
Biomimetic Membranes for Multi-Redox Center Proteins


Abstract
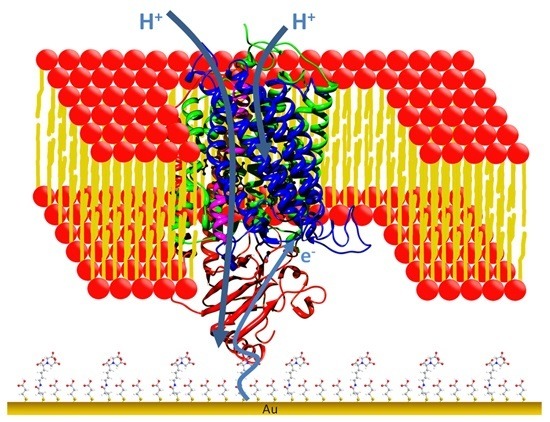
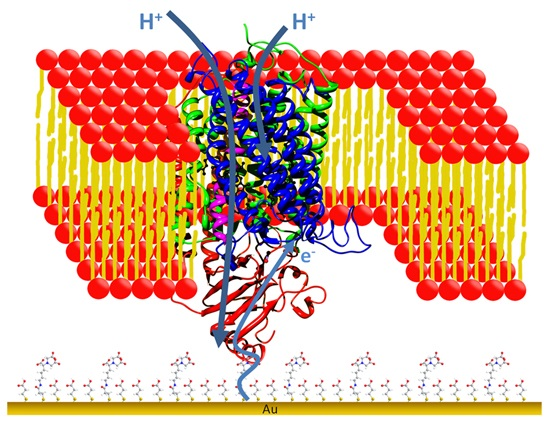
:
1. Introduction
2. Results and Discussion
2.1. Multi-Redox Center Proteins Immobilized on Planar Surfaces
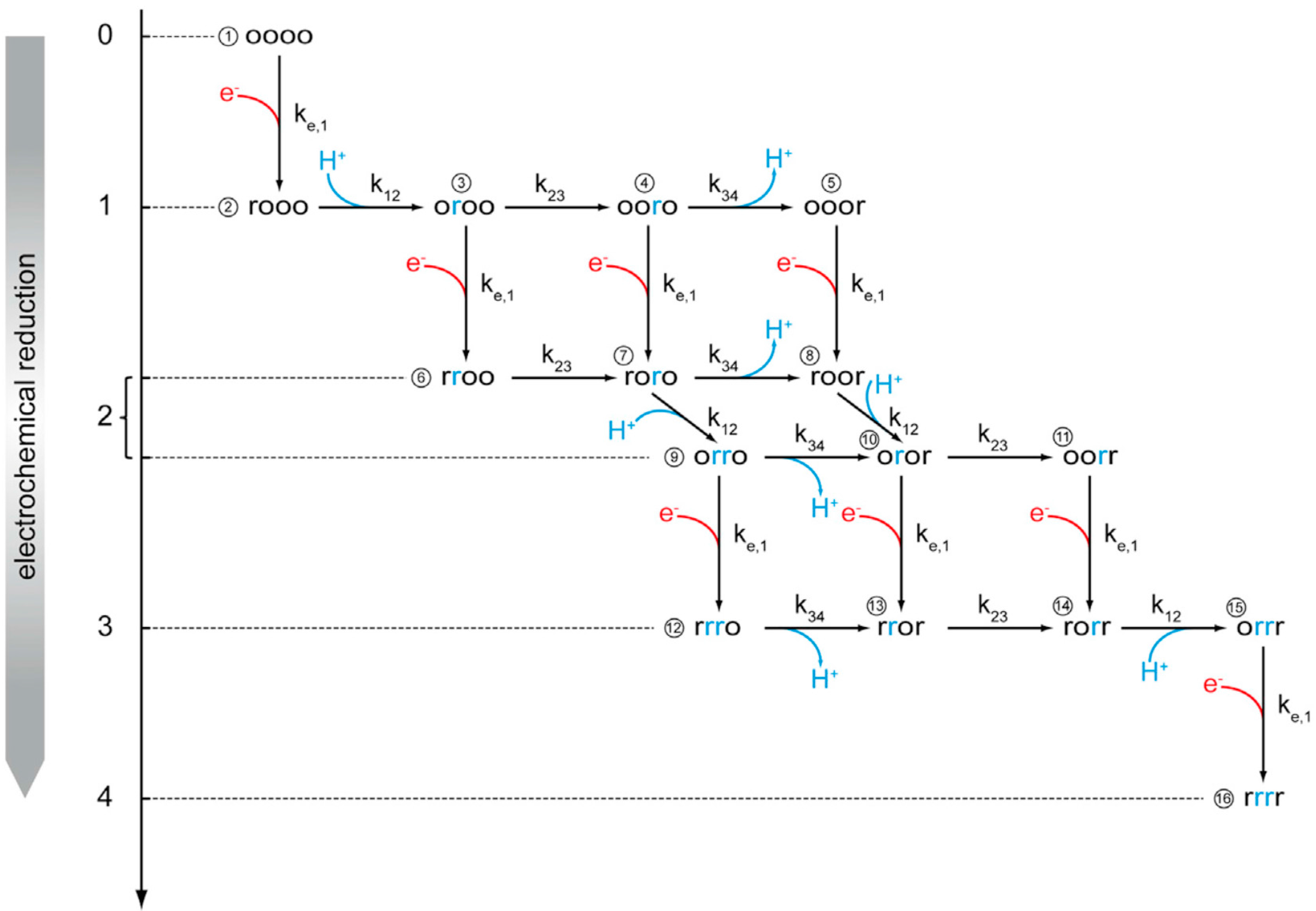
2.1.1. Time Resolved tr-SEIRAS of Cytochrome c Oxidase (CcO) from R. sphaeroides
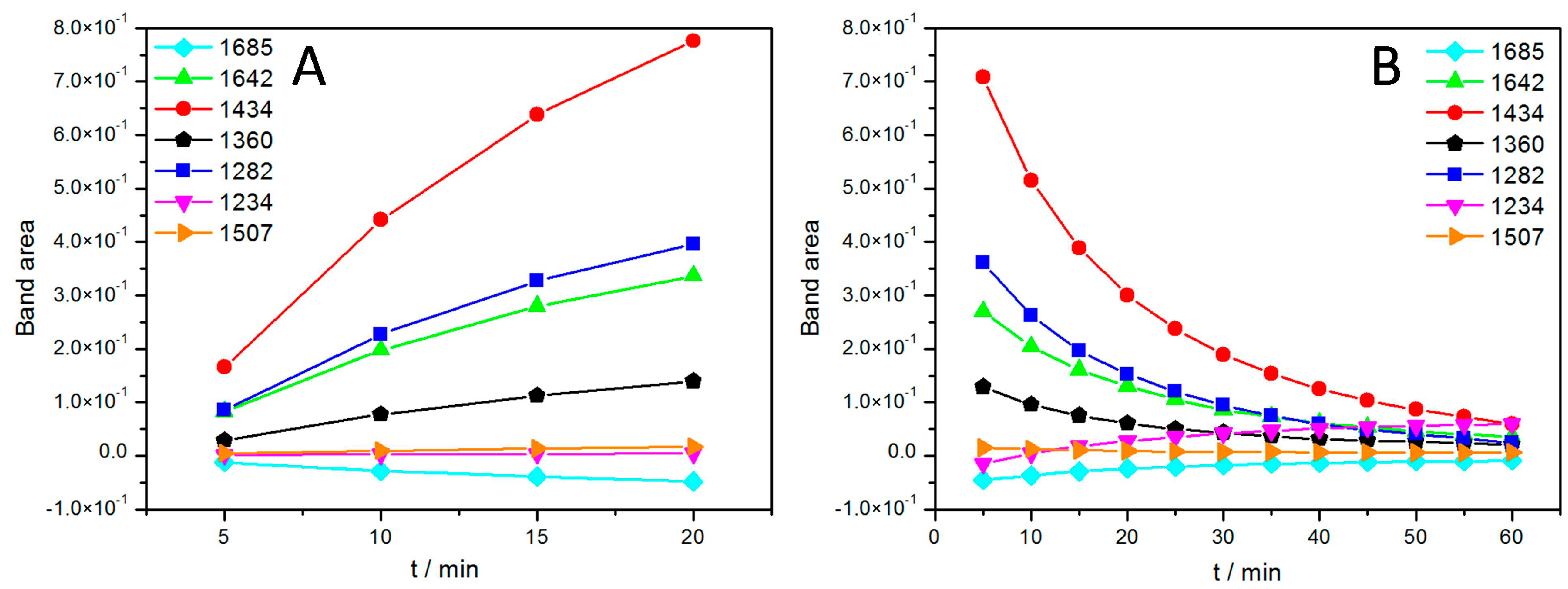
2.1.2. Photosynthetic Reaction Centers from R. sphaeroides
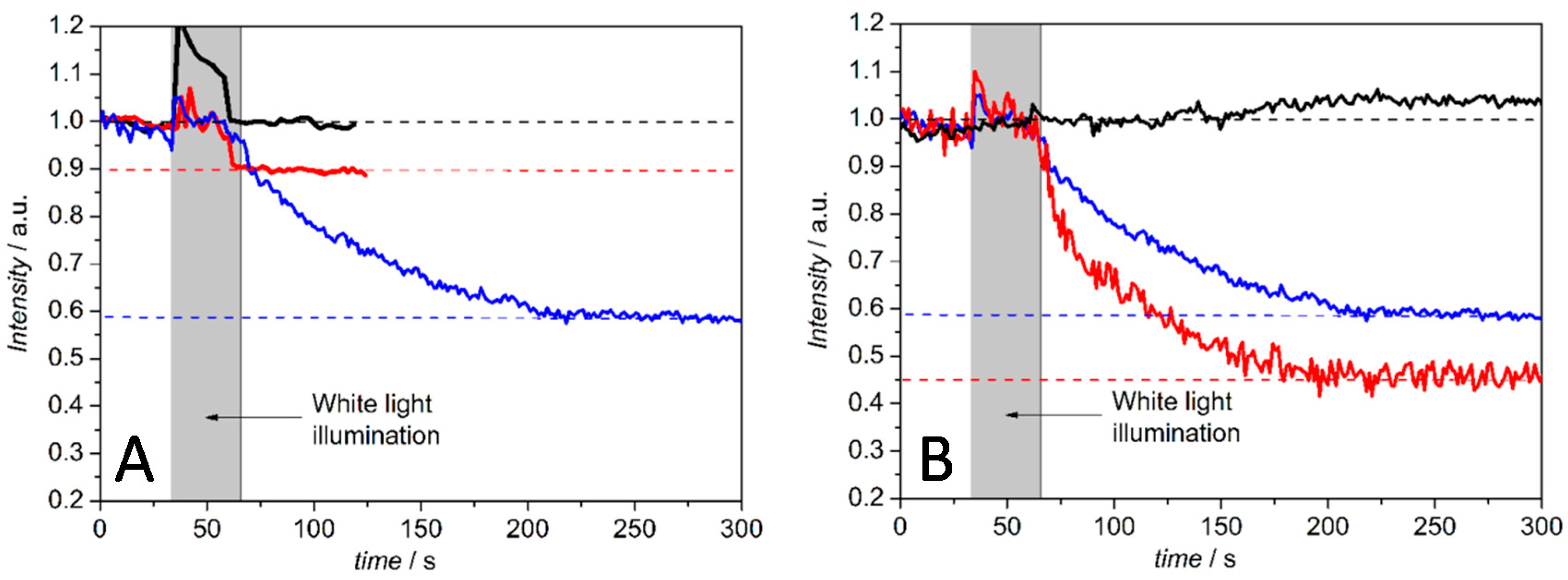
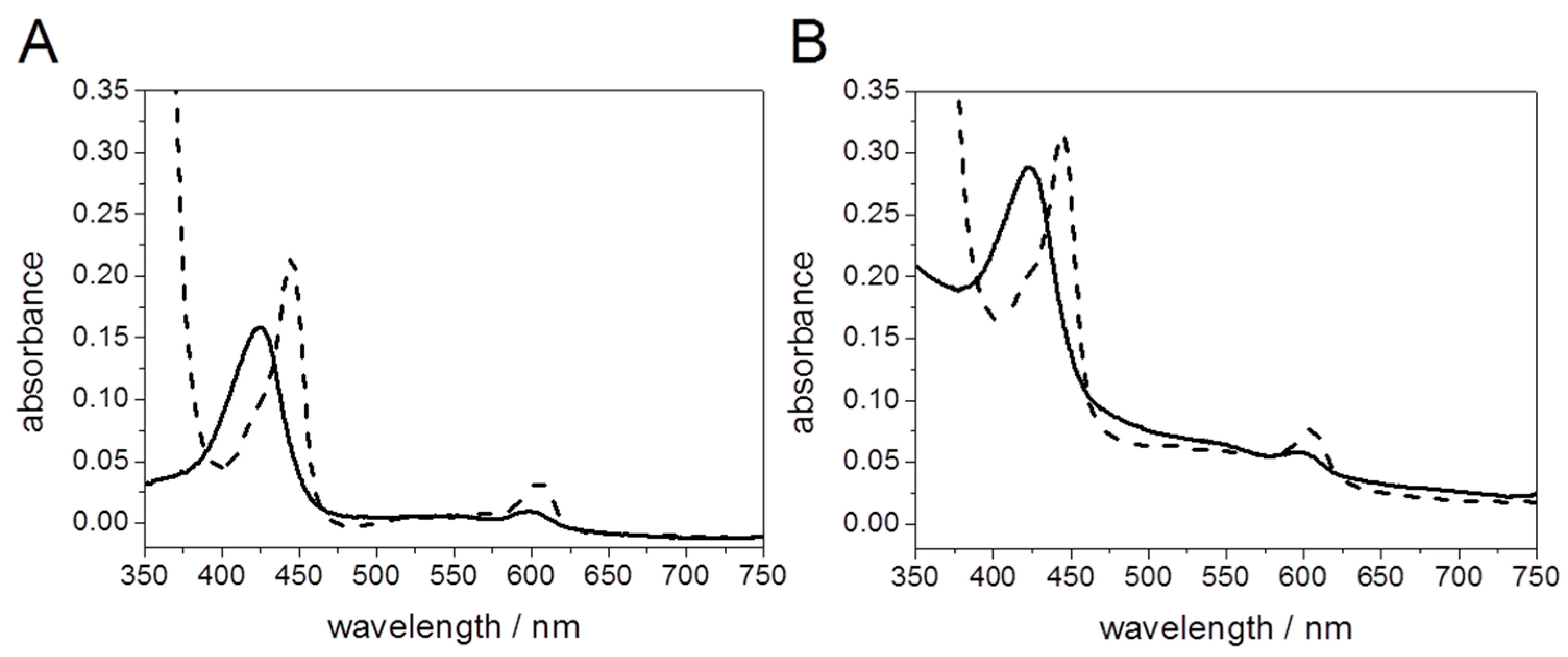
Light Activation of Reaction Centers (RCs) Followed by SEIRAS
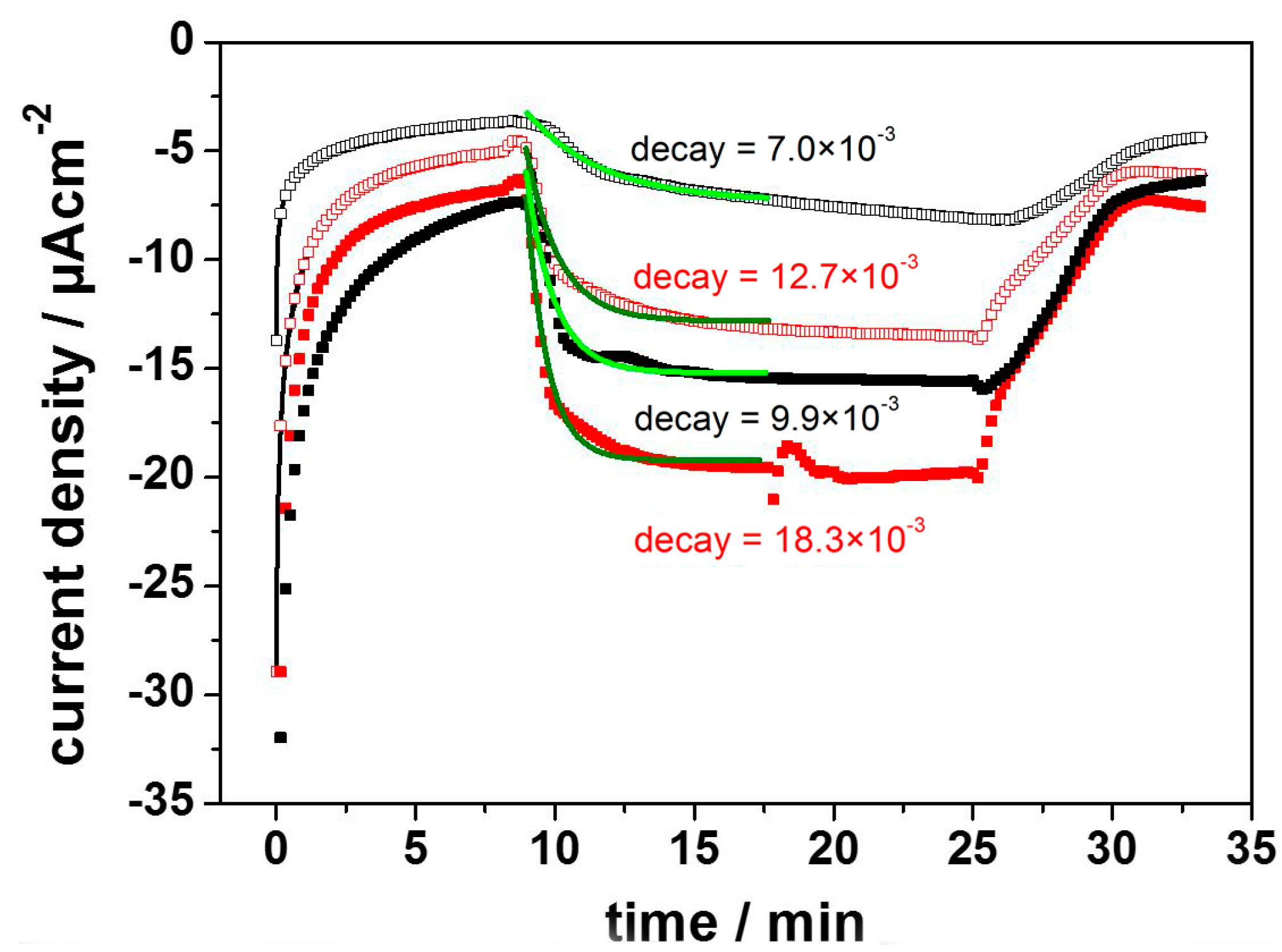
Direct Electron Transfer to Photosynthetic Reaction Centers from R. sphaeroides
2.2. From Flat Surfaces to Particles
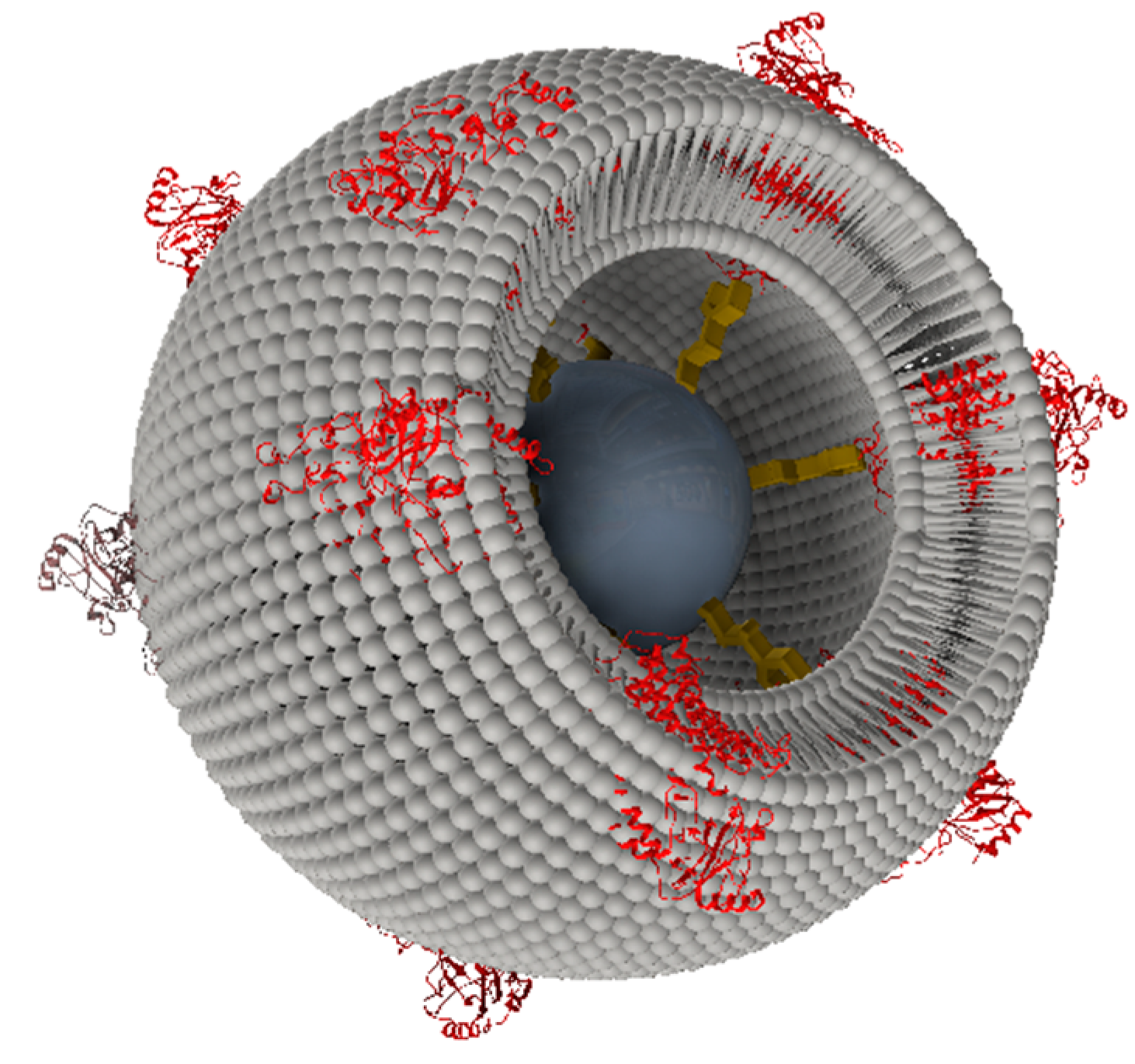
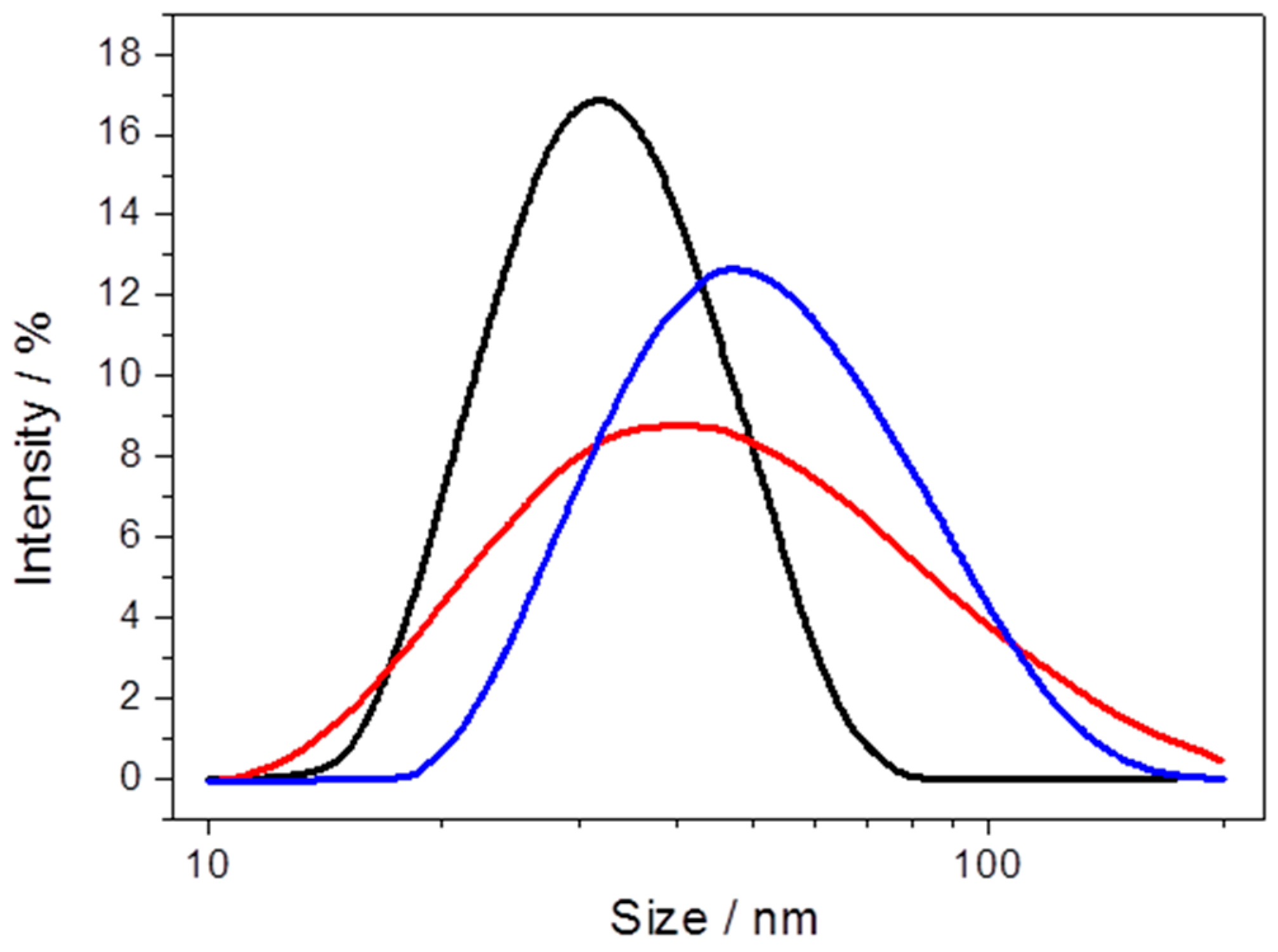
2.2.1. Cytochrome c Oxidase from P. denitrificans Incorporated in Proteo-Lipobeads (PLBs) Based on Agarose Beads
2.2.2. Cytochrome c Oxidase from P. denitrificans Incorporated in PLBs Based on Silica Nanoparticles
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jeuken, L.J.C. Electrodes for integral membrane enzymes. Nat. Prod. Rep. 2009, 26, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, F.A. Insights from protein film voltammetry into mechanisms of complex biological electron-transfer reactions. J. Chem. Soc. Dalton Trans. 2002. [Google Scholar] [CrossRef]
