
Key Immune Events of the Pathomechanisms of Early Cardioembolic Stroke: Multi-Database Mining and Systems Biology Approach


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Network Summary
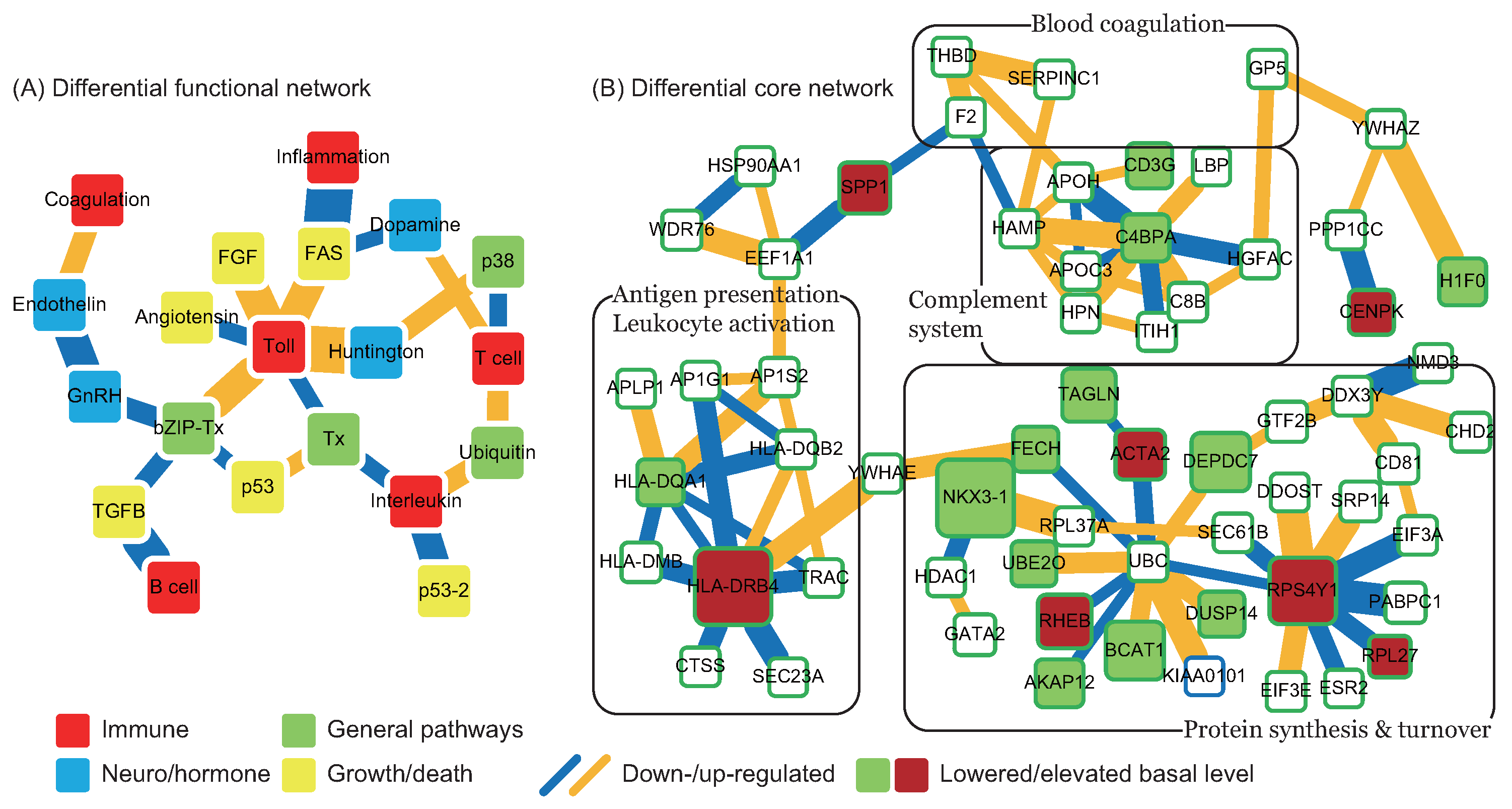
2.2. Changes in Functions and Proteins Immediately after CE Strokes
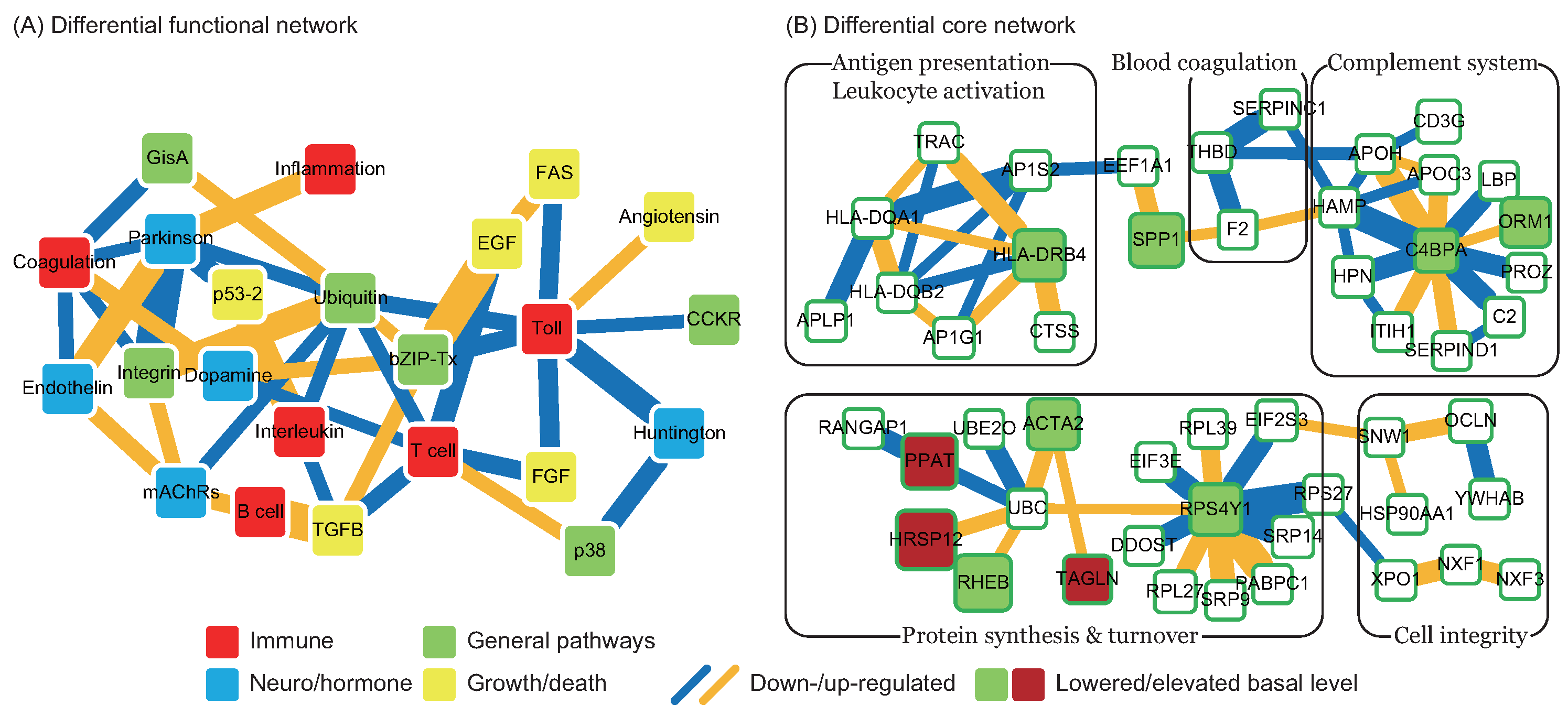
2.3. Changes in Functions and Proteins after Tissue Plasminogen Activator Treatment
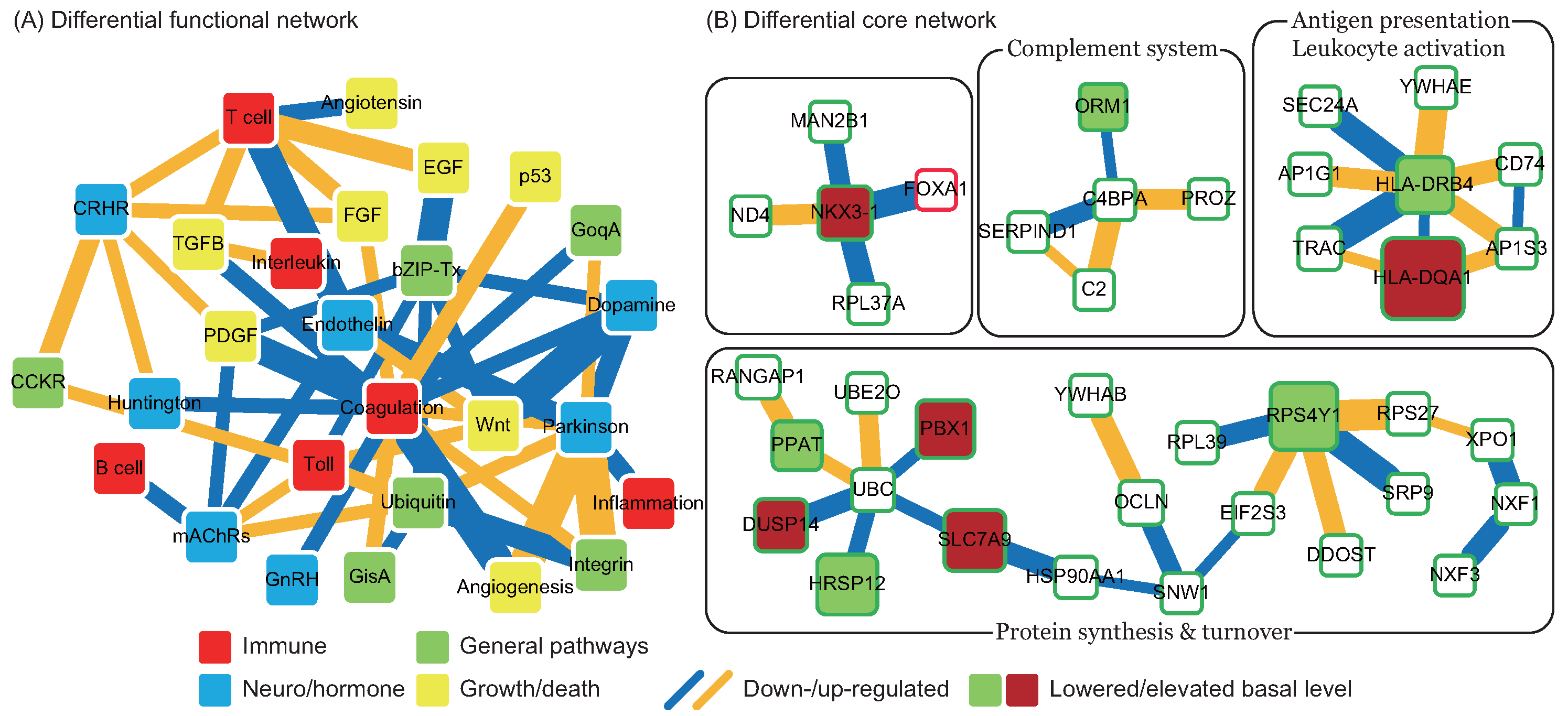
2.4. Changes in Functions and Proteins in Early tPA Treatment
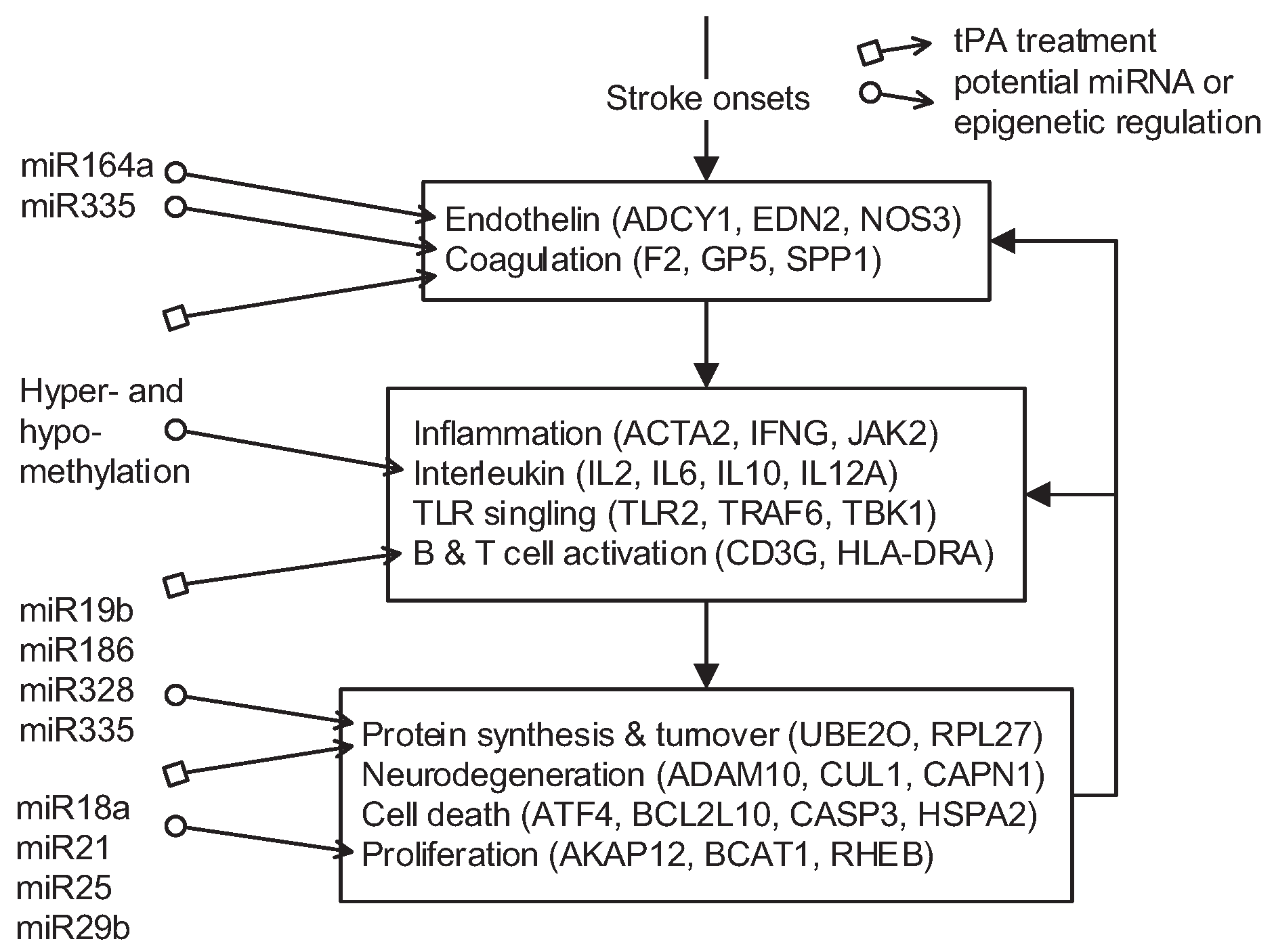
2.5. Pathomechanisms and Potential Drug Targets
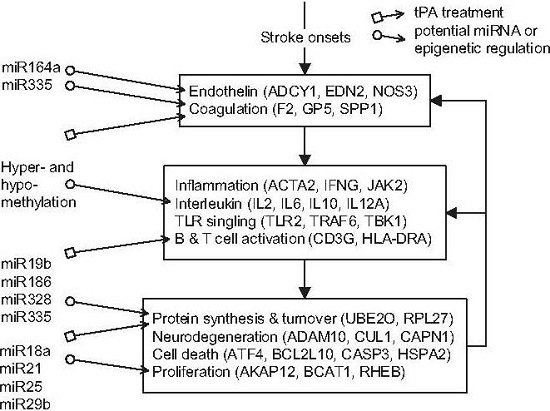
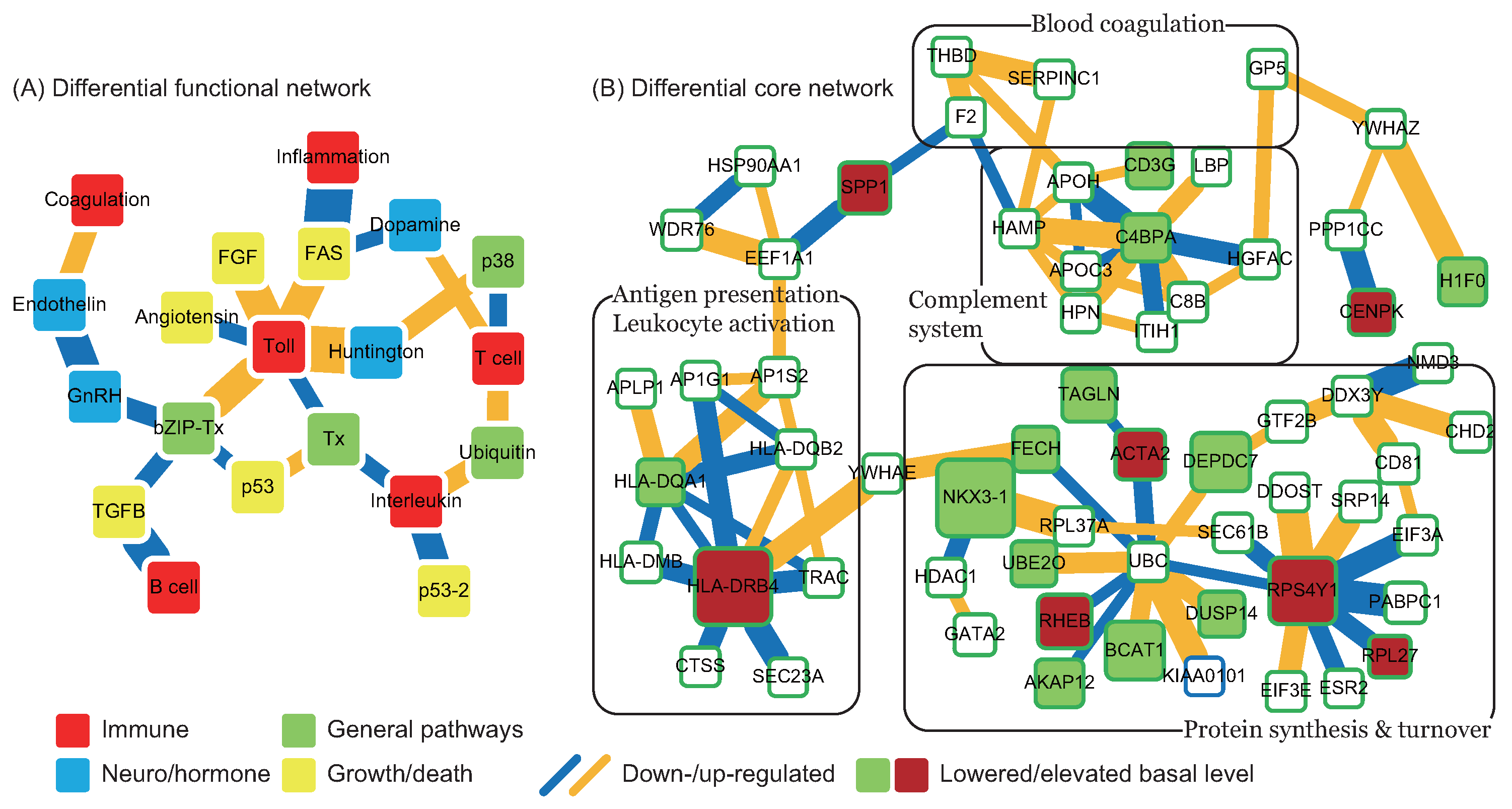
