
A Comparative Study of Molecular Structure, pKa, Lipophilicity, Solubility, Absorption and Polar Surface Area of Some Antiplatelet Drugs


Abstract
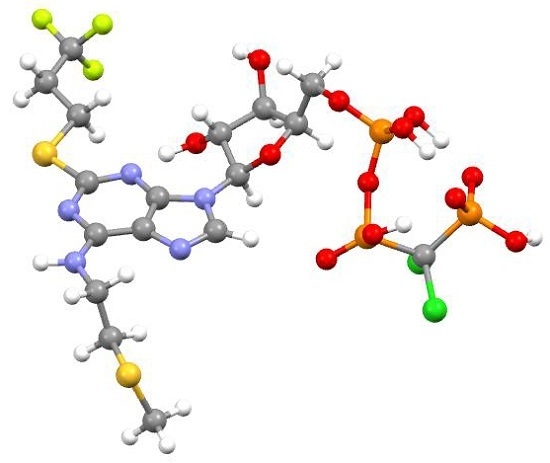
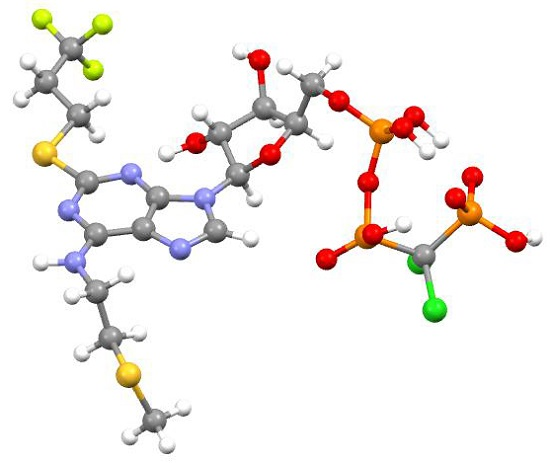
:
1. Introduction
2. Results and Discussion
2.1. Molecular Structures
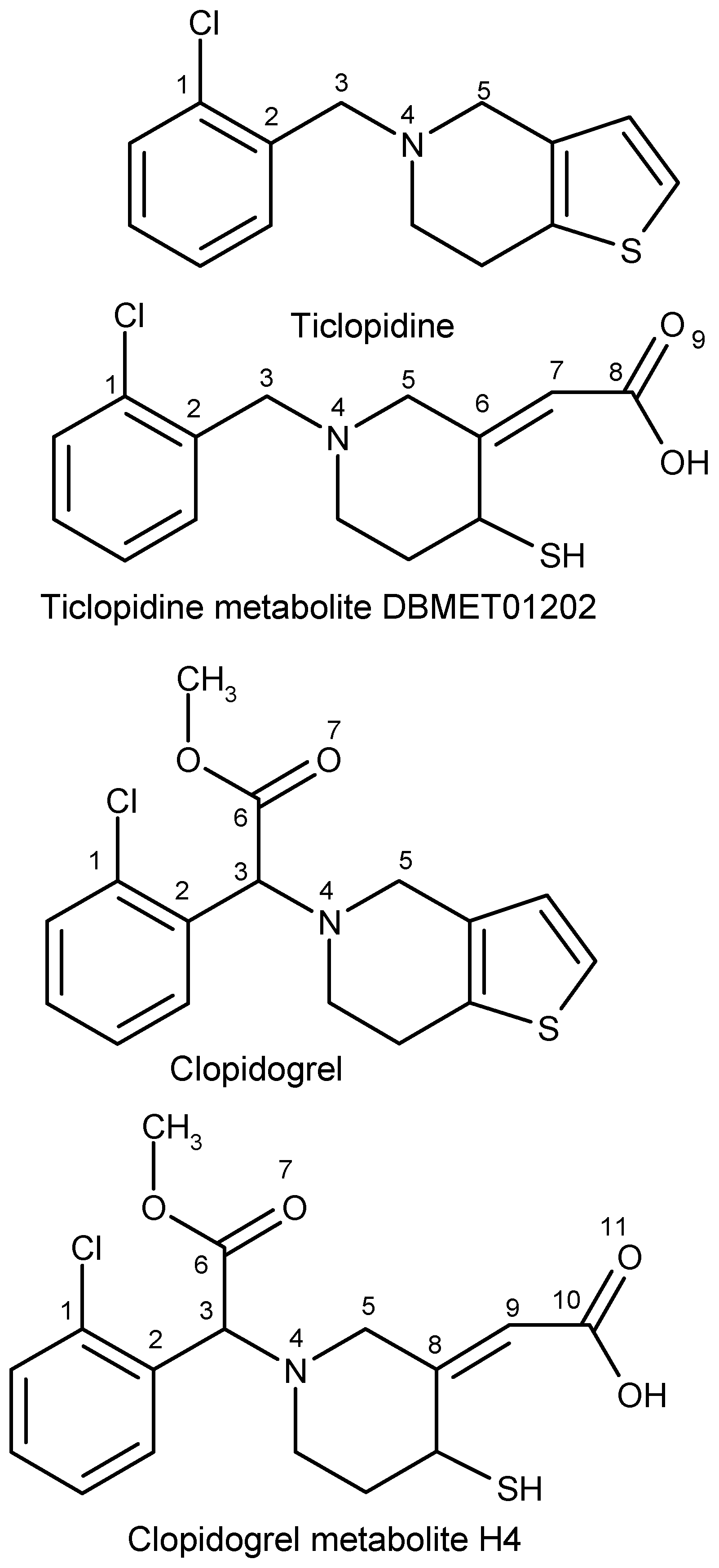
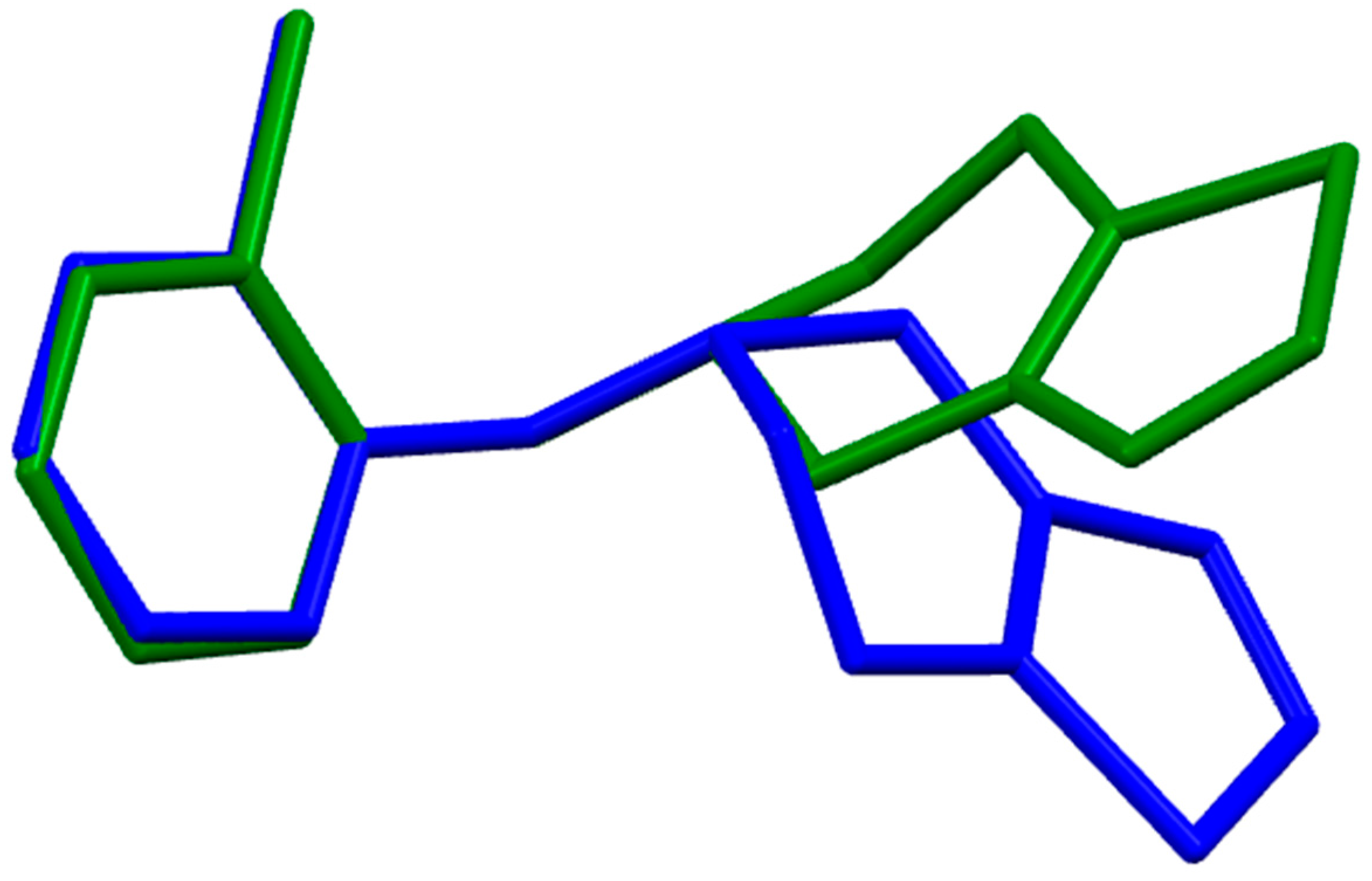
2.2. Ticlopidine
2.2.1. Clopidogrel
2.2.2. Prasugrel
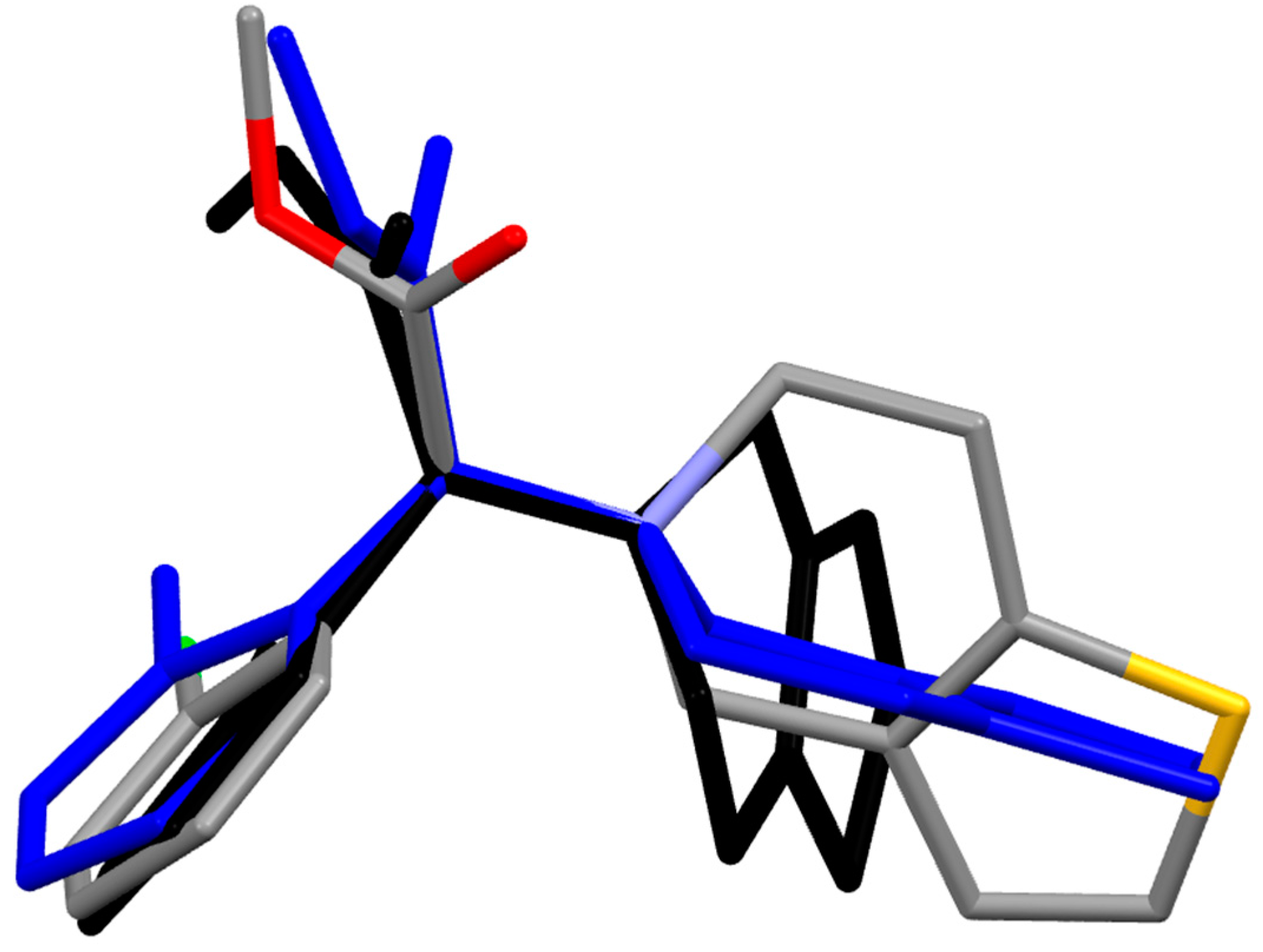
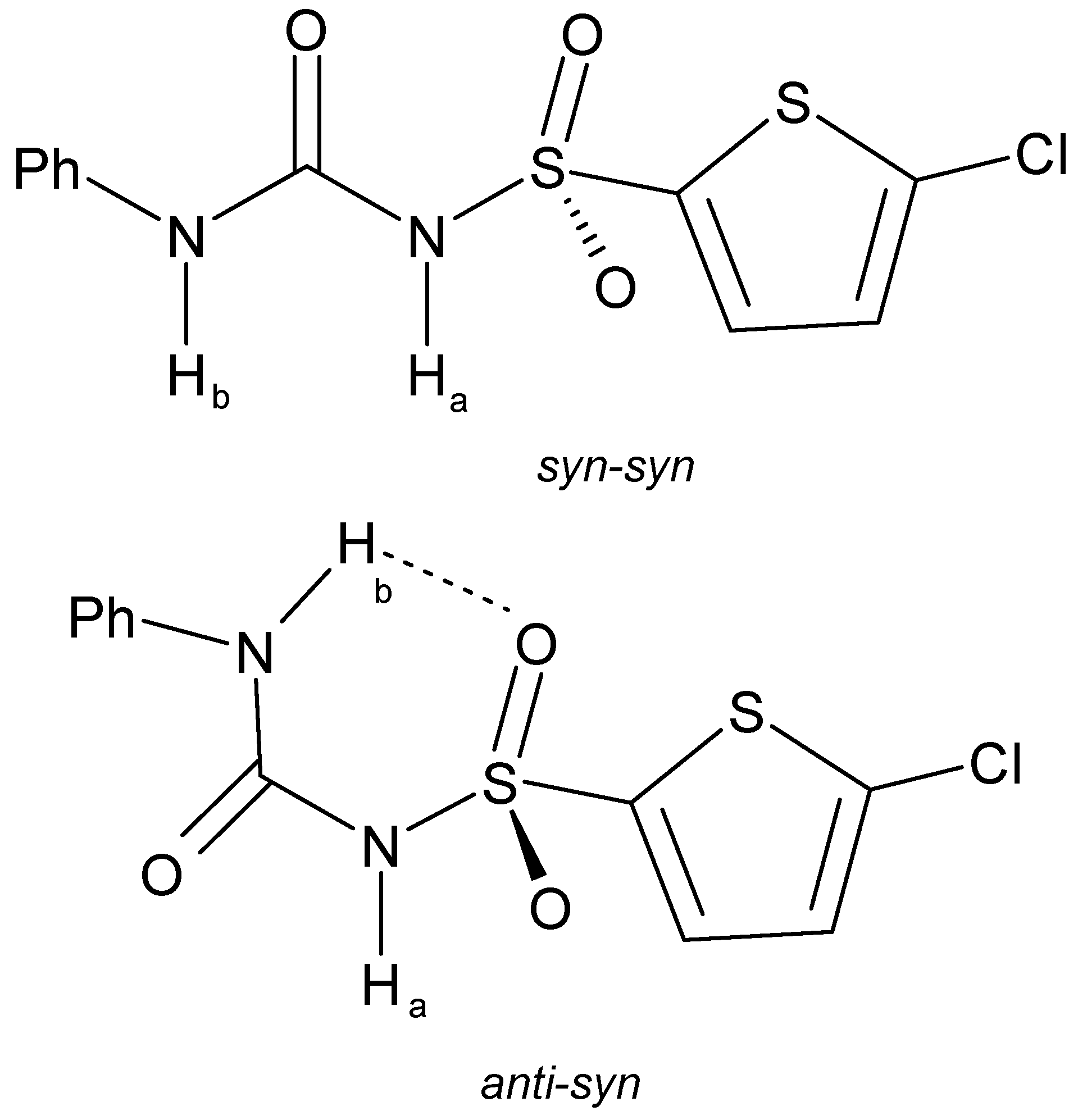
