
Evolving Diversity of Hepatitis C Viruses in Yunnan Honghe, China


Abstract
:
1. Introduction
2. Results
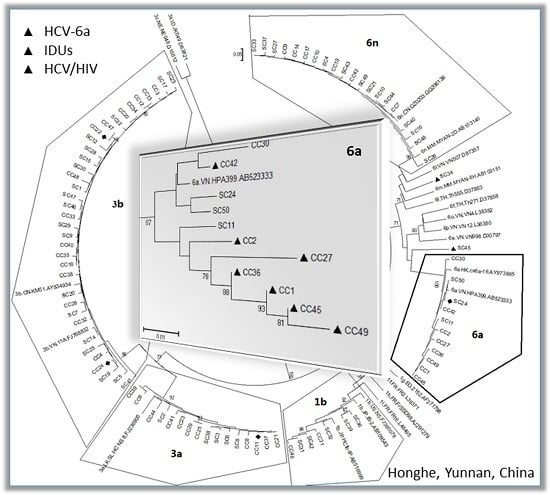
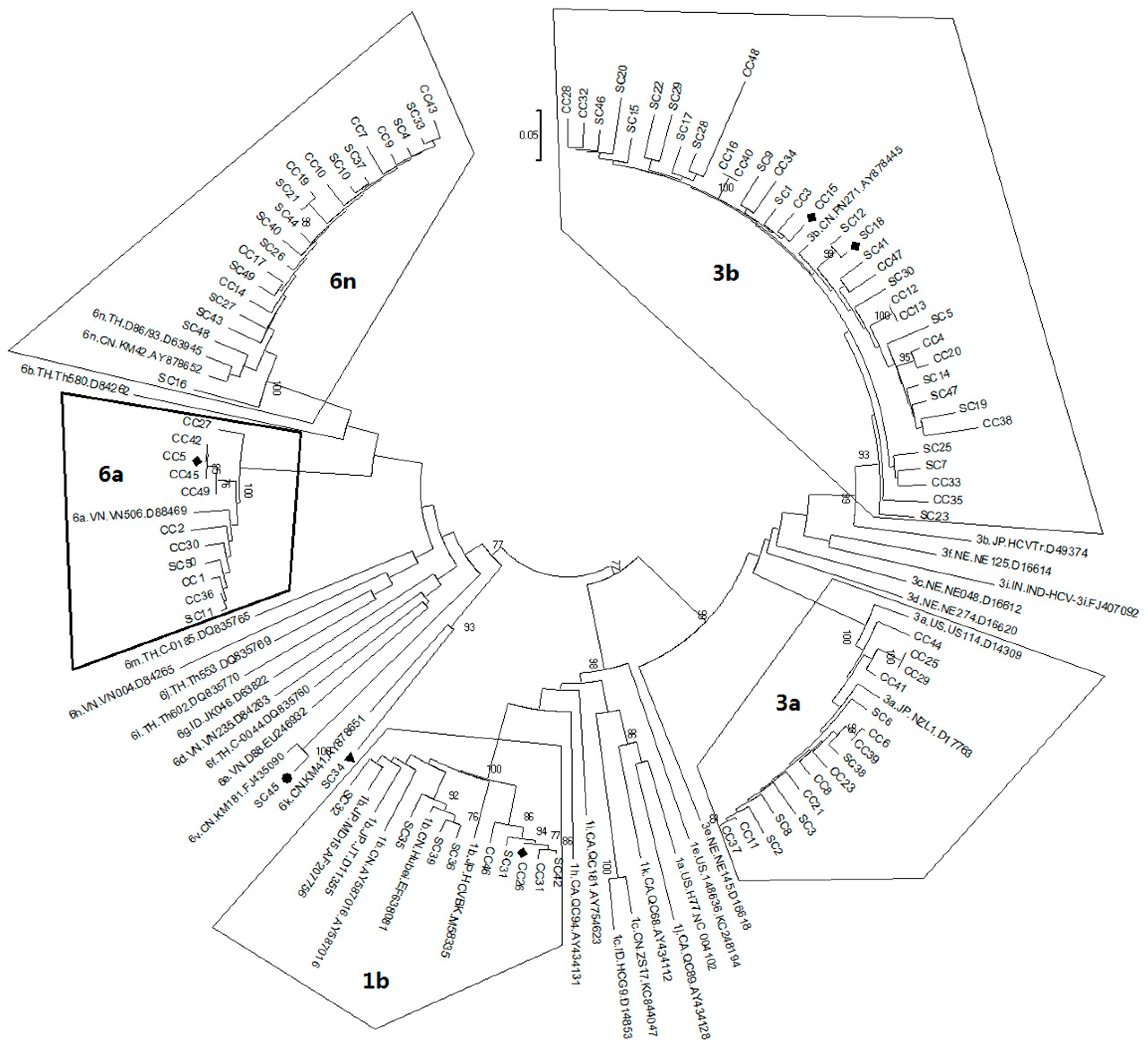
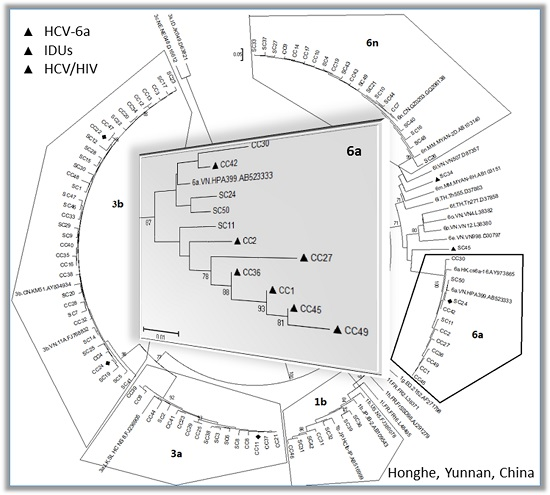
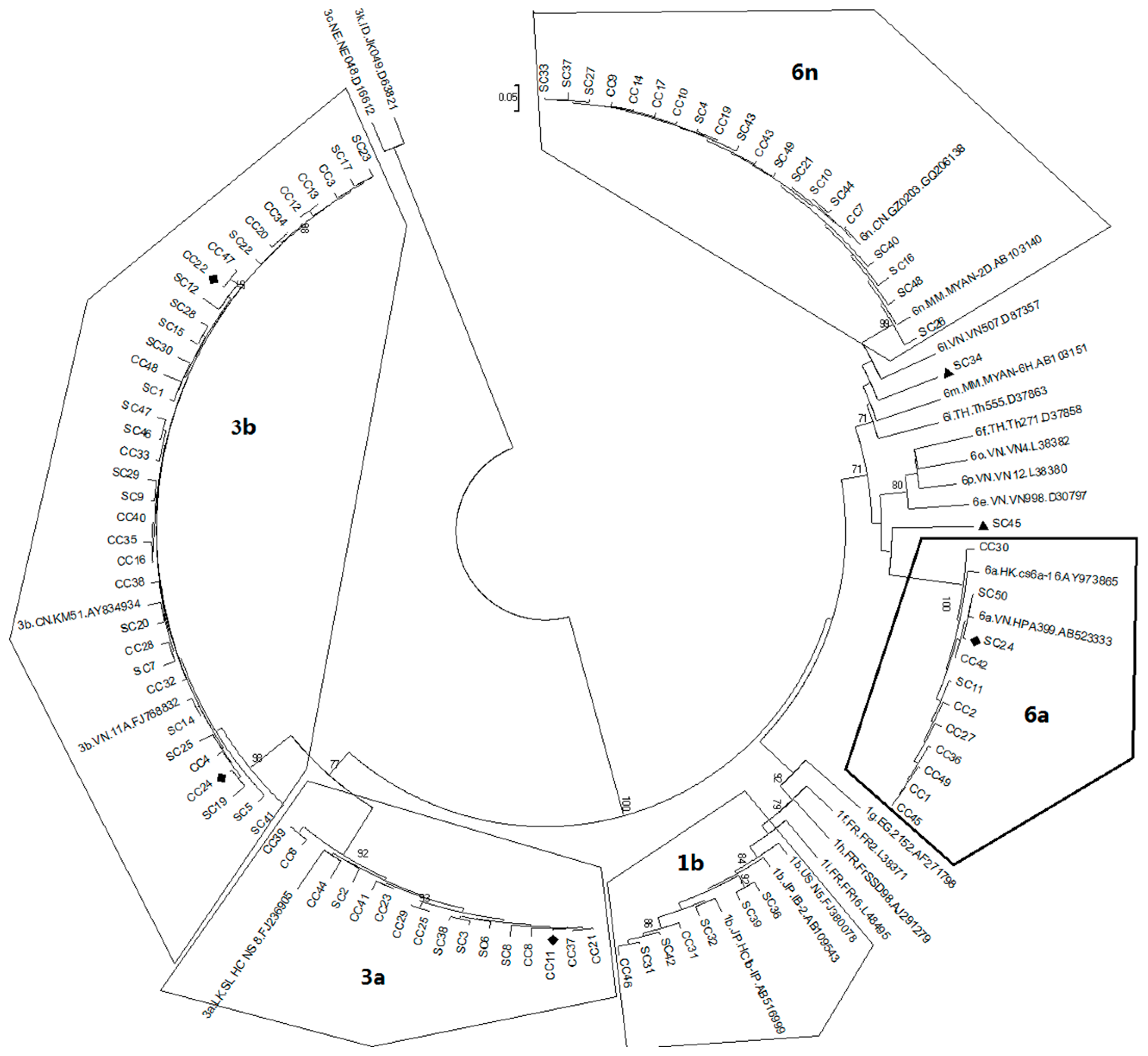
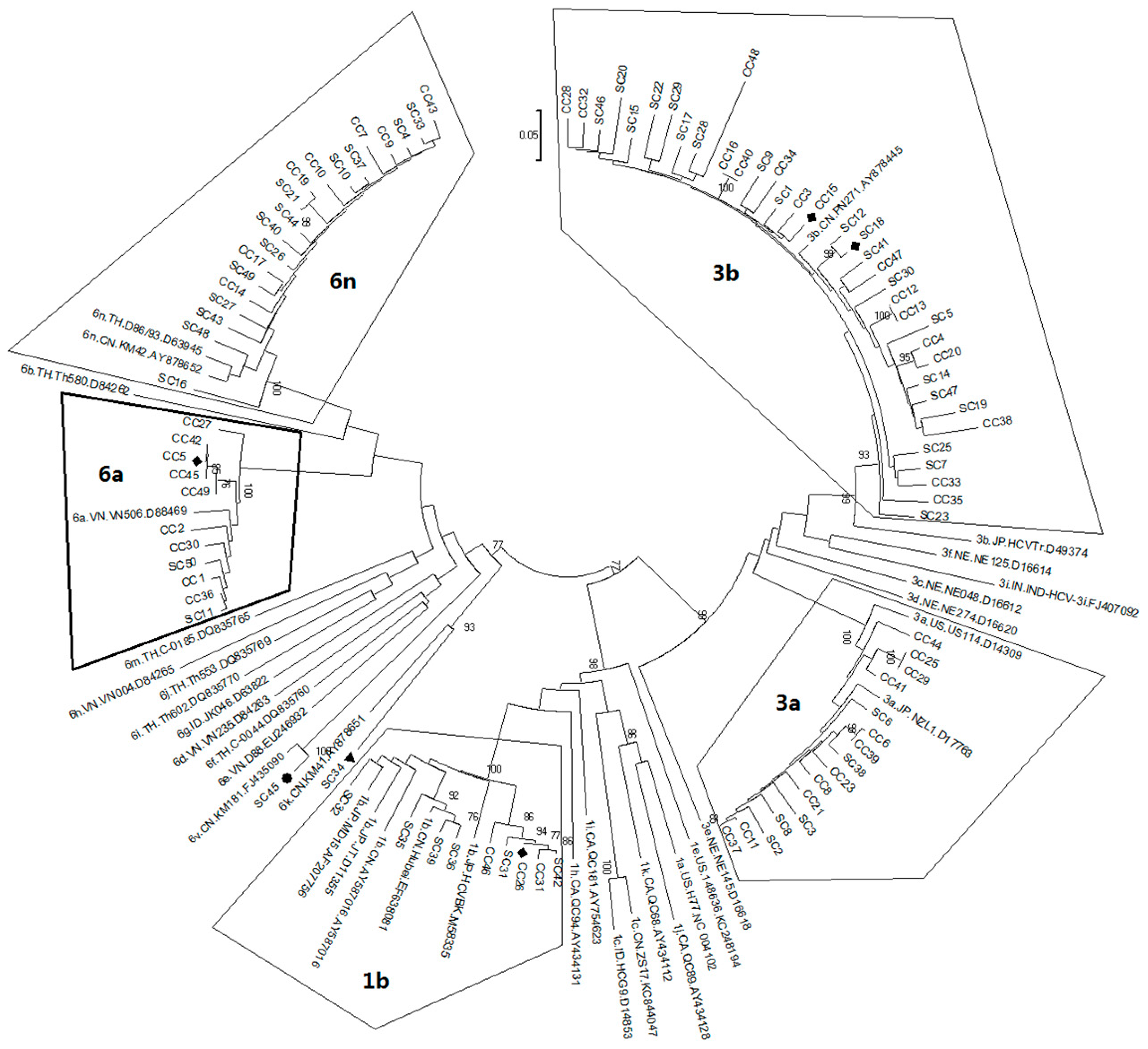
2.1. Diversity and Prevalence of Hepatitis C virus (HCV) Genotypes and Subtypes in Honghe
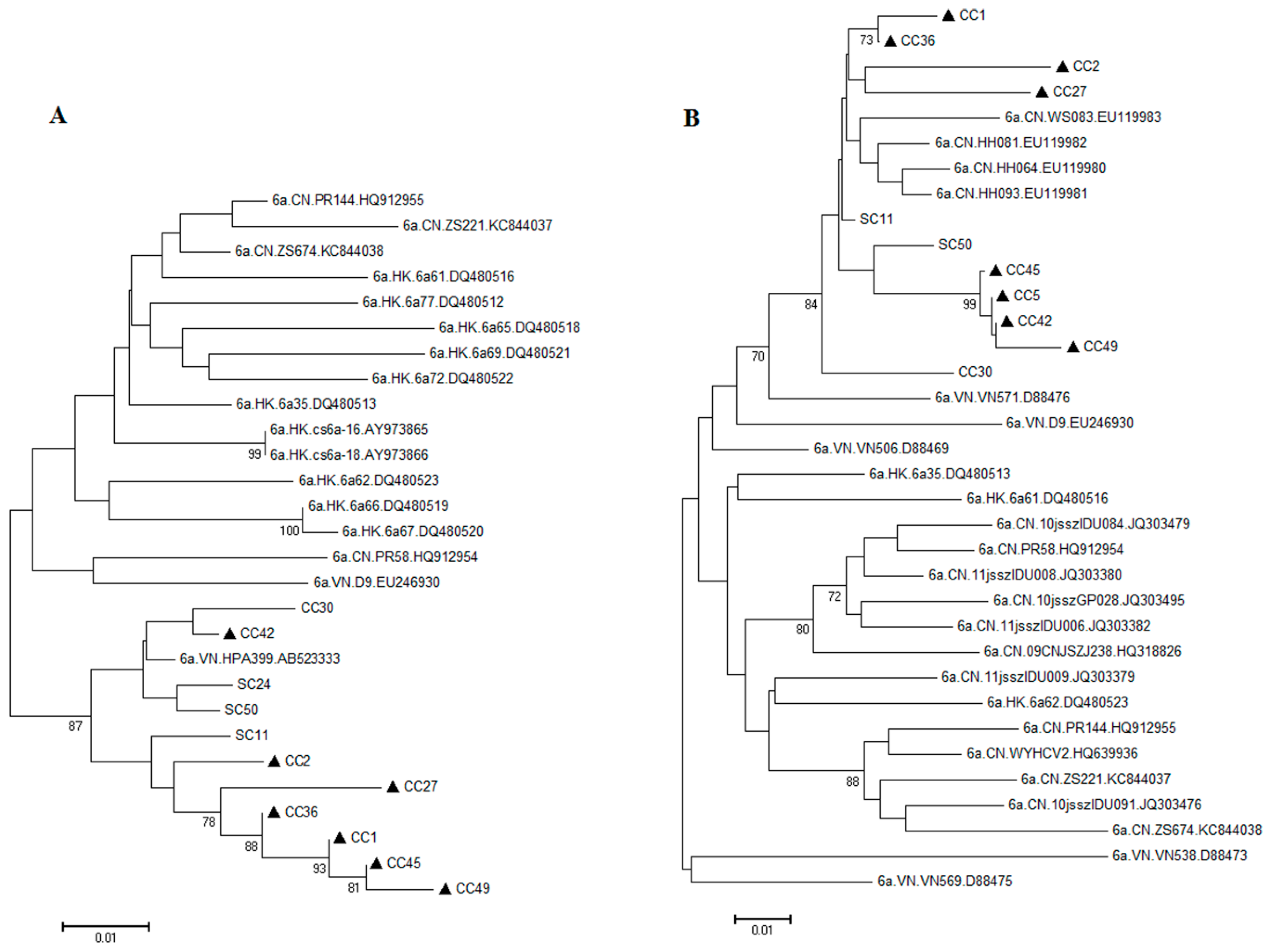
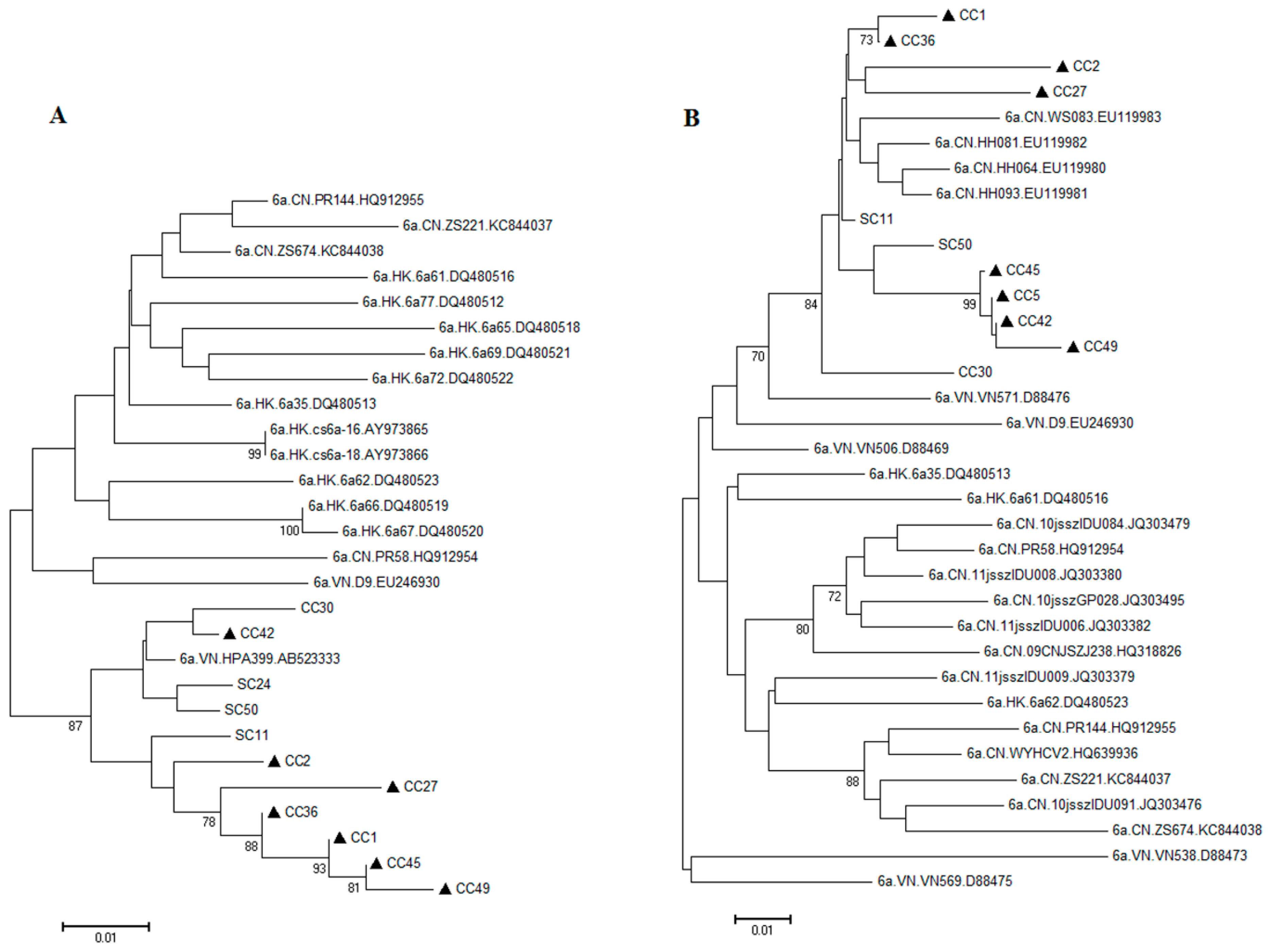
2.2. Possible Rise and Dissemination of HCV-6a in Honghe from Vietnam or Adjacent Areas