- Gorbikova, E.A.; Belevich, N.P.; Wikstrom, M.; Verkhovsky, M.I. Time-resolved ATR-FTIR spectroscopy of the oxygen reaction in the D124N mutant of cytochrome c oxidase from Paracoccus denitrificans. Biochemistry 2007, 46, 13141–13148. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Sackmann, E. Polymer-supported membranes as models of the cell surface. Nature 2005, 437, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Terrettaz, S.; Mayer, M.; Vogel, H. Highly electrically insulating tethered lipid bilayers for probing the function of ion channel proteins. Langmuir 2003, 19, 5567–5569. [Google Scholar] [CrossRef]
- Giess, F.; Friedrich, M.G.; Heberle, J.; Naumann, R.L.; Knoll, W. The protein-tethered lipid bilayer: A novel mimic of the biological membrane. Biophys. J. 2004, 87, 3213–3220. [Google Scholar] [CrossRef] [PubMed]
- Ataka, K.; Giess, F.; Knoll, W.; Naumann, R.; Haber-Pohlmeier, S.; Richter, B.; Heberle, J. Oriented attachment and membrane reconstitution of his-tagged cytochrome c oxidase to a gold electrode: In situ monitoring by surface-enhanced infrared absorption spectroscopy. J. Am. Chem. Soc. 2004, 126, 16199–16206. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zaitseva, E.; Schmidt, M.; Siebert, F.; Engelhard, M.; Schlesinger, R.; Ataka, K.; Vogel, R.; Heberle, J. Resolving voltage-dependent structural changes of a membrane photoreceptor by surface-enhanced IR difference spectroscopy. Proc. Natl. Acad. Sci. USA 2008, 105, 12113–12117. [Google Scholar] [CrossRef] [PubMed]
- Kriegel, S.; Uchida, T.; Osawa, M.; Friedrich, T.; Hellwig, P. Biomimetic environment to study E-coli complex I through surface-enhanced IR absorption spectroscopy. Biochemistry 2014, 53, 6340–6347. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.G.; Robertson, J.W.F.; Walz, D.; Knoll, W.; Naumann, R.L.C. Electronic wiring of a multi-redox site membrane protein in a biomimetic surface architecture. Biophys. J. 2008, 94, 3698–3705. [Google Scholar] [CrossRef] [PubMed]