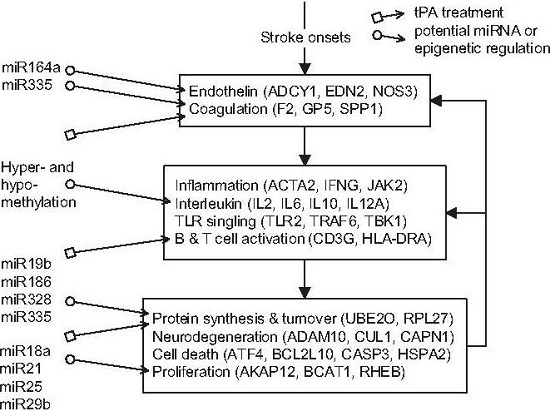
 in Figure 4), and the pathomechanism of CE stroke is also subject to interference. Although this interference can briefly relieve the symptoms caused by vessel obstruction, several neurodegenerative diseases emerge in the functional networks following tPA treatment.
in Figure 4), and the pathomechanism of CE stroke is also subject to interference. Although this interference can briefly relieve the symptoms caused by vessel obstruction, several neurodegenerative diseases emerge in the functional networks following tPA treatment. ). In addition to miRNA regulation of protein basal levels, methylation regulation is a potential mechanism that can change protein basal levels after stroke onset. Although studies of methylation in strokes have indicated a range of changes in protein basal level [33], the targets of methylation regulation have not yet been the subject of a dedicated study. Proteins that have large basal level changes and are not miRNA targets (ACTA2 [34], C4BPA [35], CD3G [36], CENPK [37], DEPDC7 [38], FECH [39], HLA-DQA1 [40], HLA-DRB4 [41], and NKX3-1 [42]) can be potential targets of methylation regulations. Based on the specific targets of miRNA regulations and the position of the target proteins in the core networks, several potential drug targets can be selected (* in Table 2). SPP1 in blood coagulation bridges the complement systems and antigen presentation, and the connection can activate the subsequent inflammation and immune responses. In addition, SPP1 can also activate IFNγ and IL12, making it a potential treatment candidate. Another potential target is RPS4Y1, a male-specific protein that may play a role in males’ higher susceptibility to stroke. Finally, the possibility that the targets of miRNA and methylation regulation are identical cannot be ruled out and there are other factors may cause the changes of protein basal levels, such as differential gene regulations through transcription factors. The mechanisms of miRNA and methylation regulations after CE stroke onset require further investigation in future studies.
