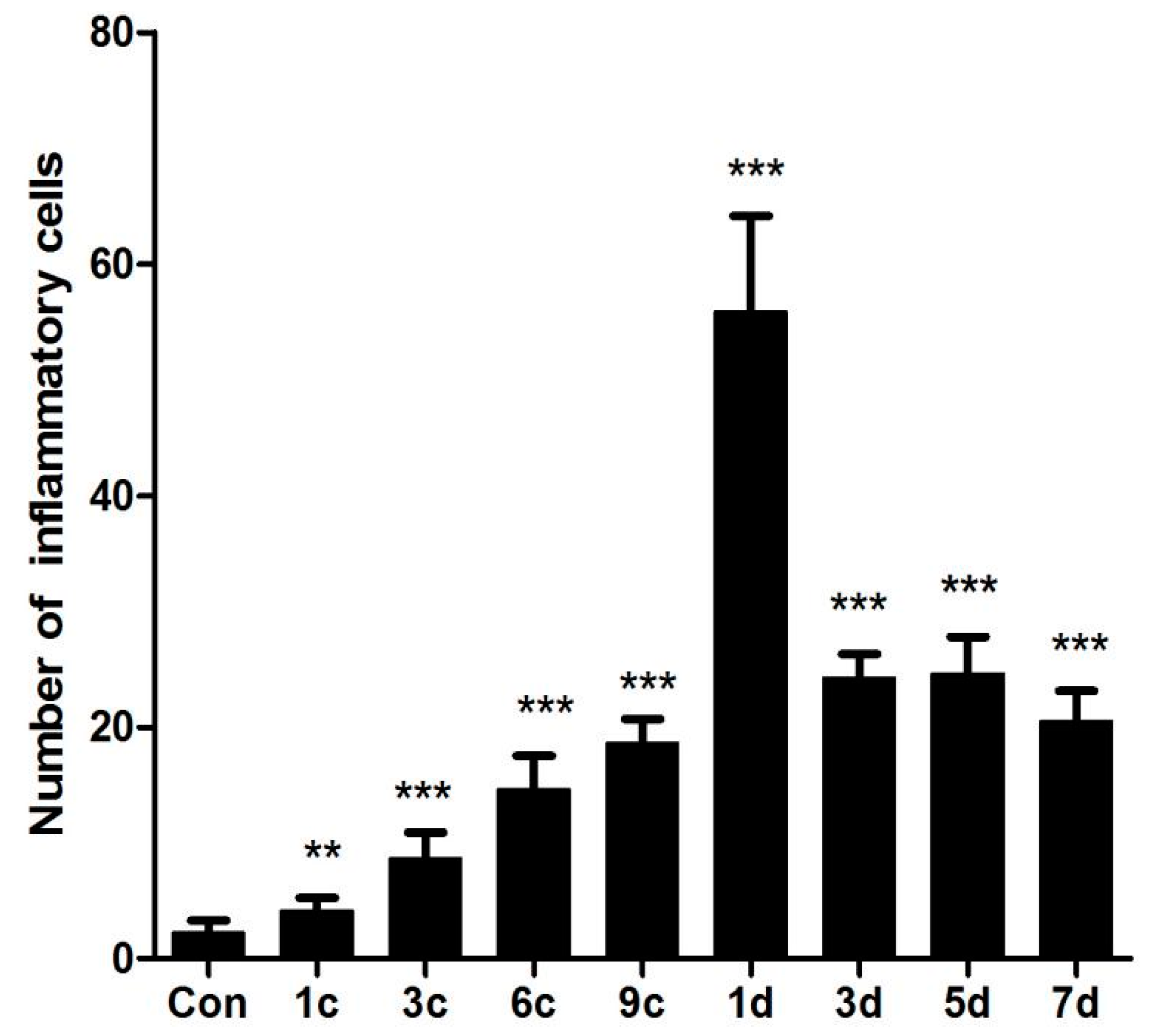
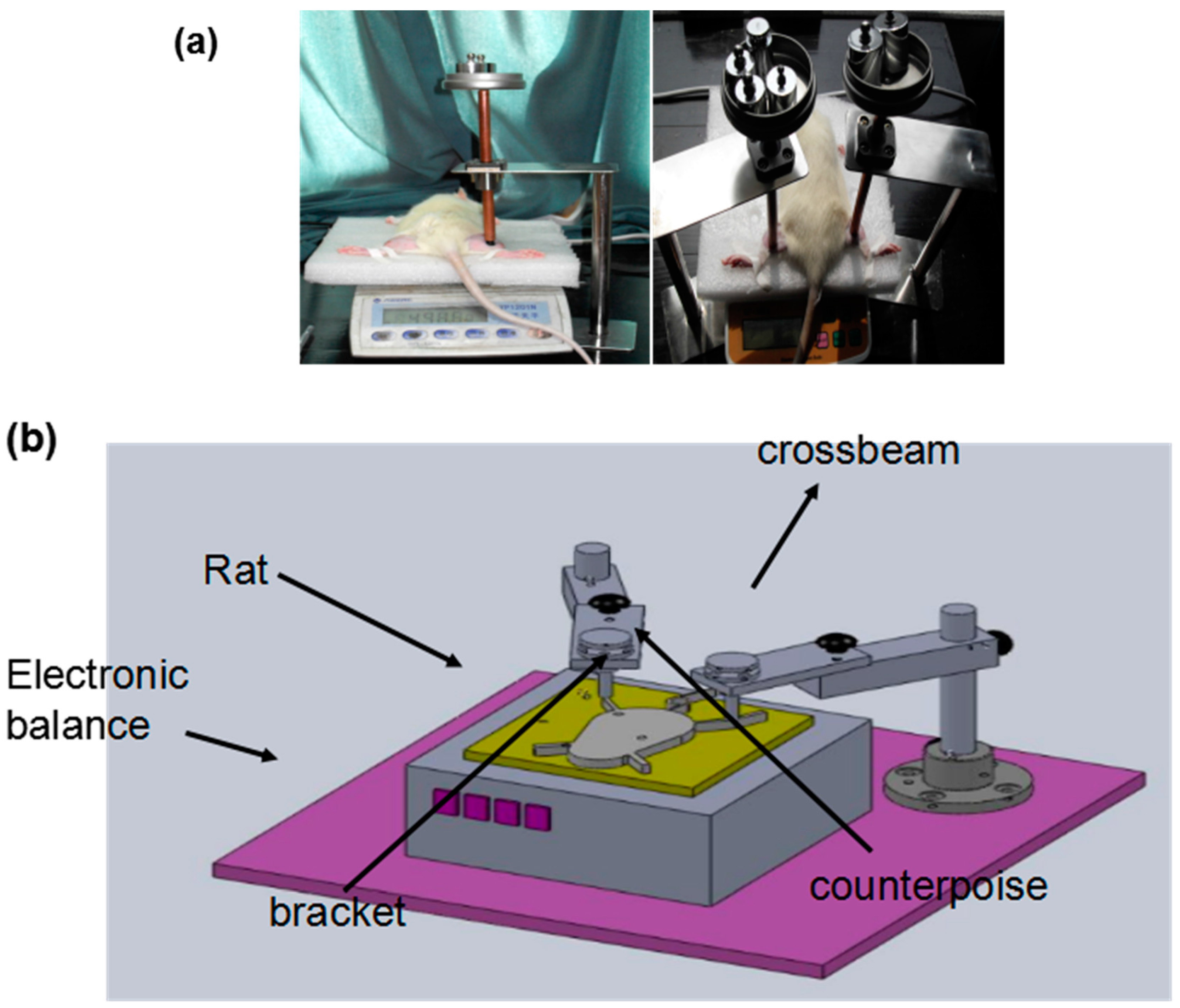
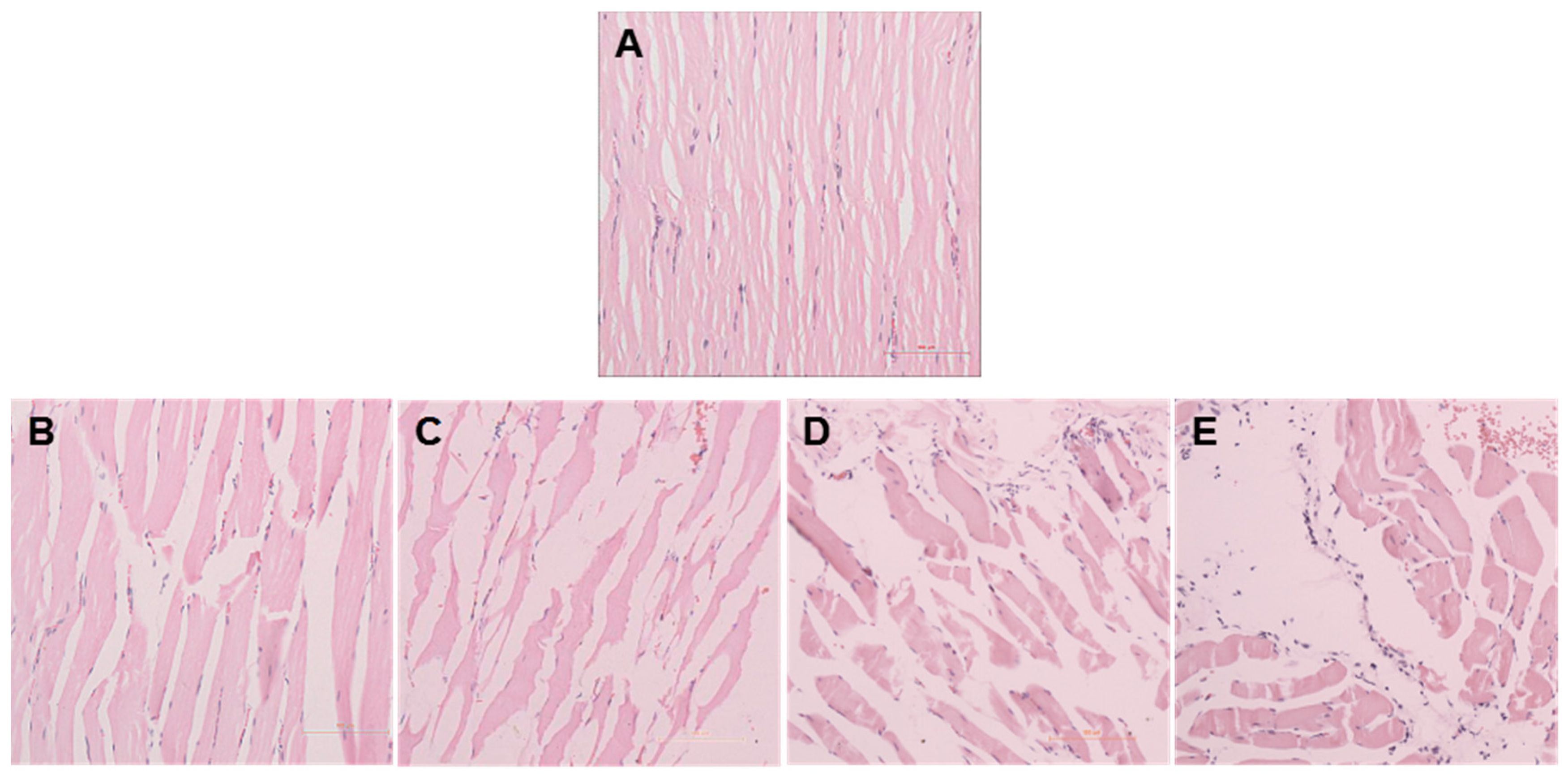
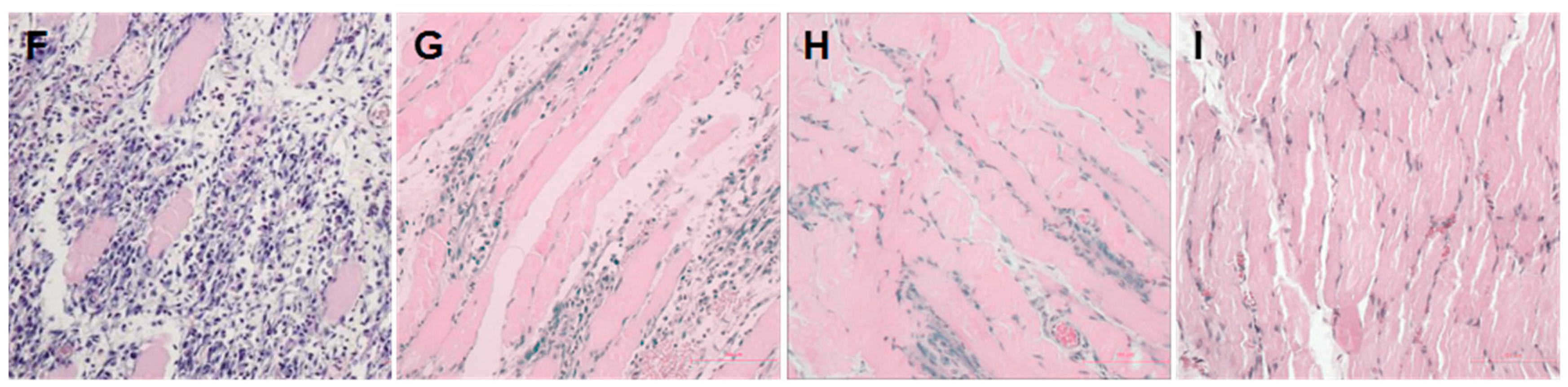
2.1. Establishment of a Pressure Ulcer Model
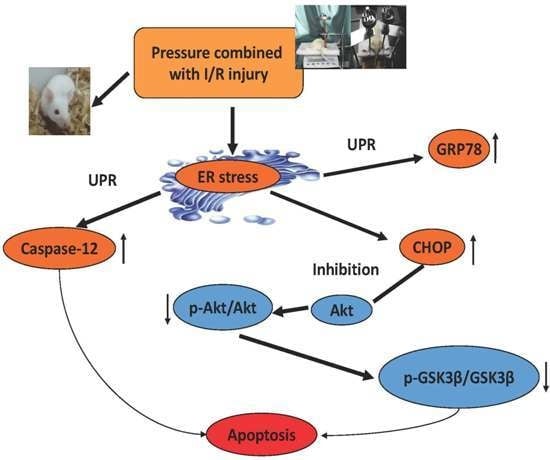
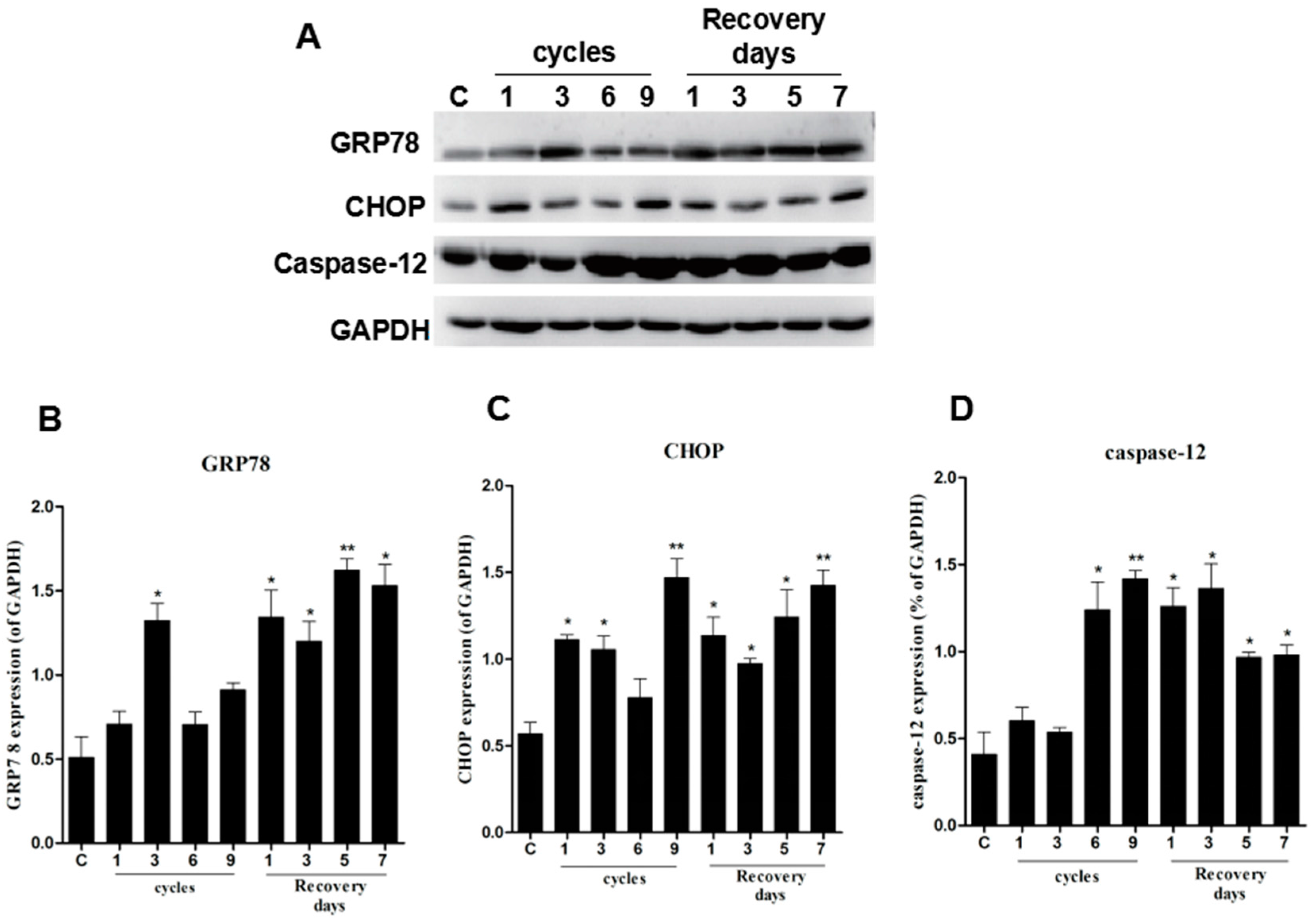
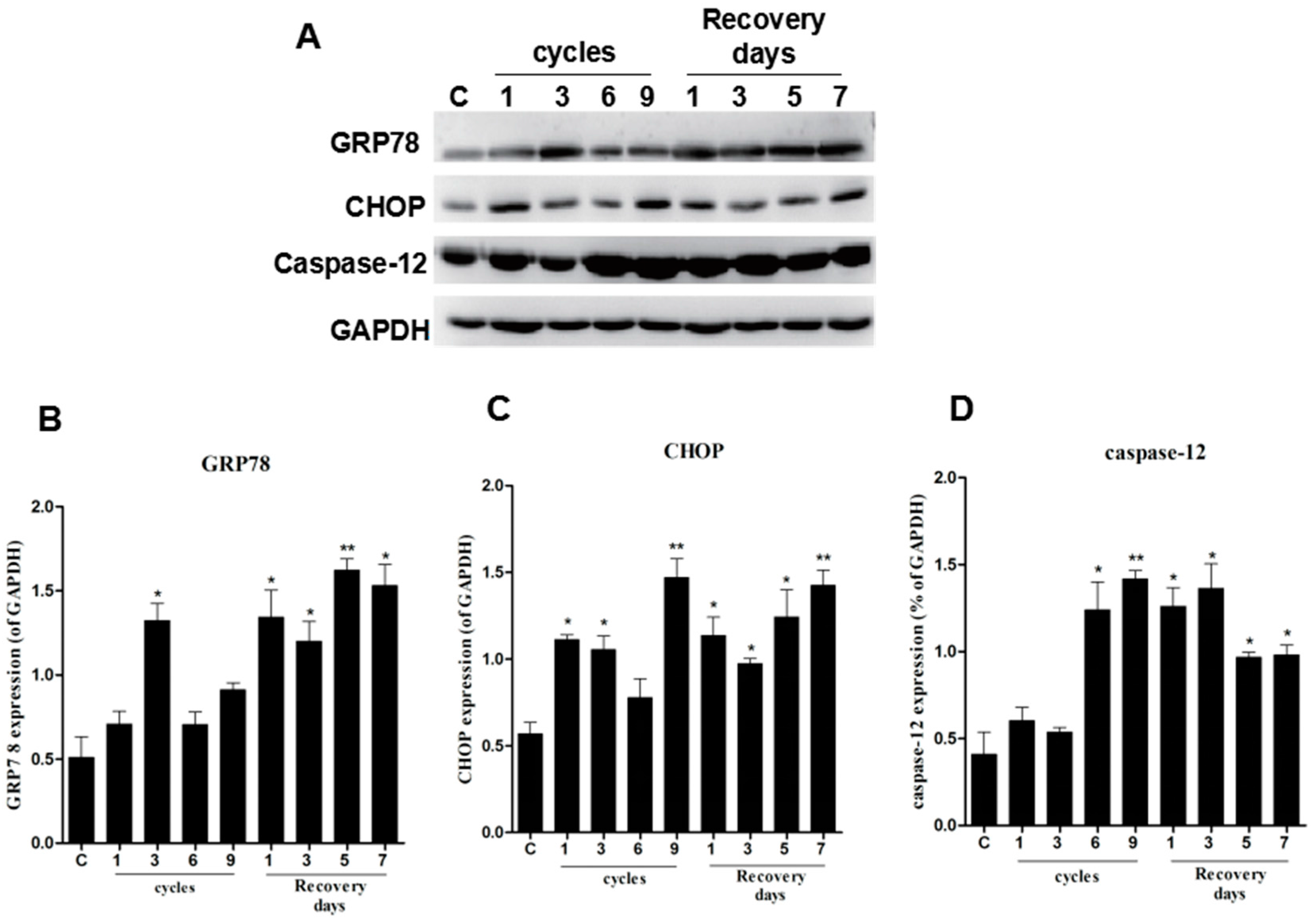
2.2. ER Stress Was Observed in the Pressure Ulcer Model
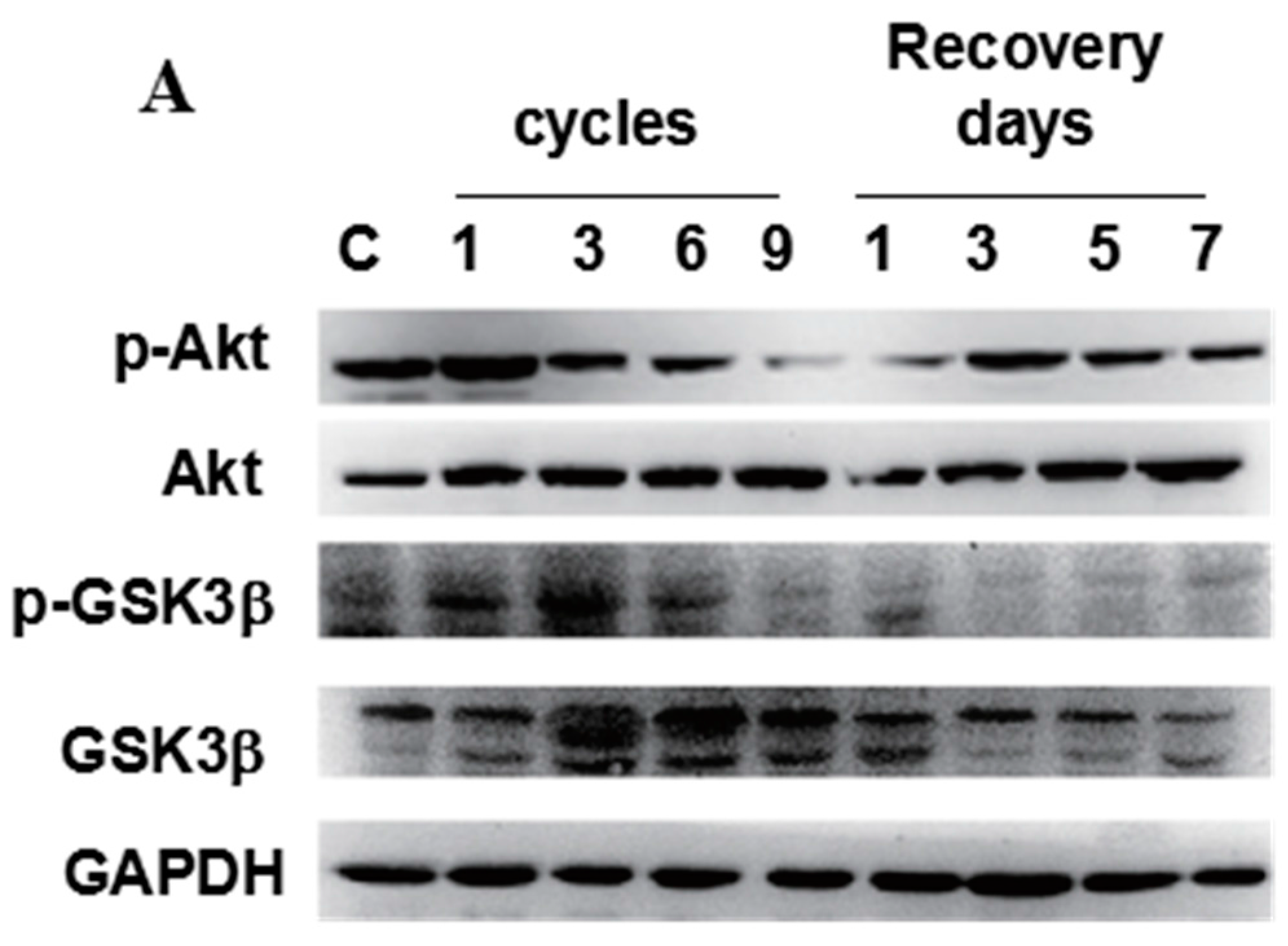
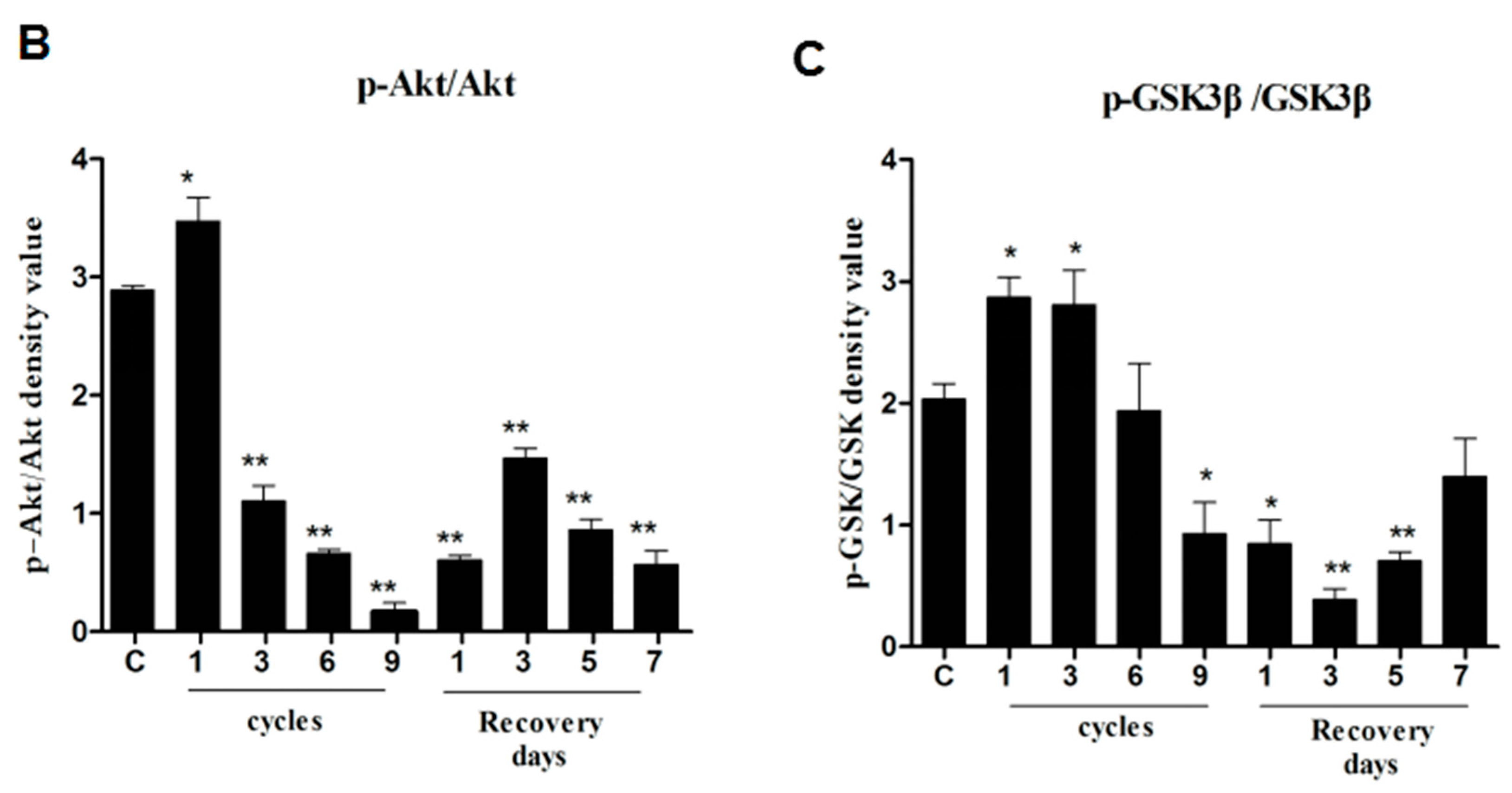
2.3. Correlations between CHOP, Phospho-Akt, and Phospho-GSK3β
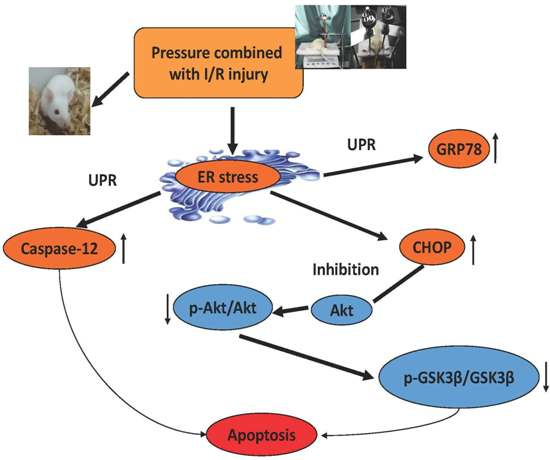
3. Discussion
4. Materials and Methods
4.1. Animals
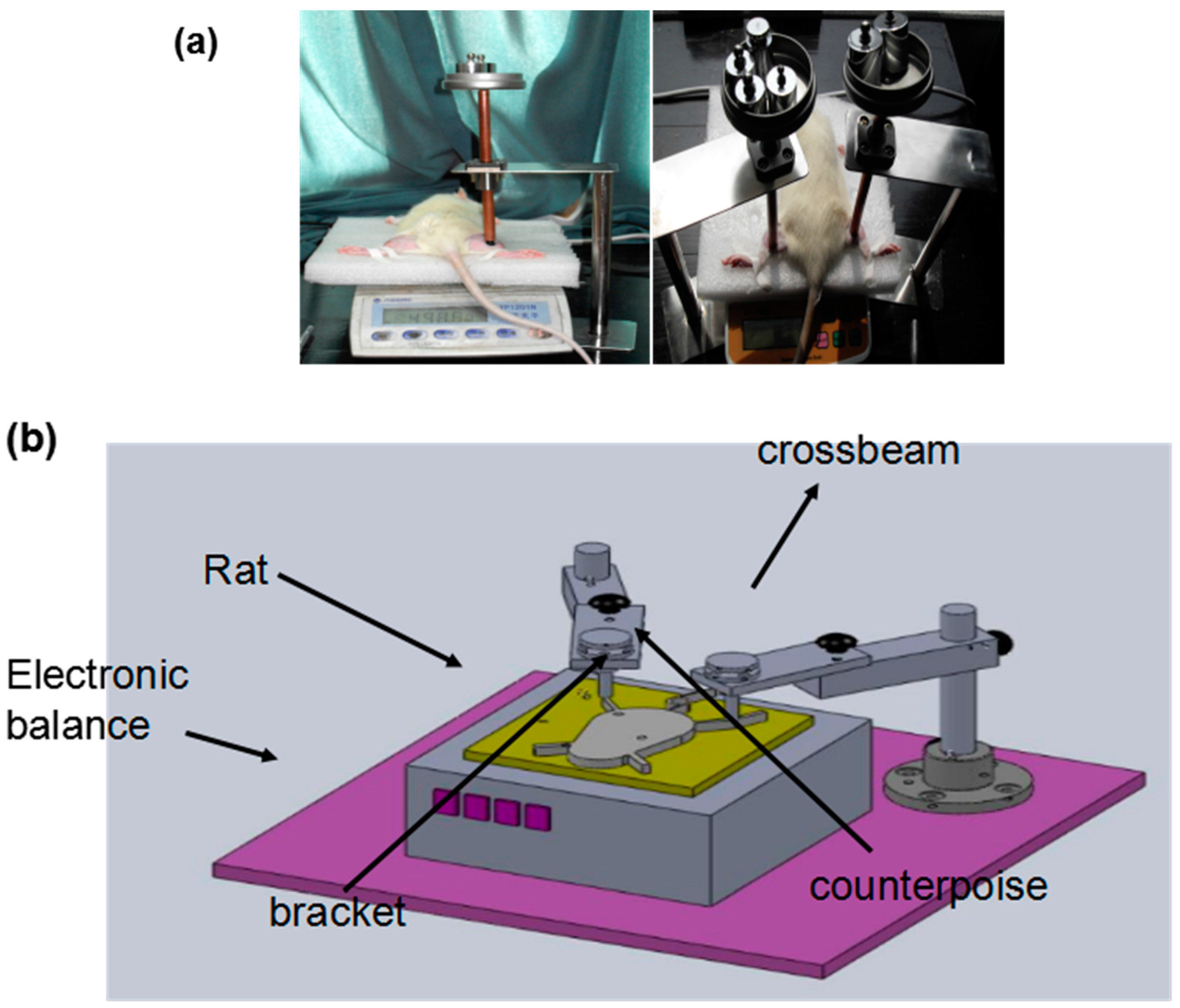
4.2. Animal Model for Deep Pressure Ulcers
4.3. Histological Analysis
4.4. Western Blotting Analysis
4.5. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DTI | Deep tissue injury |
| Akt/GSK3β | Serine/threonine Kinase /Glycogen synthase kinase 3β |
| ER | Endoplasmic reticulum |
| CHOP | C/EBP homologous protein |
| IPostC | Ischemic postconditioning |
| I/R | Ischemia/reperfusion |
| MPTP | Mitochondrial permeability transition pores |
| GRP78 | Glucose regulated protein 78 |