- Nowak, C.; Santonicola, M.G.; Schach, D.; Zhu, J.P.; Gennis, R.B.; Ferguson-Miller, S.; Baurecht, D.; Walz, D.; Knoll, W.; Naumann, R.L.C. Conformational transitions and molecular hysteresis of cytochrome c oxidase: Varying the redox state by electronic wiring. Soft Matter 2010, 6, 5523–5532. [Google Scholar] [CrossRef]
- Nowak, C.; Schach, D.; Gebert, J.; Grosserueschkamp, M.; Gennis, R.B.; Ferguson-Miller, S.; Knoll, W.; Walz, D.; Naumann, R.L.C. Oriented immobilization and electron transfer to the cytochrome c oxidase. J. Solid State Electrochem. 2011, 15, 105–114. [Google Scholar] [CrossRef]
- Nowak, C.; Laredo, T.; Gebert, J.; Lipkowski, J.; Gennis, R.B.; Ferguson-Miller, S.; Knoll, W.; Naumann, R.L.C. 2D-SEIRA spectroscopy to highlight conformational changes of the cytochrome c oxidase induced by direct electron transfer. Metallomics 2011, 3, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Naumann, R.L.C.; Nowak, C.; Knoll, W. Proteins in biomimetic membranes: Promises and facts. Soft Matter 2011, 7, 9535–9548. [Google Scholar] [CrossRef]
- Hexter, R.M.; Albrecht, M.G. Metal surface Raman spectroscopy: Theoriy. Spectrochim. Acta A 1979, 35, 233–251. [Google Scholar] [CrossRef]
- Moskovits, M. Surface roughness and the enhanced intensity of Raman scattering by molecules adsorbed on metals. J. Chem. Phys. 1978, 69, 4159–4161. [Google Scholar] [CrossRef]
- Srajer, J.; Schwaighofer, A.; Nowak, C. Surface-enhanced Raman spectroscopy for biomedical diagnostics and imaging. Biomed. Spectrosc. Imaging 2013, 2, 51–71. [Google Scholar]