). In addition to miRNA regulation of protein basal levels, methylation regulation is a potential mechanism that can change protein basal levels after stroke onset. Although studies of methylation in strokes have indicated a range of changes in protein basal level [33], the targets of methylation regulation have not yet been the subject of a dedicated study. Proteins that have large basal level changes and are not miRNA targets (ACTA2 [34], C4BPA [35], CD3G [36], CENPK [37], DEPDC7 [38], FECH [39], HLA-DQA1 [40], HLA-DRB4 [41], and NKX3-1 [42]) can be potential targets of methylation regulations. Based on the specific targets of miRNA regulations and the position of the target proteins in the core networks, several potential drug targets can be selected (* in Table 2). SPP1 in blood coagulation bridges the complement systems and antigen presentation, and the connection can activate the subsequent inflammation and immune responses. In addition, SPP1 can also activate IFNγ and IL12, making it a potential treatment candidate. Another potential target is RPS4Y1, a male-specific protein that may play a role in males’ higher susceptibility to stroke. Finally, the possibility that the targets of miRNA and methylation regulation are identical cannot be ruled out and there are other factors may cause the changes of protein basal levels, such as differential gene regulations through transcription factors. The mechanisms of miRNA and methylation regulations after CE stroke onset require further investigation in future studies.3. Material and Methods
3.1. Microarray Data for Early Cardioembolic Stroke
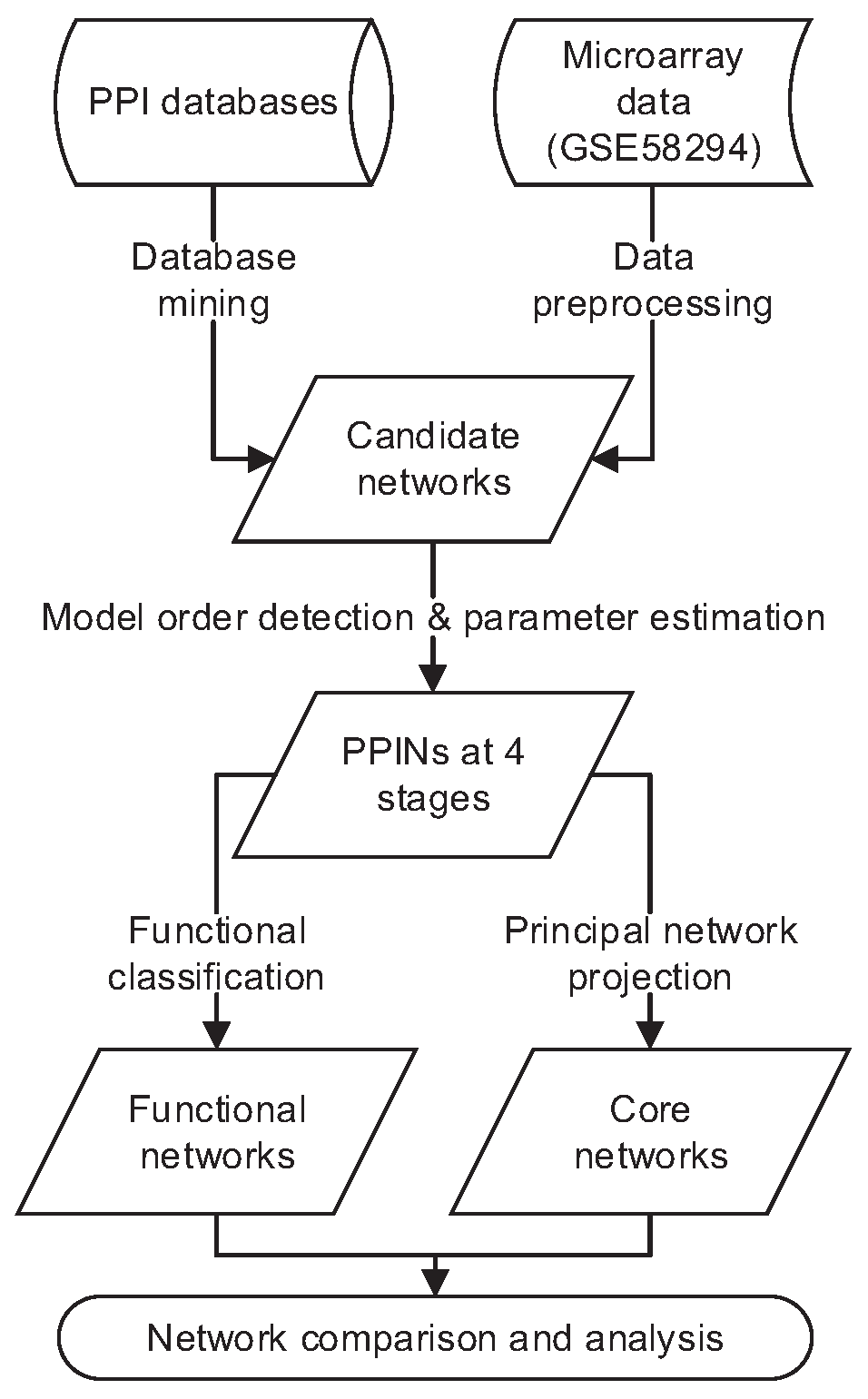
3.2. Network Construction
3.2.1. Candidate Network Construction via Multi-Database Mining
3.2.2. Protein Interaction Model
3.2.3. Model Order Detection and Identification
| Require: candidate network, gene expression profiles at a specific stage |
| for all protein i in the candidate network do |
| expression profiles of protein i |
| expression profiles of all proteins interacting with protein i in the candidate network |
| function AICstepwise() |
| Start with forward selection and after each candidate (other than the first one) is added to the model, perform backward elimination to see if any of the selected candidates can be eliminated without increasing the AIC value. |
| return |
| end function |
| function Ttest() |
| Calculate p-value for each interaction activity in and delete if . |
| return |
| end function |
| end for |
3.3. Network Analysis
3.3.1. Functional Networks
3.3.2. Core Networks
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Iosif, R.E.; Ekdahl, C.T.; Ahlenius, H.; Pronk, C.J.H.; Bonde, S.; Kokaia, Z.; Jacobsen, S.E.W.; Lindvall, O. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J. Neurosci. 2006, 26, 9703–9712. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.C.; Fang, Z.B.; Li, L.; Luo, L.F. Using zebrafish as the model organism to understand organ regeneration. Sci. China Life Sci. 2015, 58, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Kyritsis, N.; Kizil, C.; Zocher, S.; Kroehne, V.; Kaslin, J.; Freudenreich, D.; Iltzsche, A.; Brand, M. Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science 2012, 338, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Stamova, B.; Jickling, G.C.; Ander, B.P.; Zhan, X.H.; Liu, D.Z.; Turner, R.; Ho, C.; Khoury, J.C.; Bushnell, C.; Pancioli, A.; et al. Gene Expression in Peripheral Immune Cells following Cardioembolic Stroke Is Sexually Dimorphic. PLoS ONE 2014, 9, e102550. [Google Scholar] [CrossRef] [PubMed]
- Macrez, R.; Ali, C.; Toutirais, O.; Le Mauff, B.; Defer, G.; Dirnagl, U.; Vivien, D. Stroke and the immune system: From pathophysiology to new therapeutic strategies. Lancet Neurol. 2011, 10, 471–480. [Google Scholar] [CrossRef]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.G.; Muruve, D.A.; Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Fann, D.Y.W.; Lee, S.Y.; Manzanero, S.; Chunduri, P.; Sobey, C.G.; Arumugam, T.V. Pathogenesis of acute stroke and the role of inflammasomes. Ageing Res. Rev. 2013, 12, 941–966. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Chen, B.S. Integrated cellular network of transcription regulations and protein-protein interactions. BMC Syst. Biol. 2010, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-S.; Wu, C.-C. Systems Biology : An Integrated Platform for Bioinformatics, Systems Synthetic Biology and Systems Metabolic Engineering; Nova Publishers: New York, NY, USA, 2014; pp. 47–63. [Google Scholar]
- Coucha, M.; Li, W.G.; Ergul, A. The effect of endothelin receptor a antagonism on basilar artery endothelium-dependent relaxation after ischemic stroke. Life Sci. 2012, 91, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Shahpouri, M.M.; Mousavi, S.A.; Khorvash, F.; Mousavi, S.M.; Hoseini, T. Anticoagulant therapy for ischemic stroke: A review of literature. J. Res. Med. Sci. 2012, 17, 396–401. [Google Scholar] [PubMed]