2.2.3. Elinogrel
2.2.4. Ticagrelor
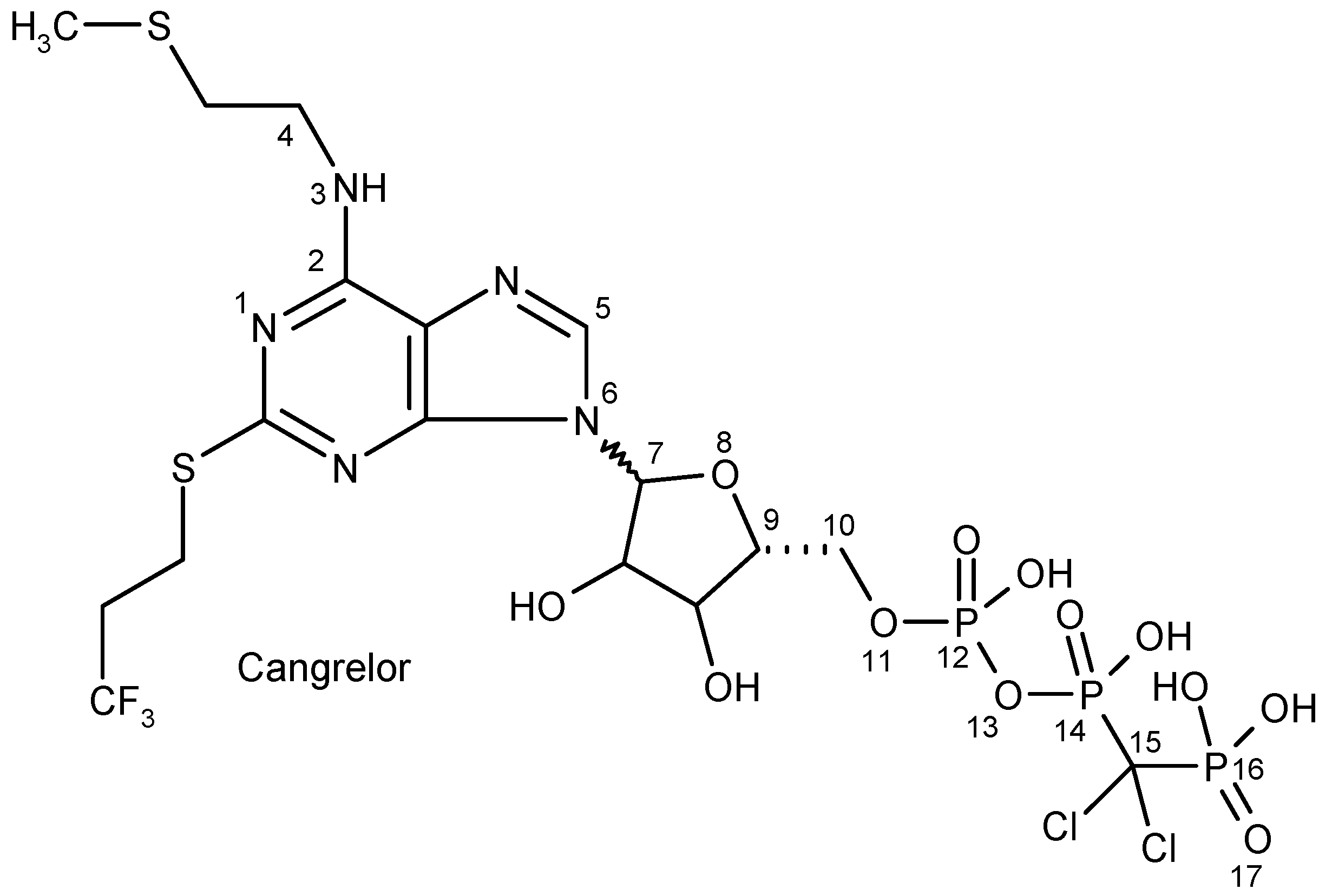
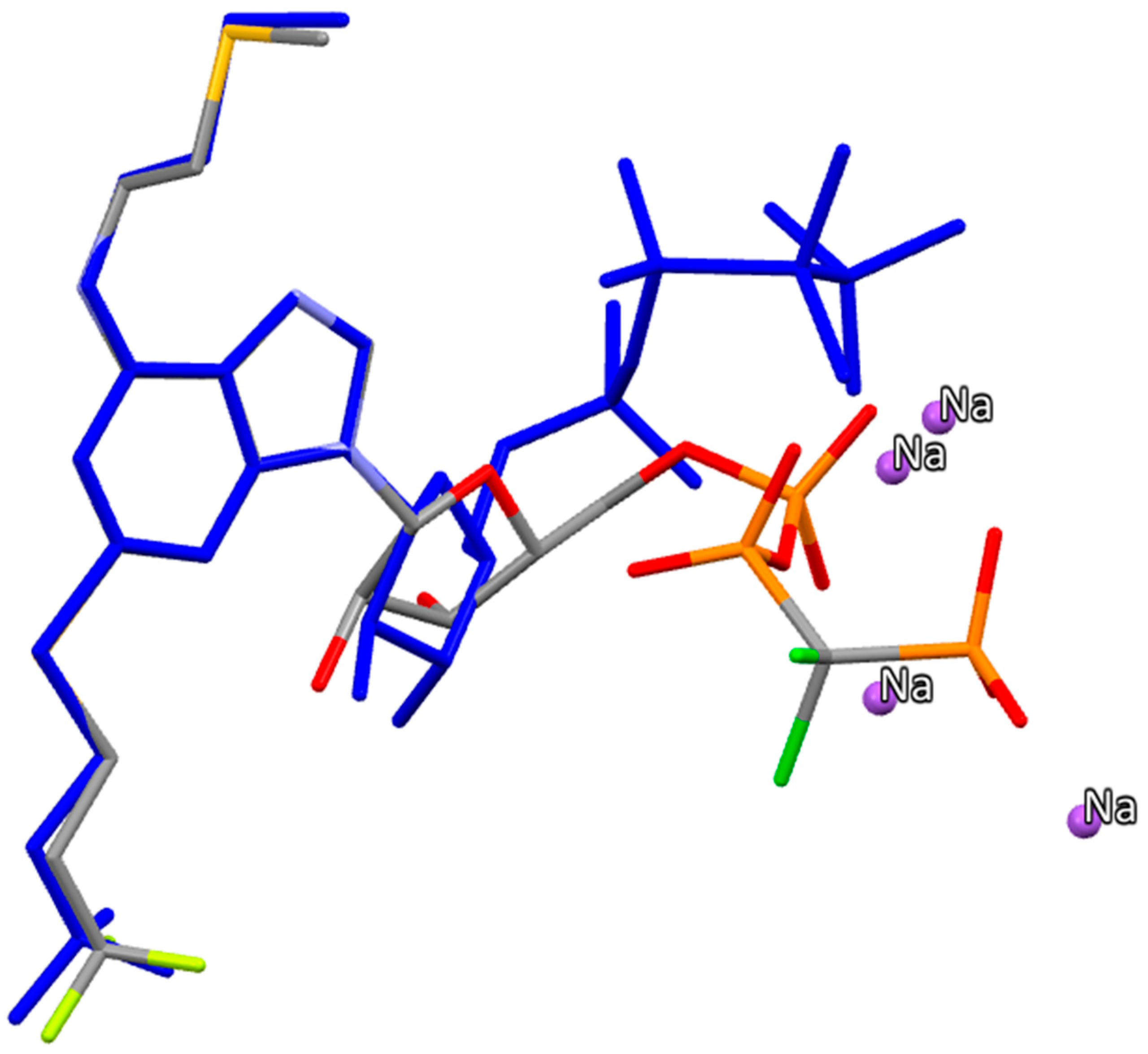
2.2.5. Cangrelor
2.3. Acidity and Basicity
2.4. Lipophilicity, Solubility, Absorption, Polar Surface Area and “Rule of Five” Properties
2.5. Selection Criteria for Antiplatelets Targeting the P2Y12 Platelet Receptor
3. Computational Methods
4. Conclusions
- (i)
- The density functional Becke3LYP method has been applied to study the molecular structure of ten species representing prodrugs and drugs acting at the P2Y12 platelet receptor. The fully-optimized most stable conformers of the thienopyridine drugs ticlopidine, clopidogrel and ticlopidine in both gas-phase and water solution are conformers with a mutual gauche orientation of phenyl and thienopyridine rings.
- (ii)
- Of the indirect irreversible thienopyridine drugs, ticlopidine is the most basic molecule. The basicity decreases after its metabolization in the liver. Therapeutically-active metabolites of ticlopidine, clopidogrel and prasugrel are organic acids with pKas in the range of 3–3.4, and at pH = 7.4, they exist in the dissociated form only. The acidic sulfonamide group of the direct antiplatelet agent elinogrel is completely dissociated at physiological pH. Both ticagrelor and its active metabolite are present at pH = 7.4 in the neutral undissociated form. Cangrelor contains three acidic phosphate groups with pKas in the range of 0.33–0.59, which are completely dissociated at blood pH.