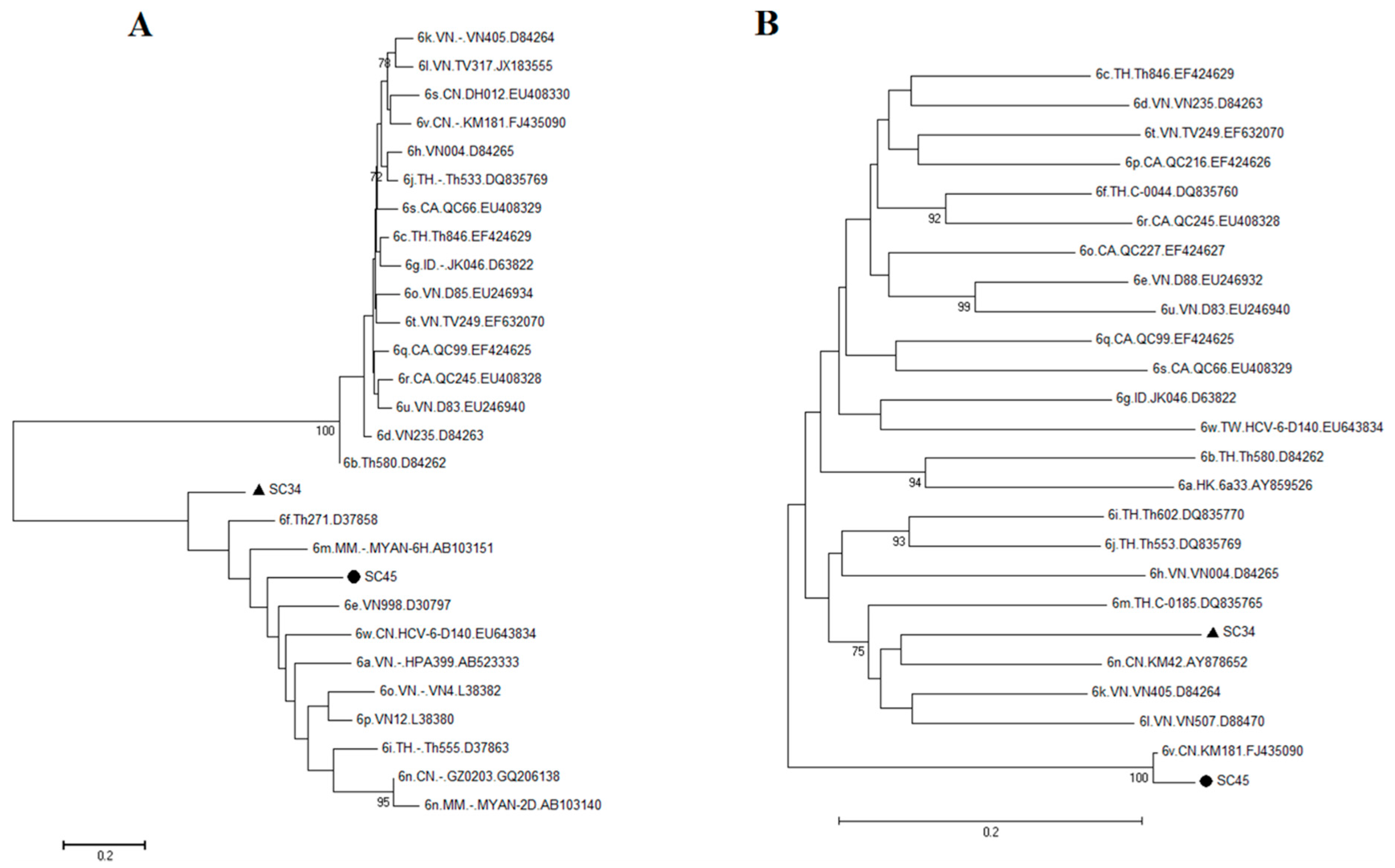
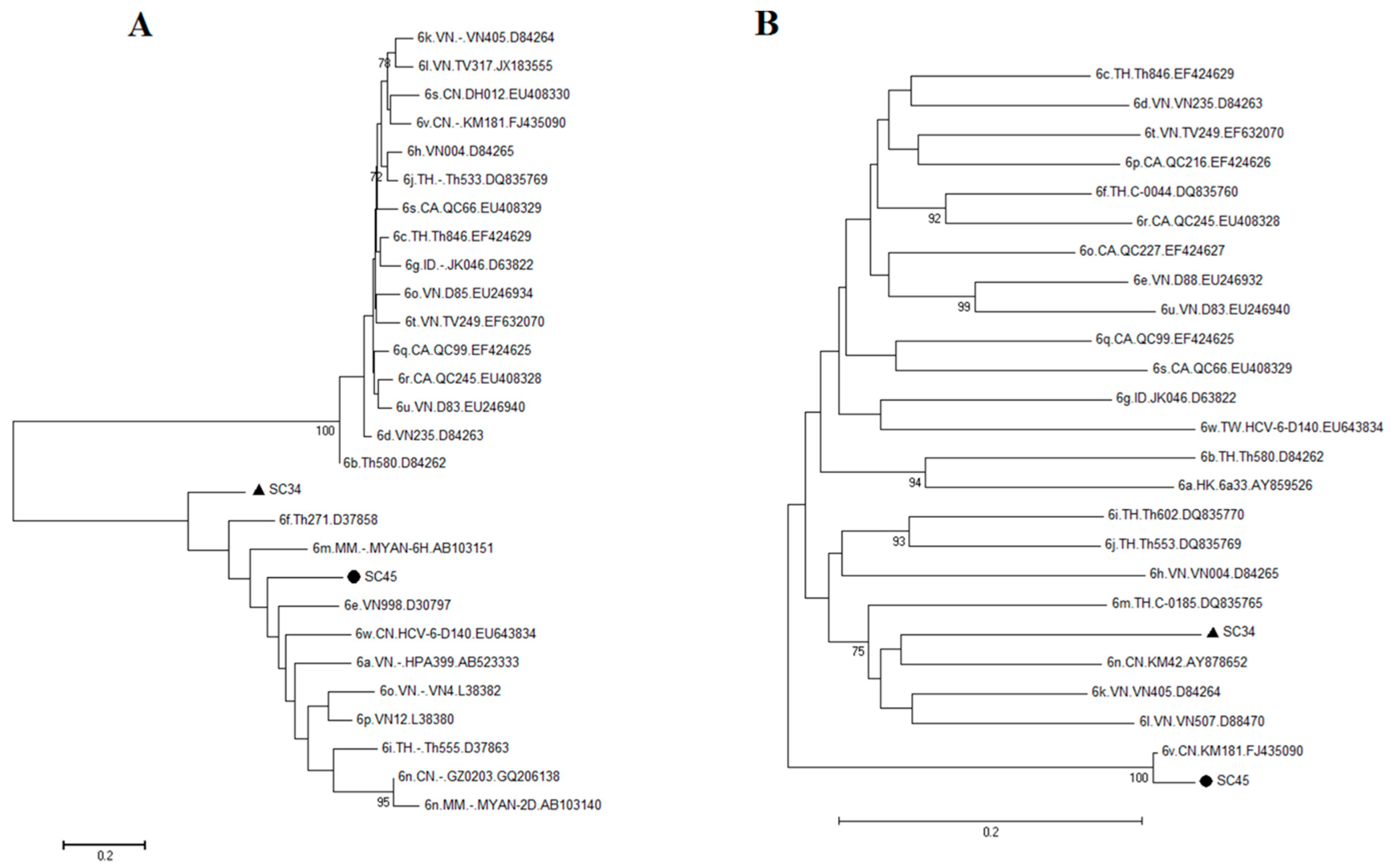
2.3. Possible New HCV Variants of HCV-6 in Honghe
3. Discussion
4. Materials and Methods
4.1. Study Cohort
4.2. Nucleic Acid Extraction and Reverse Transcription
4.3. PCR Amplification
4.4. HCV Nucleotide Sequencing and Phylogenetic Analysis
4.5. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mohd Hanafiah, K.; Groeger, J.; Flaxman, A.D.; Wiersma, S.T. Global epidemiology of hepatitis C virus infection: New estimates of age-specific