- Swardfager, W.; Winer, D.A.; Herrmann, N.; Winer, S.; Lanctot, K.L. Interleukin-17 in post-stroke neurodegeneration. Neurosci. Biobehav. R. 2013, 37, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Gorelick, P.B. Hypertension, angiotensin, and stroke: Beyond blood pressure. Stroke 2004, 35, 348–350. [Google Scholar] [CrossRef] [PubMed]
- Wolf, W.A.; Martin, J.L.; Kartje, G.L.; Farrer, R.G. Evidence for fibroblast growth factor-2 as a mediator of amphetamine-enhanced motor improvement following stroke. PLoS ONE 2014, 9, e108031. [Google Scholar] [CrossRef] [PubMed]
- Issa, R.; AlQteishat, A.; Mitsios, N.; Saka, M.; Krupinski, J.; Tarkowski, E.; Gaffney, J.; Slevin, M.; Kumar, S.; Kumar, P. Expression of basic fibroblast growth factor mrna and protein in the human brain following ischaemic stroke. Angiogenesis 2005, 8, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Heidecker, B.; Lamirault, G.; Kasper, E.K.; Wittstein, I.S.; Champion, H.C.; Breton, E.; Russell, S.D.; Hall, J.; Kittleson, M.M.; Baughman, K.L.; et al. The gene expression profile of patients with new-onset heart failure reveals important gender-specific differences. Eur. Heart J. 2010, 31, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Atkin, G.; Paulson, H. Ubiquitin pathways in neurodegenerative disease. Front. Mol. Neurosci. 2014, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- Brott, T.; Broderick, J.; Kothari, R.; ODonoghue, M.; Barsan, W.; Tomsick, T.; Spilker, J.; Miller, R.; Sauerbeck, L.; Farrell, J.; et al. Intracerebral hemorrhage after intravenous t-PA therapy for ischemic stroke. Stroke 1997, 28, 2109–2118. [Google Scholar]
- Marchetti, B.; Pluchino, S. Wnt your brain be inflamed? Yes, it wnt! Trends Mol. Med. 2013, 19, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Su, E.J.; Fredriksson, L.; Geyer, M.; Folestad, E.; Cale, J.; Andrae, J.; Gao, Y.; Pietras, K.; Mann, K.; Yepes, M.; et al. Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat. Med. 2008, 14, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Prentice, R.L.; Paczesny, S.J.; Aragaki, A.; Amon, L.M.; Chen, L.; Pitteri, S.J.; McIntosh, M.; Wang, P.; Busald, T.B.; Hsia, J.; et al. Novel proteins associated with risk for coronary heart disease or stroke among postmenopausal women identified by in-depth plasma proteome profiling. Genome Med. 2010, 2, 48. [Google Scholar] [CrossRef] [PubMed]
- Shen, N.; Yan, Z.; He, P. A study of the hereditary susceptibility of HLA-DQA1 to essential hypertension, athrothrombotic brain infarction and lacunar stroke. Zhonghua Yi Xue Za Zhi 2001, 81, 352–355. [Google Scholar] [PubMed]
- Madden, B.; Chebl, R.B. Hemi orolingual angioedema after TPA administration for acute ischemic stroke. West. J. Emerg. Med. 2015, 16, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Ayala, P.; Uchida, M.; Akiyoshi, K.; Cheng, J.; Hashimoto, J.; Jia, T.P.; Ronnekleiv, O.K.; Murphy, S.J.; Wiren, K.M.; Hurn, P.D. Androgen receptor overexpression is neuroprotective in experimental stroke. Transl. Stroke Res. 2011, 2, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Arboleda-Velasquez, J.F.; Zhou, Z.; Shin, H.K.; Louvi, A.; Kim, H.H.; Savitz, S.I.; Liao, J.K.; Salomone, S.; Ayata, C.; Moskowitz, M.A.; et al. Linking notch signaling to ischemic stroke. Procc. Natl. Acad. Sci. USA 2008, 105, 4856–4861. [Google Scholar] [CrossRef] [PubMed]
- Marumo, T.; Takagi, Y.; Muraki, K.; Hashimoto, N.; Miyamoto, S.; Tanigaki, K. Notch signaling regulates nucleocytoplasmic olig2 translocation in reactive astrocytes differentiation after ischemic stroke. Neurosci. Res. 2013, 75, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Hankey, G.J.; Eikelboom, J.W. Homocysteine and stroke. Curr. Opin. Neurol. 2001, 14, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Tsai, T.H.; Chang, C.; Lee, T.T.; Lin, C.; Cheng, I.H.J.; Sun, M.C.; Chuang, Y.J.; Chen, B.S. On the crucial cerebellar wound healing-related pathways and their cross-talks after traumatic brain injury in Danio rerio. PLoS ONE 2014, 9, e97902. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.Y.; Aromolaran, K.A.; Zukin, R.S. Epigenetic mechanisms in stroke and epilepsy. Neuropsychopharmacology 2013, 38, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.D.; Lin, F.M.; Wu, W.Y.; Liang, C.; Huang, W.C.; Chan, W.L.; Tsai, W.T.; Chen, G.Z.; Lee, C.J.; Chiu, C.M.; et al. Mirtarbase: A database curates experimentally validated microRNA–Target interactions. Nucleic Acids Res. 2011, 39, D163–D169. [Google Scholar] [CrossRef] [PubMed]