- Osawa, M. Surface-enhanced infrared absorption. Near Field Opt. Surf. Plasmon. Polaritons 2001, 81, 163–187. [Google Scholar]
- Schwaighofer, A.; Steininger, C.; Hildenbrandt, D.M.; Srajer, J.; Nowak, C.; Knoll, W.; Naumann, R.L.C. Time-resolved surface-enhanced IR-absorption spectroscopy of direct electron transfer to cytochrome c oxidase from R. sphaeroides. Biophys. J. 2013, 105, 2706–2713. [Google Scholar] [CrossRef] [PubMed]
- Schwaighofer, A.; Ferguson-Miller, S.; Naumann, R.L.C.; Knoll, W.; Nowak, C. Phase-sensitive detection in modulation excitation spectroscopy applied to potential induced electron transfer in cytochrome c oxidase. Appl. Spectrosc. 2014, 68, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Srajer, J.; Schwaighofer, A.; Hildenbrandt, D.M.; Kibrom, A.; Naumann, R.L.C. A kinetic model of proton transport in a multiredox centre protein: Cytochrome c oxidase. Prog. React. Kinet. Mech. 2013, 38, 32–47. [Google Scholar] [CrossRef]
- Schach, D.; Nowak, C.; Gennis, R.B.; Ferguson-Miller, S.; Knoll, W.; Walz, D.; Naumann, R.L.C. Modeling direct electron transfer to a multi-redox center protein: Cytochrome c oxidase. J. Electroanal. Chem. 2010, 649, 268–276. [Google Scholar] [CrossRef]
- Buhrow, L.; Ferguson-Miller, S.; Kuhn, L.A. From static structure to living protein: Computational analysis of cytochrome c oxidase main-chain flexibility. Biophys. J. 2012, 102, 2158–2266. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.A.; Qin, L.; Ferguson-Miller, S. Crystallographic and online spectral evidence for role of conformational change and conserved water in cytochrome oxidase proton pump. Proc. Natl. Acad. Sci. USA 2011, 108, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Ferguson-Miller, S.; Hiser, C.; Liu, J. Gating and regulation of the cytochrome c oxidase proton pump. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Nedelkovski, V.; Schwaighofer, A.; Wraight, C.A.; Nowak, C.; Naumann, R.L.C. Surface-enhanced infrared absorption spectroscopy (SEIRAS) of light-activated photosynthetic reaction centers from Rhodobacter sphaeroides reconstituted in a biomimetic membrane system. J. Phys. Chem. 2013, 117, 16357–16363. [Google Scholar]
- Breton, J. Steady-state FTIR spectra of the photoreduction of QA and QB in Rhodobacter sphaeroides reaction centers provide evidence against the presence of a proposed transient electron acceptor X between the two quinones. Biochemistry 2007, 46, 4459–4465. [Google Scholar] [CrossRef] [PubMed]
- Mezzetti, A.; Leibl, W. Investigation of ubiquinol formation in isolated photosynthetic reaction centers by rapid-scan Fourier transform IR spectroscopy. Eur. Biophys. J. Biophys. Lett. 2005, 34, 921–936. [Google Scholar] [CrossRef] [PubMed]
- Mezzetti, A.; Leibl, W.; Breton, J.; Nabedryk, E. Photoreduction of the quinone pool in the bacterial photosynthetic membrane: Identification of infrared marker bands for quinol formation. FEBS Lett. 2003, 537, 161–165. [Google Scholar] [CrossRef]
- Mezzetti, A.; Blanchet, L.; de Juan, A.; Leibl, W.; Ruckebusch, C. Ubiquinol formation in isolated photosynthetic reaction centres monitored by time-resolved differential FTIR in combination with 2D correlation spectroscopy and multivariate curve resolution. Anal. Bioanal. Chem. 2011, 399, 1999–2014. [Google Scholar] [CrossRef] [PubMed]