| Caspase-12 | Cysteine aspartate-specific protease 12 |
References
- Black, J.; Baharestani, M.M.; Cuddigan, J.; Dorner, B.; Edsberg, L.; Langemo, D.; Posthauer, M.E.; Ratliff, C.; Taler, G. National Pressure Ulcer Advisory Panel’s updated pressure ulcer staging system. Adv. Skin Wound Care 2007, 20, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Stekelenburg, A.; Gawlitta, D.; Bader, D.L.; Oomens, C.W. Deep tissue injury: How deep is our understanding? Arch. Phys. Med. Rehabil. 2008, 89, 1410–1413. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Phillips, L.G. MOC-PSSM CME article: Pressure sores. Plast. Reconstr. Surg. 2008, 121, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Stekelenburg, A.; Oomens, C.W.; Strijkers, G.J.; Nicolay, K.; Bader, D.L. Compression-induced deep tissue injury examined with magnetic resonance imaging and histology. J. Appl. Physiol. 2006, 100, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Gefen, A. Bioengineering models of deep tissue injury. Adv. Skin Wound Care 2008, 21, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, J. Should we include deep tissue injury in pressure ulcer staging systems? The NPUAP debate. J. Wound Care 2005, 14, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.; Yue, S.; Fu, Y.; Zhu, J.; Wang, X.; Busuttil, R.W.; Zhai, Y. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia reperfusion injury. Am. J. Transplant. 2014, 14, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Weymann, A.; Sabashnikov, A.; Patil, N.P.; Konertz, W.; Modersohn, D.; Dohmen, P.M. Eprosartan improves cardiac function in swine working heart model of ischemia-reperfusion injury. Med. Sci. Monit. Basic Res. 2014, 20, 55–62. [Google Scholar] [PubMed]
- Loerakker, S.; Manders, E.; Strijkers, G.J.; Nicolay, K.; Baaijens, F.P.; Bader, D.L.; Oomens, C.W. The effects of deformation, ischemia, and reperfusion on the development of muscle damage during prolonged loading. J. Appl. Physiol. 2011, 111, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Siu, P.M.; Tam, E.W.; Teng, B.T.; Pei, X.M.; Ng, J.W.; Benzie, I.F.; Mak, A.F. Muscle apoptosis is induced in pressure-induced deep tissue injury. J. Appl. Physiol. 