- Koutsis, G.; Siasos, G.; Spengos, K. The emerging role of microrna in stroke. Curr. Top. Med. Chem. 2013, 13, 1573–1588. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, I.A.; Mehler, M.F. Emerging role of epigenetics in stroke part 1: DNA methylation and chromatin modifications. Arch. Neurol-Chicago 2010, 67, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Satish, L.; Gallo, P.H.; Baratz, M.E.; Johnson, S.; Kathju, S. Reversal of TGF-β1 stimulation of α-smooth muscle actin and extracellular matrix components by cyclic amp in dupuytren’s-derived fibroblasts. BMC Musculoskelet. Disord. 2011, 12, 113. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.L.; Weichel, B.; Ohm, J.E.; Zhang, K. An integrative analysis of DNA methylation and RNA-seq data for human heart, kidney and liver. BMC Syst. Biol. 2011, 5, S4. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, B.F.; Wotton, D.; Soong, T.W.; Owen, M.J. DNase hypersensitivity and methylation of the human CD3G and D-genes during T-cell development. Immunogenetics 1990, 31, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Sabato, S.; Hellman, A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol 2013, 14, R21. [Google Scholar] [CrossRef] [PubMed]
- Cordova-Palomera, A.; Fatjo-Vilas, M.; Palma-Gudiel, H.; Blasco-Fontecilla, H.; Kebir, O.; Fananas, L. Further evidence of depdc7 DNA hypomethylation in depression: A study in adult twins. Eur. Psychiat. 2015, 30, 715–718. [Google Scholar] [CrossRef] [PubMed]
- Onaga, Y.; Ido, A.; Uto, H.; Hasuike, S.; Kusumoto, K.; Moriuchi, A.; Numata, M.; Nagata, K.; Hori, T.; Hayashi, K.; et al. Hypermethylation of the wild-type ferrochelatase allele is closely associated with severe liver complication in a family with erythropoietic protoporphyria. Biochem. Biophys. Res. Commun. 2004, 321, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Majumder, P.; Boss, J.M. DNA methylation dysregulates and silences the HLA-DQ locus by altering chromatin architecture. Genes Immun. 2011, 12, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Shiina, T.; Hosomichi, K.; Inoko, H.; Kulski, J.K. The HLA genomic loci map: Expression, interaction, diversity and disease. J. Hum. Genet. 2009, 54, 15–39. [Google Scholar] [CrossRef] [PubMed]
- Asatiani, E.; Huang, W.X.; Wang, A.; Ortner, E.R.; Cavalli, L.R.; Haddad, B.R.; Gelmann, E.P. Deletion, methylation, and expression of the NKX3.1 suppressor gene in primary human prostate cancer. Cancer Res. 2005, 65, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Betel, D.; Hogue, C.W.V. Bind: The biomolecular interaction network database. Nucleic Acids Res. 2003, 31, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. Biogrid: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [PubMed]
- Xenarios, I.; Salwinski, L.; Duan, X.Q.J.; Higney, P.; Kim, S.M.; Eisenberg, D. Dip, the database of interacting proteins: A research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002, 30, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Peri, S.; Navarro, J.D.; Amanchy, R.; Kristiansen, T.Z.; Jonnalagadda, C.K.; Surendranath, V.; Niranjan, V.; Muthusamy, B.; Gandhi, T.K.B.; Gronborg, M.; et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003, 13, 2363–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.R.; Jurisica, I. Online predicted human interaction database. Bioinformatics 2005, 21, 2076–2082. [Google Scholar] [CrossRef] [PubMed]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The mintact project-intact as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [PubMed]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. Mint, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef] [PubMed]
- McDowall, M.D.; Scott, M.S.; Barton, G.J. Pips: Human protein-protein interaction prediction database. Nucleic Acids Res. 2009, 37, D651–D656. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.M.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.Y.; Minguez, P.; Bork, P.; von Mering, C.; et al. String v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.E.; Murray, W.; Wright, M.H. Practical Optimization; Academic Press: London, UK, 1981; p. 401. [Google Scholar]
- Akaike, H. New look at statistical-model identification. IEEE Trans. Autom. Control 1974, Ac19, 716–723. [Google Scholar] [CrossRef]
- Mi, H.Y.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the panther classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef] [PubMed]

 ) and miRNA regulation ( ) can interfere with the progression of stroke and potential targeted functions and proteins.
) and miRNA regulation ( ) can interfere with the progression of stroke and potential targeted functions and proteins.
) and miRNA regulation ( ) can interfere with the progression of stroke and potential targeted functions and proteins.