antibody to HCV seroprevalence. Hepatology 2013, 57, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Messina, J.P.; Humphreys, I.; Flaxman, A.; Brown, A.; Cooke, G.S.; Pybus, O.G.; Barnes, E. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 2015, 61, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Gattoni, A.; Parlato, A.; Vangieri, B.; Bresciani, M.; Petraccaro, M. Chronic hepatitis C in the advanced adult and elderly subjects. Minerva Gastroenterol. Dietol. 2009, 55, 145–157. [Google Scholar] [PubMed]
- Kato, N. Genome of human hepatitis C virus (HCV): Gene organization, sequence diversity, and variation. Microb. Comp. Genomics 2000, 5, 129–151. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, N.; Moratorio, G.; Cristina, J.; Moreno, P. Hepatitis C virus genetic variability and evolution. World J. Hepatol. 2015, 7, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Bukh, J.; Combet, C.; Deléage, G.; Enomoto, N.; Feinstone, S.; Halfon, P.; Inchauspé, G.; Kuiken, C.; Maertens, G. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 2005, 42, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Bukh, J.; Miller, R.H.; Purcell, R.H. Biology and genetic heterogeneity of hepatitis C virus. Clin. Exp. Rheumatol. 1995, 13 (Suppl. 13), S3–S7. [Google Scholar] [PubMed]
- Murphy, D.G.; Willems, B.; Deschenes, M.; Hilzenrat, N.; Mousseau, R.; Sabbah, S. Use of sequence analysis of the NS5B region for routine genotyping of hepatitis C virus with reference to C/E1 and 5′ untranslated region sequences. J. Clin. Microbiol. 2007, 45, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.; Qin, H.; Song, J. Intrinsically