- (iii)
- A trend in the drug lipophilicity was also observed. It is lowest for the intravenous agents cangrelor and elinogrel. The studied antiplatelet agents are only slightly soluble in water. Prodrug activation in thienopyridines results in the lowering of lipophilicity in comparison with parent prodrugs and in improving their solubility characteristics.
- (iv)
- Polar surface area, owing to the heterogeneous character of these antiplatelet drugs, shows very large intervals of values (Table 5). Thienopyridine prodrugs, like ticlopidine, clopidogrel and prasugrel, with the lowest PSA values, exhibit the largest absorption.
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rosamond, W.; Flegal, K.; Friday, G.; Furie, K.; Go, A.; Greenlund, K.; Haase, N.; Ho, M.; Howard, V.; Kissela, B.; et al.; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2007 update: A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2007, 115, e69–e171. [Google Scholar] [CrossRef] [PubMed]
- Michelson, A.D. Antiplatelet therapies for the treatment of cardiovascular disease. Nat. Rev. Drug Discov. 2010, 9, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Capodanno, D.; Ferreiro, J.L.; Angiolillo, D.J. Antiplatelet therapy: New pharmacological agents and changing paradisms. J. Thromb. Haemost 2013, 11, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Davi, G.; Patrono, C. Platelet activation and atherothrombosis. N. Engl. J. Med. 2007, 357, 2482–1494. [Google Scholar] [CrossRef] [PubMed]
- Angiolillo, D.L.; Bhatt, D.J.; Gurbel, P.A.; Jennings, L.K. Advances in antiplatelet therapy: Agents in clinical development. Am. J. Cardiol. 2009, 103, 40A–51A. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.-Z.; Kang, L.Y.; Gao, X.M.; Shang, H.C.; Zhang, J.H.; Zhang, B.L. Strategies for antiplatelet targets and agents. Thromb. Res. 2008, 123, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Birkeland, K.; Parra, D.; Rosenstein, R. Antiplatelet therapy in acute coronary syndromes: Focus on ticagrelor. J. Blood Med. 2010, 1, 197–219. [Google Scholar] [PubMed]
- Bickelhaupt, F.M.; Baerends, E.J. Kohn-sham density functional theory: Predicting and understanding chemistry. In Reviews in Computational Chemistry; Lipkowitz, K.B., Boyd, D.B., Eds.; Wiley-VCH: New York, NY, USA, 2000; Volume 15, pp. 1–86. [Google Scholar]
- Parr, R.G.; Wang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1994. [Google Scholar]
- Neumann, R.; Nobes, R.H.; Handy, N.C. Exchange functionals and potentials. Mol. Phys. 1996, 87, 1–36. [Google Scholar] [CrossRef]
- Carloni, P.; Alber, F. Quantum Medicinal Chemistry; Wiley-VCH GmbH & Co. KGaA: Weinheim, Germany, 2003. [Google Scholar]
- Mucs, D.; Bryce, R.A. The application of quantum mechanics in structure-based drug design. Expert Opin. Drug Discov. 2013, 8, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Gay, S.C.; Roberts, A.G.; Maekawa, K.; Talakad, J.C.; Hong, W.X.; Zhang, Q.; Stout, C.D.; Halpert, J.R. Structures of cytochrome P450 2B4 complexed with the antiplatelet drugs ticlopidine and clopidogrel. Biochemistry 2010, 49, 8709–8720. [Google Scholar] [CrossRef] [PubMed]
- Enjalbert, R.; Galy, J.; Géhénot, A.; Rao, C.; Maire, G. Structure of ticlopidine hydrochloride—A platelet antiaggregating agent. Acta Cryst. 1992, C48, 1043–1045. [Google Scholar] [CrossRef]
- Shah, M.B.; Jang, H.-H.; Zhang, Q.; Stout, C.D.; Halpert, J.R. X-ray crystal structure of the cytochrome P450 2B4 active site mutant F297A in complex with clopidogrel: Insights into compensatory rearrangements of the binding pocket. Arch. Biochem. Biophys. 2013, 530, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Chernyshev, V.V.; Pirogov, S.V.; Shishkina, I.N.; Velikodny, Y.A. Monoclinic form I of clopidogrel hydrogen sulfate from powder diffraction data. Acta Cryst. 2010, E66, o2101–o2102. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-M.; Zhao, J.; Xu, G. Prasugrel, a new medicine for preventing blockages in the arteries. Acta Cryst. 2010, E66, o1354. [Google Scholar] [CrossRef] [PubMed]
- Maffrand, J.-P. The story of clopidogrel and its predecessor, ticlopidine: Could these major antiplatelet and antithrombotic drugs be discovered and developed today? C. R. Chim. 2012, 15, 737–743. [Google Scholar] [CrossRef]
- Yoneda, K.; Iwamura, R.; Kishi, H.; Mizukami, Y.; Mogami, K.; Kobayashi, S. Identification of the active metabolite of ticlopidine from rat in vitro metabolites. Br. J. Pharmacol. 2004, 142, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Higgs, M.A.; Sass, R.L. The crystal structure of acrylic acid. Acta Cryst. 1963, 16, 657–661. [Google Scholar] [CrossRef]
- Elsinghorst, P. W. Quantitative determination of clopidogrel and its metabolites in biological samples: A mini-review. J. Chomatogr. B 2013, 917-918, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Masenenia, S.; Donzellia, M.; Taegtmeyera, A.B.; Brechta, K.; Krähenbühla, S. Toxicity of clopidogrel and ticlopidine on human myeloid progenitor cells: Importance of metabolites. Toxicology 2012, 299, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Pereillo, J.M.; Maftouh, M.; Andrieu, A.; Uzabiaga, M.F.; Fedeli, O.; Savi, P.; Pascal, M.; Herbert, J.M.; Maffrand, J.P.; Picard, C. Structure and stereochemistry of the active metabolite of clopidogrel. Drug Metab. Dispos. 2002, 30, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, A.; Castro, B.; Germain, J.S. Polymorphic form of clopidogrel hydrogen sulphate. US Patent No. 6504030 B1, 7 January 2003. [Google Scholar]