) and miRNA regulation ( ) can interfere with the progression of stroke and potential targeted functions and proteins.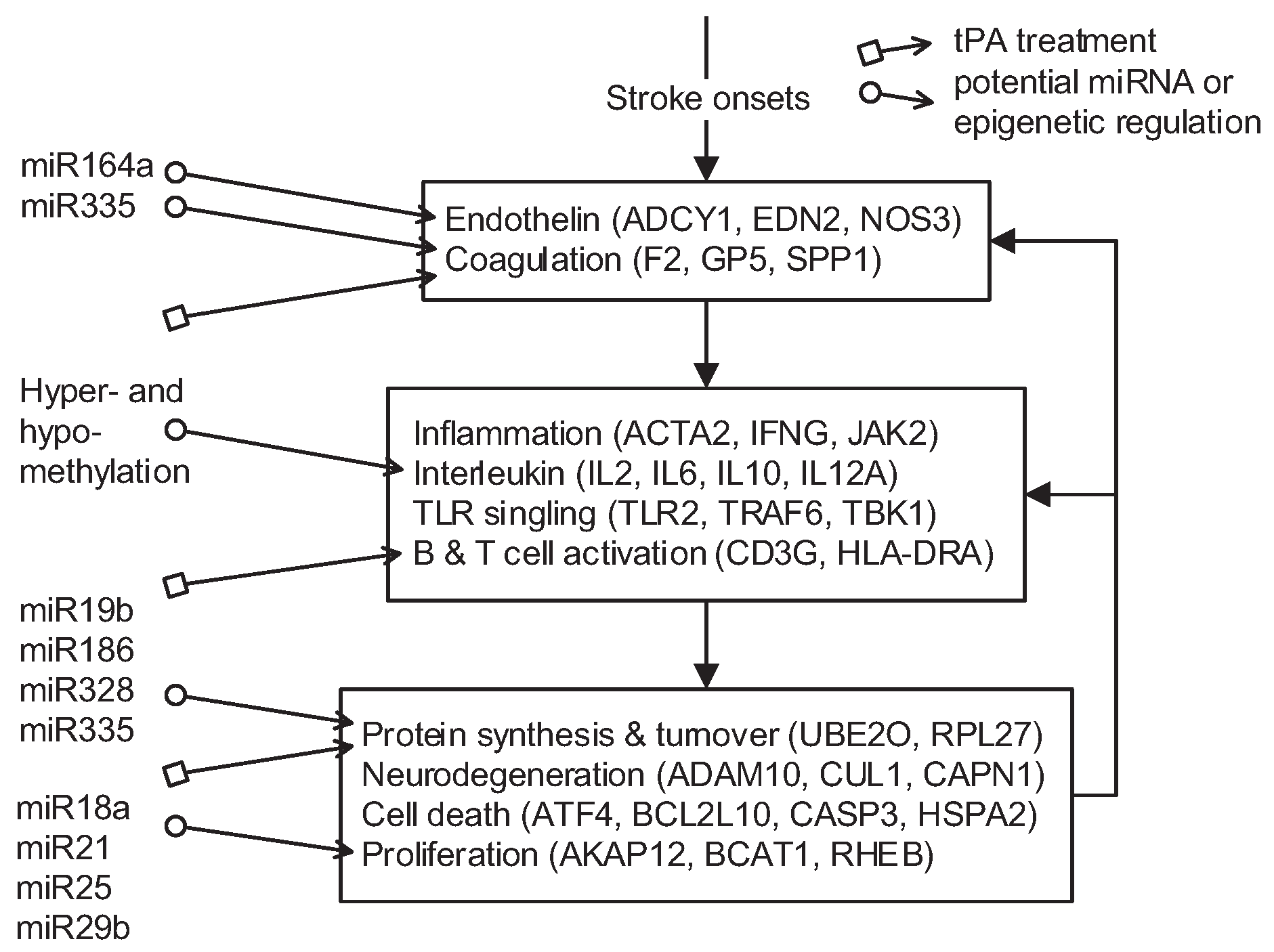


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stage | Number of Proteins | Number of Interactions |
|---|---|---|
| Control (C) | 11,554 | 91,729 |
| ≤3 h (I) | 9433 | 52,295 |
| 5 h (II) | 9432 | 52,774 |
| 24 h (III) | 9339 | 51,989 |
| Target Protein | Regulation Type | Function of Target Protein | Literature Validation | |
|---|---|---|---|---|
| Lowered level | BCAT1 | miR-21, 25, 140, 146a | cell growth | [31,32] and references therein |
| AKAP12 | miR-29b-1, 181a, 183, 335 | cell growth | [31,32] and references therein | |
| DUSP14 | miR-16, 26b | signaling pathway | [31,32] and references therein | |
| FECH | miR-16, 25, 124 | heme synthesis | [31,32] and references therein | |
| H1F0 | miR-181a, 494 | histones | [31,32] and references therein | |
| TAGLN | miR-26b, 149 | undetermined | [31,32] and references therein | |
| UBE2O | miR-328, 335 | protein synthesis & turnover | [31,32] and references therein | |
| RPS4Y1* | miR-19b | protein synthesis & turnover | [31,32] and references therein | |
| SPP1* | miR-146a, 335 | coagulation | [31,32] and references therein | |
| C4BPA | hypermethylation | complement system | [35] | |
| CD3G | hypermethylation | complement system | [36] | |
| DEPDC7 | hypermethylation | protein synthesis & turnover | [38] | |
| FECH | hypermethylation | protein synthesis & turnover | [39] | |
| HLA-DQA1 | hypermethylation | leukocyte activation | [40] | |
| NKX3-1 | hypermethylation | protein synthesis & turnover | [42] | |
| Elevated level | RHEB | miR-18a, 155 | cell growth | [31,32] and references therein |
| RPL27 | miR-186 | protein synthesis & turnover | [31,32] and references therein | |
| ACTA2 | hypomethylation | inflammation | [34] | |
| CENPK | hypomethylation | cell growth | [37] | |
| HLA-DRB4 | hypomethylation | Leukocyte activation | [41] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-C.; Chen, B.-S. Key Immune Events of the Pathomechanisms of Early Cardioembolic Stroke: Multi-Database Mining and Systems Biology Approach. Int. J. Mol. Sci. 2016, 17, 305. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030305
Wu C-C, Chen B-S. Key Immune Events of the Pathomechanisms of Early Cardioembolic Stroke: Multi-Database Mining and Systems Biology Approach. International Journal of Molecular Sciences. 2016; 17(3):305. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030305
Chicago/Turabian StyleWu, Chia-Chou, and Bor-Sen Chen. 2016. "Key Immune Events of the Pathomechanisms of Early Cardioembolic Stroke: Multi-Database Mining and Systems Biology Approach" International Journal of Molecular Sciences 17, no. 3: 305. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030305