- Brudler, R.; Gerwert, K. Step-scan FTIR spectroscopy resolves the QA−QB −> QAQB− transition in Rb-sphaeroides R26 reaction centres. Photosynth. Res. 1998, 55, 261–266. [Google Scholar] [CrossRef]
- Arrondo, J.L.R.; Castresana, J.; Valpuesta, J.M.; Goni, F.M. Structure and thermal-denaturation of crystalline and noncrystalline cytochrome-oxidase as studied by infrared-spectroscopy. Biochemistry 1994, 33, 11650–11655. [Google Scholar] [CrossRef] [PubMed]
- Gebert, J.; Reiner-Rozman, C.; Steininger, C.; Nedelkovski, V.; Nowak, C.; Wraight, C.; Naumann, R.L.C. Electron Transfer to Light-Activated Photosynthetic Reaction Centers from Rhodobacter sphaeroides Reconstituted in a Biomimetic Membrane System. J. Phys. Chem. C 2015, 119, 890–895. [Google Scholar] [CrossRef]
- Lebedev, N.; Trammell, S.A.; Spano, A.; Lukashev, E.; Griva, I.; Schnur, J. Conductive wiring of immobilized photosynthetic reaction center to electrode by cytochrome c. J. Am. Chem. Soc. 2006, 128, 12044–12045. [Google Scholar] [CrossRef] [PubMed]
- Trammell, S.A.; Griva, I.; Spano, A.; Tsoi, S.; Tender, L.M.; Schnur, J.; Lebedev, N. Effects of distance and driving force on photoinduced electron transfer between photosynthetic reaction centers and gold electrodes. J. Phys. Chem. C 2007, 111, 17122–17130. [Google Scholar] [CrossRef]
- Trammell, S.A.; Wang, L.Y.; Zullo, J.M.; Shashidhar, R.; Lebedev, N. Orientated binding of photosynthetic reaction centers on gold using Ni-NTA self-assembled monolayers. Biosens. Bioelectron. 2004, 19, 1649–1655. [Google Scholar] [CrossRef] [PubMed]