2009, 107, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Vecchione, C.; Patrucco, E.; Marino, G.; Barberis, L.; Poulet, R.; Aretini, A.; Maffei, A.; Gentile, M.T.; Storto, M.; Azzolino, O.; et al. Protection from angiotensin II-mediated vasculotoxic and hypertensive response in mice lacking PI3Kγ. J. Exp. Med. 2005, 201, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Hyoda, K.; Hosoi, T.; Horie, N.; Okuma, Y.; Ozawa, K.; Nomura, Y. PI3K-Akt inactivation induced CHOP expression in endoplasmic reticulum-stressed cells. Biochem. Biophys. Res. Commun. 2006, 340, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.S.; Woods, A.J.; Dale, T.C.; van der Sluijs, P.; Norman, J.C. Protein kinase B/Akt acts via glycogen synthase kinase 3 to regulate recycling of αvβ3 and α5β1 integrins. Mol. Cell. Biol. 2004, 24, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Stoica, B.A.; Movsesyan, V.A.; Knoblach, S.M.; Faden, A.I. Ceramide induces neuronal apoptosis through mitogen-activated protein kinases and causes release of multiple mitochondrial proteins. Mol. Cell. Neurosci. 2005, 29, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Han, Z.; Couvillon, A.D.; Exton, J.H. Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress-induced cell death. J. Biol. Chem. 2004, 279, 49420–49429. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Wu, J.; Back, S.H.; Callaghan, M.U.; Ferris, S.P.; Iqbal, J.; Clark, R.; Miao, H.; Hassler, J.R.; Fornek, J.; et al. UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev. Cell 2008, 15, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, V.; Zeitlin, R. Cotinine: A potential new therapeutic agent against Alzheimer’s disease. CNS Neurosci. Ther. 2012, 18, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Bouten, C.V.; Oomens, C.W.; Baaijens, F.P.; Bader, D.L. The etiology of pressure ulcers: Skin deep or muscle bound? Arch. Phys. Med. Rehabil. 2003, 84, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Agam, L.; Gefen, A. Pressure ulcers and deep tissue injury: a bioengineering perspective. J. Wound Care 2007, 16, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pu, L.; Li, Z.; Hu, X.; Jiang, L. Hypoxia-inducible factor-1a gene expression and apoptosis in ischemia-reperfusion injury a rat model of early-stage pressure ulcer. Nurs. Res. 2016, 65, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, T.; Endo, M.; Oike, Y. Endoplasmic reticulum stress-related inflammation and cardiovascular diseases. Int. J. Inflamm. 2011, 2011, 259462. [Google Scholar] [CrossRef] [PubMed]
- Teng, B.T.; Pei, X.M.; Tam, E.W.; Benzie, I.F.; Siu, P.M. Opposing responses of apoptosis and autophagy to moderate compression in skeletal muscle. Acta Physiol. 2011, 201, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Zhang, X.; Wang, Z.G.; Shi, H.X.; Wu, F.Z.; Lin, B.B.; Xu, X.L.; Wang, X.J.; Fu, X.B.; Li, Z.Y.; et al. Exogenous basic fibroblast growth factor inhibits ER stress-induced apoptosis and improves recovery from spinal cord injury. CNS Neurosci. Ther. 2013, 19, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.L.; Lun, M.; Teng, J.; Huang, J.; Blasick, T.M.; Yin, L.; Herrera, G.A.; Cheung, J.Y. Preinduced molecular chaperones in the endoplasmic reticulum protect cardiomyocytes from lethal injury. Ann. Clin. Lab. Sci. 2004, 34, 449–457. [Google Scholar] [PubMed]