- Shan, J.; Sun, H. The discovery and development of prasugrel. Expert. Opin. Drug Discov. 2013, 8, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Angiolillo, D.J.; Suryadevara, S.; Capranzano, P.; Bass, T.A. Prasugrel: A novel platelet ADP P2Y12 receptor antagonist. A review on its mechanism of action and clinical development. Expert Opin. Pharmacother. 2008, 9, 2893–2900. [Google Scholar] [CrossRef] [PubMed]
- Farid, N.A.; Smith, R.L.; Gillespie, T.A.; Rash, T.J.; Blair, P.E.; Kurihara, A.; Goldberg, M.J. The disposition of prasugrel, a novel thienopyridine, in humans. Drug Metab. Dispos. 2007, 35, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.T.; Jones, K.O.; Ponsler, G.D.; Lowery, S.M.; Perkins, E.J.; Wrighton, S.A.; Ruterbories, K.J.; Kazui, M.; Farid, N.A. The biotransformation of prasugrel, a new thienopyridine prodrug, by the human carboxylesterases 1 and 2. Drug Metab. Dispos. 2008, 36, 1227–1232. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Nagao, Y.; Honjo, T.; Iimori, H.; Goto, S.; Sano, S.; Shiro, M.; Yamaguchi, K.; Sei, Y. Intramolecular nonbonded S···O interaction in acetazolamide and thiadiazolinethione molecules in their dimeric crystalline structures and complex crystalline structures with enzymes. Tetrahedron Lett. 2004, 45, 8757–8761. [Google Scholar] [CrossRef]
- Remko, M. Molecular structure, pKa, lipophilicity, solubility and absorption of biologically active aromatic and heterocyclic sulfonamides. J. Mol. Struct. 2010, 944, 34–42. [Google Scholar] [CrossRef]
- Zhou, F.; Liu, R.; Li, P.; Zhang, H. On the properties of S···O and S···π noncovalent interactions: The analysis of geometry, interaction energy and electron density. New J. Chem. 2015, 39, 1611–1618. [Google Scholar] [CrossRef]
- Wickremsinhe, E.R.; Tian, Y.; Ruterbories, K.L.; Verburg, E.M.; Weerakkody, G.J.; Kurihara, A.; Farid, N.A. Stereoselective metabolism of prasugrel in humans using a novel chiral liquid chromatography-tandem mass spectrometry method. Drug Metab. Dispos. 2007, 35, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Lu, H. Stereoselectivity in drug metabolism. Expert Opin. Drug Metab. Toxicol. 2007, 3, 149–158. [Google Scholar] [PubMed]
- Gurbel, P.A.; Kereiakes, D.; Tantry, U.S. Elinogrel potassium. Drugs Future 2010, 35, 885–892. [Google Scholar]
- Brett, W.A.; Rademacher, P. Structure of N-methyl-N′-phenylurea. Acta Cryst. 1990, C46, 880–882. [Google Scholar] [CrossRef]
- Remko, M. Theoretical study of molecular structure, pKa, lipophilicity, solubility, absorption, and polar surface area of some hypoglycemic agents. J. Mol. Struct. Theochem 2009, 897, 73–82. [Google Scholar] [CrossRef]
- Kasetti, Y.; Patel, N.K.; Sundriyal, S.; Bharatam, P.V. Conformational polymorphism in sulfonylurea drugs: Electronic structure analysis. J. Phys. Chem. B 2010, 114, 11603–11611. [Google Scholar] [CrossRef] [PubMed]
- Gelbrich, T.; Haddow, M.F.; Griesser, U.J. Gliquidone. Acta Cryst. 2011, E67, o1343. [Google Scholar] [CrossRef] [PubMed]
- Kashino, S.; Haisa, M. The crystal and molecular structure of phenylurea. Acta Cryst. 1977, B33, 855–860. [Google Scholar] [CrossRef]
- Springthorpe, B.; Bailey, A.; Barton, P.; Birkinshaw, T.N.; Bonnert, R.V.; Brown, R.C.; Willis, P.A. From ATP to AZD6140: The discovery of an orally active reversible P2Y12 receptor antagonist for the prevention of thrombosis. Bioorg. Med. Chem. Letts. 2007, 17, 6013–6018. [Google Scholar] [CrossRef] [PubMed]
- Teng, R.; Oliver, S.; Hayes, M.A.; Butler, K. Absorption, distribution, metabolism, and excretion of ticagrelor in healthy subjects. Drug Metabol. Dispos. 2010, 38, 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Ingall, A.H.; Dixon, J.; Bailey, A.; Coombs, M.E.; Cox, D.; McInally, J.I.; Tomlinson, W. Antagonists of the platelet P2T receptor: A novel approach to antithrombotic therapy. J. Med. Chem. 1999, 42, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Shikata, K.; Ueki, T.; Mitsu, T. The crystal and molecular structure of adenosine hydrochloride. Acta Crystallogr. Sect. B 1973, 29, 31–38. [Google Scholar] [CrossRef]
- Remko, M.; Remková, A.; Broer, R. Theoretical study of molecular structure and physicochemical properties of novel factor Xa inhibitors and dual factor Xa and factor IIa inhibitors. Molecules 2016, 21, 185. [Google Scholar] [CrossRef] [PubMed]
- Remko, M.; Broer, R.; Remková, A. A comparative study of the molecular structure, lipophilicity, solubility, acidity, absorption and polar surface area of coumarinic anticoagulants and direct thrombin inhibitors. RSC Adv. 2014, 4, 8072–8084. [Google Scholar] [CrossRef]
- Xing, L.; Glen, R.C. Novel methods for the prediction of logP, pKa, and logD. J. Chem. Inf. Comput. Sci. 2002, 42, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Farid, N.A.; Kurihara, A.; Wrighton, S.A. Review: Metabolism and disposition of the thienopyridine antiplatelet drugs ticlopidine, clopidogrel, and prasugrel in humans. J. Clin. Pharmacol. 2010, 50, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.H.; Abraham, M.H.; Lee, J.; Hersey, A.; Luscombe, C.N.; Beck, G.; Sherborne, B. Cooper, Rate-limited steps of human oral absorption and QSAR studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J. Gaussian 09; Version 9.0; Gaussian Inc.: Wallingford, CT, USA, 2011. [Google Scholar]
- Becke, D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. A 1965, 140, 1133–1138. [Google Scholar] [CrossRef]
- Hehre, W.J.; Radom, L.; Schleyer, P.V.R.; Pople, J.A. Ab Initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Klamt, A.; Schűűman, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 1993, 799–805. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalman, I.G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comp. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Program SPARC. Available online: http://archemcalc.com/sparc/ (accessed on 03 November 2015).