- Frank, P.; Siebenhofer, B.; Hanzer, T.; Geiss, A.F.; Schadauer, F.; Reiner-Rozman, C.; Durham, B.; Loew, L.M.; Ludwig, B.; Richter, O.M.H.; et al. Proteo-lipobeads for the oriented encapsulation of membrane proteins. Soft Matter 2015, 11, 2906–2908. [Google Scholar] [CrossRef] [PubMed]
- Fluhler, E.; Burnham, V.G.; Loew, L.M. Spectra, membrane binding, and potentiometric responses of new charge shift probes. Biochemistry 1985, 24, 5749–5755. [Google Scholar] [CrossRef] [PubMed]
- Belevich, I.; Bloch, D.A.; Belevich, N.; Wikstrom, M.; Verkhovsky, M.I. Exploring the proton pump mechanism of cytochrome c oxidase in real time. Proc. Natl. Acad. Sci. USA 2007, 104, 2685–2690. [Google Scholar] [CrossRef] [PubMed]
- Schadauer, F.; Geiss, A.F.; Srajer, J.; Siebenhofer, B.; Frank, P.; Reiner-Rozman, C.; Ludwig, B.; Richter, O.M.; Nowak, C.; Naumann, R.L.C. Silica nanoparticles for the oriented encapsulation of membrane proteins into artificial bilayer lipid membranes. Langmuir 2015, 31, 2511–2516. [Google Scholar] [CrossRef] [PubMed]
- Tihova, M.; Tattrie, B.; Nicholls, P. Electron-microscopy of cytochrome-c oxidase-containing proteoliposomes—Imaging analysis of protein orientation and monomer-dimer behavior. Biochem. J. 1993, 292, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, B.; Schatz, G. A two-subunit cytochrome c oxidase (cytochrome aa3) from Paracoccus dentrificans. Proc. Natl. Acad. Sci. USA 1980, 77, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Witt, H.; Zickermann, V.; Ludwig, B. Site-directed mutagenesis of cytochrome c oxidase reveals 2 acidic residues involved in the binding of cytochrome c. Biochim. Biophys. Acta Bioenerg. 1995, 1230, 74–76. [Google Scholar] [CrossRef]