- Liao, K.; Li, J.; Wang, Z. Dihydroartemisinin inhibits cell proliferation via AKT/GSK3beta/cyclinD1 pathway and induces apoptosis in A549 lung cancer cells. Int. J. Clin. Exp. Pathol. 2014, 7, 8684–8691. [Google Scholar] [PubMed]
- Dey, G.; Bharti, R.; Dhanarajan, G.; Das, S.; Dey, K.K.; Kumar, B.N.; Sen, R.; Mandal, M. Marine lipopeptide Iturin A inhibits Akt mediated GSK3β and FoxO3a signaling and triggers apoptosis in breast cancer. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Mozaffari, M.S.; Liu, J.Y.; Schaffer, S.W. Effect of pressure overload on cardioprotection via PI3K-Akt: Comparison of postconditioning, insulin, and pressure unloading. Am. J. Hypertens. 2010, 23, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.L.; Shen, T.; Shao, L.L.; Ma, H.; Wang, J.K. Ischemic postconditioning mediates cardioprotection via PI3K/GSK-3β/β-catenin signaling pathway in ischemic rat myocardium. Shock 2012, 38, 165–169. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Correlated Factors | r a | p |
|---|---|---|
| p-Akt and CHOP | −0.63 | < 0.01 |
| p-Akt and cleaved caspase-12 | −0.72 | < 0.01 |
| p-GSK3β and CHOP | −0.28 | > 0.05 |