- Hilal, S.; Karickhoff, S.W.; Carreira, L.A. A Rigorous test for SPARC’s chemical reactivity models: Estimation of more than 4300 ionization pKas. Quant. Struct. Act. Relatsh. 1995, 14, 348–355. [Google Scholar] [CrossRef]
- Carreira, L.A.; Hilal, S.; Karickhoff, S.W. Estimation of chemical reactivity parameters and physical properties of organic molecules using SPARC. In Theoretical and Computational Chemistry, Quantitative Treatment of Solute/Solvent Interactions; Politzer, P., Murray, J.S., Eds.; Elsevier Publishers: Amsterdam, The Netherlands, 1994. [Google Scholar]
- Hilal, S.; Karickhoff, S.W.; Carreira, L.A. Prediction of the vapor pressure boiling point, heat of vaporization and diffusion coefficient of organic compounds. QSAR Comb. Sci. 2003, 22, 565–574. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dihedral Angle a | X-Ray from PDB | X-Ray, Solid State Structure of Drug | B3LYP | B3LYP-CPCM | |
|---|---|---|---|---|---|
| Ticlopidine | pdb.3KW4 | ||||
| α[C(1)–C(2)–C(3)–N(4)] | 79.2 b | −98.07 c | 74.13 | 75.37 | |
| β[C(2)–C(3)–N(4)–C(5)] | 179.5 b | 66.32 c | 69.62 | 69.01 | |
| Ticlopidine metabolite | |||||
| α[C(1)–C(2)–C(3)–N(4)] | 178.43 | ||||
| β[C(2)–C(3)–N(4)–C(5)] | −71.86 | ||||
| γ[C(5)–C(6)–C(7)–C(8)] | 175.34 | ||||
| δ[C(6)–C(7)–C(8)–O(9)] | −8.10 | ||||
| Clopidogrel | Pdb.4H1N | Pdb.3ME6 | |||
| α[C(1)–C(2)–C(3)–N(4)] | −122.0 d | −104.6 e | −117.4 f | −127.92 | −128.36 |
| β[C(2)–C(3)–N(4)–C(5)] | 87.5 d | 159.3 e | 60.7 f | 59.80 | 57.57 |
| γ[C(1)–C(2)–C(3)–C(6)] | 95.9 d | 115.4 e | 122.7 f | 110.59 | 109.94 |
| δ[C(2)–C(3)–C(6)–O(7)] | −134.9 d | 77.3 e | 107.8 f | 87.29 | 87.82 |
| Clopidogrel metabolite | |||||
| α[C(1)–C(2)–C(3)–N(4)] | −134.75 | ||||
| β[C(2)–C(3)–N(4)–C(5)] | 63.03 | ||||
| γ[C(1)–C(2)–C(3)–C(6)] | 101.24 | ||||
| δ[C(2)–C(3)–C(6)–O(7)] | −103.28 | ||||
| ε[C(5)–C(8)–C(9)–C(10)] | 175.05 | ||||
| ζC(8)–C(9)–C(10)]–O(11)] | −8.37 | ||||
| Prasugrel | |||||
| α[C(1)–C(2)–C(3)–N(4)] | −128.70 g | −129.99 | −124.68 | ||
| β[C(2)–C(3)–N(4)–C(5)] | 59.54 g | 58.59 | 54.90 | ||
| γ[C(1)–C(2)–C(3)–C(6)] | 109.27 g | 106.99 | 112.12 | ||
| δ[C(2)–C(3)–C(6)–O(7)] | 104.03 g | 98.87 | 98.59 | ||
| ε[C(8)–C(9)–O(10)–C(11)] | 165.16 g | 179.96 | 179.50 | ||
| ζ[C(9)–O(10)–C(11)–O(12)] | 2.96 g | −0.35 | 0.19 | ||
| R(C=O…S) | 2.7242 g | 2.7835 | 2.7870 | ||
| Prasugrel metabolite | |||||
| α[C(1)–C(2)–C(3)–N(4)] | −129.58 | −125.23 | |||
| β[C(2)–C(3)–N(4)–C(5)] | 59.65 | 56.59 | |||
| γ[C(1)–C(2)–C(3)–C(6)] | 107.48 | 111.74 | |||
| δ[C(2)–C(3)–C(6)–O(7)] | 99.23 | 98.79 | |||
| ε[C(5)–C(8)–C(9)–C(10)] | 175.23 | 176.20 | |||
| ζC(8)–C(9)–C(10)]–O(11)] | −8.25 | −7.71 | |||
| Elinogrel | |||||
| α[C(1)–N(2)–C(3)–C(4)] | 87.30 | 89.53 | |||
| β[C(5)–C(6)–N(7)–C(8)] | −1.14 | −2.10 | |||
| γ[C(6)–N(7)–C(8)–N(9)] | 177.18 | 178.51 | |||
| δ[N(7)–C(8)–N(9)–S(10)] | −35.94 | −29.38 | |||
| ε[C(8)–N(9)–S(10)–C(11)] | −65.63 | −71.85 | |||
| ζ[N(9)–S(10)–C(11)–S(12)] | −85.49 | −81.97 | |||
| N–H…O=S | 2.016 | 2.001 | |||
| S=O…S | 3.185 | 3.221 | |||
| Ticagrelor | |||||
| α[N(1)–C(2)–N(3)–C(4)] | 5.18 | 2.96 | |||
| β[C(2)–N(3)–C(4)–C(5)] | −151.08 | −152.62 | |||
| γ[N(6)–N(7)–C(8)–C(9)] | 124.16 | 122.87 | |||
| Ticagrelor metabolite | |||||
| α[N(1)–C(2)–N(3)–C(4)] | 6.18 | 4.51 | |||
| β[C(2)–N(3)–C(4)–C(5)] | −153.39 | −154.98 | |||
| γ[N(6)–N(7)–C(8)–C(9)] | 127.01 | 121.08 | |||
| Cangrelor | |||||
| α[N(1)–C(2)–N(3)–C(4)] | −179.47 | 176.59 | |||
| β[C(5)–N(6)–C(7)–O(8)] | −52.23 | −44.70 | |||
| γ[N(6)–C(7)–O(8)–C(9)] | −167.89 | −163.29 | |||
| δ[C(7)–O(8)–C(9)–C(10)] | 150.31 | 142.76 | |||
| ε[O(8)–C(9)–C(10)–O(11)] | 56.23 | 52.38 | |||
| ζ[C(9)–C(10)–O(11)–P(12)] | 56.13 | 59.20 | |||
| η[C(10)–O(11)–P(12)–O(13)] | −75.81 | −84.16 | |||
| ν[O(11)–P(12)–O(13)–P(14)] | −118.54 | −119.70 | |||
| θ[P(12)–O(13)–P(14)–C(15)] | −88.98 | −88.60 | |||
| μ[O(13)–P(14)–C(15)–P(16)] | 47.72 | 48.71 | |||
| ξ[P(14)–C(15)–P(16)–O(17)] | 37.35 | 36.18 | |||
| Cangrelor Tetrasodium | |||||
| α[N(1)–C(2)–N(3)–C(4)] | 175.92 | 175.10 | |||
| β[C(5)–N(6)–C(7)–O(8)] | −18.98 | −13.45 | |||
| γ[N(6)–C(7)–O(8)–C(9)] | −168.76 | −164.60 | |||
| δ[C(7)–O(8)–C(9)–C(10)] | 168.33 | 164.39 | |||
| ε[O(8)–C(9)–C(10)–O(11)] | 65.33 | 66.26 | |||