- Nordlund, G.; Ng, J.B.S.; Bergstrom, L.; Brzezinski, P.A. Membrane-reconstituted multisubunit functional proton pump on mesoporous silica particles. ACS Nano 2009, 3, 2639–2646. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
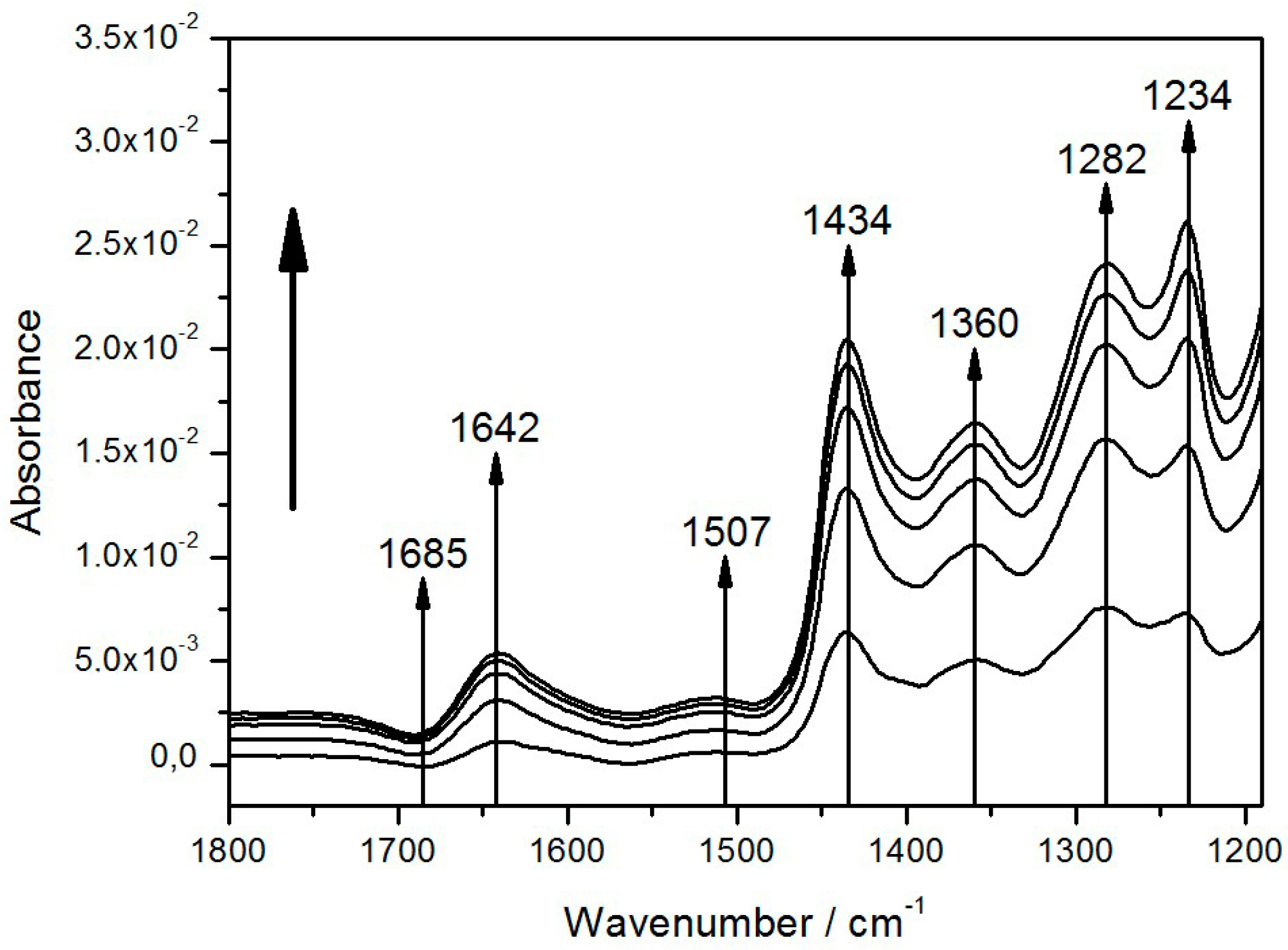
| Band (cm−1) | Tentative Assignment | ||
|---|---|---|---|
| Experimental | Literature | Species | Component |
| 1282 | 1282 | P+ | (complex) |
| 1360 | 1355, 1365 | QA | –CH3 |
| 1434 | 1433 | QH2 | |
| 1643 | 1640, 1641, 1642 [31] | quinone Q, QB | C=O |
| 1685 | 1682, 1683 | 9-keto group of P | C=O |
| 3400 | 3485 | QB−/QB or P+ | H2O |
| 3629 | 3632 | QB−/QB or P+ | H2O |
| Parameter | CcO, Solubilized | CcO, in Proteobeads | CcO, in PLBs | |
|---|---|---|---|---|
| −Val./FCCP | +Val./FCCP | |||
| KM (µM) | 5.8 ± 0.4 | 1.6 ± 0.4 | 2.6 ± 0.4 | 2.7 ± 0.6 |
| kcat (s−1) | 264.1 ± 7.4 | 32.4 ± 0.5 | 11.9 ± 0.8 | 33.4 ± 8.8 |
| Number of preparations | 3 | 3 | 3 | |
| Number of measurements | 3 | 3 | 3 | 3 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naumann, R.L.C.; Geiss, A.F.; Steininger, C.; Knoll, W. Biomimetic Membranes for Multi-Redox Center Proteins. Int. J. Mol. Sci. 2016, 17, 330. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030330
Naumann RLC, Geiss AF, Steininger C, Knoll W. Biomimetic Membranes for Multi-Redox Center Proteins. International Journal of Molecular Sciences. 2016; 17(3):330. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030330
Chicago/Turabian StyleNaumann, Renate L. C., Andreas F. Geiss, Christoph Steininger, and Wolfgang Knoll. 2016. "Biomimetic Membranes for Multi-Redox Center Proteins" International Journal of Molecular Sciences 17, no. 3: 330. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030330