| p-GSK3β cleaved caspase-12 | −0.76 | < 0.01 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, F.-F.; Pan, Y.-Y.; Xie, H.-H.; Wang, X.-H.; Shi, H.-X.; Xiao, J.; Zhang, H.-Y.; Chang, H.-T.; Jiang, L.-P. Pressure Combined with Ischemia/Reperfusion Injury Induces Deep Tissue Injury via Endoplasmic Reticulum Stress in a Rat Pressure Ulcer Model. Int. J. Mol. Sci. 2016, 17, 284. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030284
Cui F-F, Pan Y-Y, Xie H-H, Wang X-H, Shi H-X, Xiao J, Zhang H-Y, Chang H-T, Jiang L-P. Pressure Combined with Ischemia/Reperfusion Injury Induces Deep Tissue Injury via Endoplasmic Reticulum Stress in a Rat Pressure Ulcer Model. International Journal of Molecular Sciences. 2016; 17(3):284. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030284
Chicago/Turabian StyleCui, Fei-Fei, Ying-Ying Pan, Hao-Huang Xie, Xiao-Hui Wang, Hong-Xue Shi, Jian Xiao, Hong-Yu Zhang, Hao-Teng Chang, and Li-Ping Jiang. 2016. "Pressure Combined with Ischemia/Reperfusion Injury Induces Deep Tissue Injury via Endoplasmic Reticulum Stress in a Rat Pressure Ulcer Model" International Journal of Molecular Sciences 17, no. 3: 284. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030284