| ζ[C(9)–C(10)–O(11)–P(12)] | 92.66 | 108.14 | |||
| η[C(10)–O(11)–P(12)–O(13)] | −80.04 | −69.87 | |||
| ν[O(11)–P(12)–O(13)–P(14)] | −54.85 | −76.47 | |||
| θ[P(12)–O(13)–P(14)–C(15)] | −154.46 | −135.71 | |||
| μ[O(13)–P(14)–C(15)–P(16)] | 60.37 | 63.52 | |||
| ξ[P(14)–C(15)–P(16)–O(17)] | 160.82 | 169.00 | |||
| Drug | ΔECPCM, kJ/mol | μ, Debye (D) |
|---|---|---|
| Ticlopidine | −20.4 | 2.19 |
| Ticlopidine active metabolite | −35.2 | 3.46 |
| (S)-Clopidogrel | −29.2 | 1.75 |
| (S)-Clopidogrel active metabolite | −48.8 | 3.88 |
| (S)-Prasugrel | −39.6 | 3.94 |
| (S)-Prasugrel active metabolite | −46.2 | 2.86 |
| Elinogrel | −96.9 | 11.01 |
| Ticagrelor | −76.75 | 5.36 |
| Ticagrelor active metabolite | −73.7 | 4.32 |
| Cangrelor | −91.9 | 2.23 |
| Cangrelor tetrasodium | −1316.2 | 14.44 |
| Position 1 | Stereoisomer | ΔE, kJ/mol | ΔECPCM, kJ/mol | μ 2, Debye (D) |
|---|---|---|---|---|
| a,b | S,S | 0 | 0 | 3.55 |
| a,b | R,S | 41.8 | 32.5 | 4.56 |
| a,b | S,R | 18.9 | 17.4 | 3.83 |
| a,b | R,R | 51.8 | 40.8 | 3.19 |
| Drug | pKa | % Ionized Form | ||
|---|---|---|---|---|
| Acid Function | Basic Function | Acid Function | Basic Function | |
| Ticlopidine | 7.85 | 74 | ||
| Ticlopidine active metabolite | 3.41; 9.43 | 7.26 | 100; 1 | 42 |
| Clopidogrel | 4.61 | 0.2 | ||
| Clopidogrel active metabolite | 3.01; 9.15 | 4.43 | 100; 2 | 0.1 |
| Prasugrel | 5.50 | 1 | ||
| Prasugrel active metabolite | 3.29; 9.19 | 5.06 | 100; 1.5 | 0.5 |
| Elinogrel | 4.26; 11.37 | 100; 0 | ||
| Ticagrelor | 13.48 | 2.28 | 0 | 0 |
| Ticagrelor active metabolite | 13.27 | 2.59 | 0 | 0 |
| Cangrelor | 0.33–0.59 | 100 | ||
| Drug | LogP b, Exp | ClogP (XLOGP2) | logD (pH = 7.4) | ClogS (AClogS) | %ABS | logS Exp | Volume Å3 | PSA Å2 | NROTB | n ON Accept- ors | n OHNH Donors | Formula Weight (Da) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ticlopidine | 2.9 | 2.93 | −3.55 (74.70 mg/L) | 107.9 | 228.85 | 3.24 | 2 | 1 | 0 | 263.78 | ||
| Ticlopidine active metabolite | 2.39 | −0.87 | −3.05 (0.27 g/L) | 95.0 | 255.64 | 40.53 | 3 | 3 | 1 | 297.81 | ||
| Clopidogrel | 2.5 | 2.50 | −3.22 (0.19 g/L) | 98.8 | 273.40 | 29.54 | 4 | 3 | 0 | 321.82 | ||
| Clopidogrel active metabolite | 1.96 | −1.20 | −2.72 (0.68 g/L) | 85.9 | 300.19 | 66.84 | 4 | 5 | 1 | 355.84 | ||
| Prasugrel | 3.53 | 1.97 | −4.08 (30.80 mg/L) | 92.9 | 323.37 | 46.61 | 6 | 4 | 0 | 373.44 | ||
| Prasugrel active metabolite | 1.65 | −1.07 | −3.13 (0.26 g/L) | 89.1 | 305.63 | 57.61 | 5 | 4 | 1 | 349.43 | ||
| Elinogrel | 1.89 | −1.80 | −6.55 (0.15 mg/L) | 59.9 | 392.53 | 142.16 | 5 | 10 | 4 | 523.95 (viol.) | ||
| Ticagrelor | 1.95 | −5.21 (3.24 mg/L) | 61.2 | −4.71 (10 mg/L) | 438.27 | 138.45 | 10 | 10 | 4 | 522.56 (viol.) | ||
| Ticagrelor active metabolite | 2.36 | −5.29 (2.47 mg/L) | 64.4 | 395.68 | 129.21 | 7 | 4 | 9 (viol.) | 458.52 | |||
| Cangrelor | −0.41 | −5.86 | −3.89 (99.10 mg/L) | 20.7 | 541.61 | 255.92 | 16 | 17 (viol.) | 7 (viol.) | 776.37 (viol.) |
| Molecular Descriptor | Prodrug | Drug (Active Metabolite) |
|---|---|---|
| Molecular weight (Mw) | 260–530 | 300–450 |
| Octanol/water partition coefficient (clogP) | 1.9–2.9 | 1.7–2.4 |
| Aqueous solubility (clogS) | −6.5–(−3.2) | −5.3–(−2.7) |
| Polar surface area (PSA, Å2) | 3–140 | 40–130 |
| Percent of oral absorption (%ABS) | 60–107 | 65–95 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Remko, M.; Remková, A.; Broer, R. A Comparative Study of Molecular Structure, pKa, Lipophilicity, Solubility, Absorption and Polar Surface Area of Some Antiplatelet Drugs. Int. J. Mol. Sci. 2016, 17, 388. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030388
Remko M, Remková A, Broer R. A Comparative Study of Molecular Structure, pKa, Lipophilicity, Solubility, Absorption and Polar Surface Area of Some Antiplatelet Drugs. International Journal of Molecular Sciences. 2016; 17(3):388. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030388
Chicago/Turabian StyleRemko, Milan, Anna Remková, and Ria Broer. 2016. "A Comparative Study of Molecular Structure, pKa, Lipophilicity, Solubility, Absorption and Polar Surface Area of Some Antiplatelet Drugs" International Journal of Molecular Sciences 17, no. 3: 388. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030388