unstructured domain 3 of hepatitis C Virus NS5A forms a “fuzzy complex” with VAPB-MSP domain which carries ALS-causing mutations. PLoS ONE 2012, 7, e39261. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Leveque, V.; Ma, H.; Johnson, K.A.; Klumpp, K. Assembly, purification, and pre-steady-state kinetic analysis of active RNA-dependent RNA polymerase elongation complex. J. Biol. Chem. 2012, 287, 10674–10683. [Google Scholar] [CrossRef] [PubMed]
- Strickland, G.T.; El-Kamary, S.S.; Klenerman, P.; Nicosia, A. Hepatitis C vaccine: Supply and demand. Lancet Infect. Dis. 2008, 8, 379–386. [Google Scholar] [CrossRef]
- Takada, N.; Takase, S.; Takada, A.; Date, T. Differences in the hepatitis C virus genotypes in different countries. J. Hepatol. 1993, 17, 277–283. [Google Scholar] [CrossRef]
- McOmish, F.; Yap, P.L.; Dow, B.C.; Follett, E.A.; Seed, C.; Keller, A.J.; Cobain, T.J.; Krusius, T.; Kolho, E.; Naukkarinen, R.; et al. Geographical distribution of hepatitis C virus genotypes in blood donors: An international collaborative survey. J. Clin. Microbiol. 1994, 32, 884–892. [Google Scholar] [PubMed]
- Sartori, M.; Andorno, S.; Avogadro, E.; Ballarè, M.; La Terra, G.; Leone, F.; Quaglia, V.; Fortina, G.; Aglietta, M. High prevalence of hepatitis C virus (HCV) genotype 2 in Italian patients with chronic liver disease. Ital. J. Gastroenterol. 1996, 28, 452–456. [Google Scholar] [PubMed]
- Zein, N.N.; Persing, D.H. Hepatitis C genotypes: Current trends and future implications. Mayo Clin. Proc. 1996, 71, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.D.; Liu, M.Y.; Yu, W.L.; Li, J.Q.; Peng, M.; Dai, Q.; Liu, X.; Zhou, Z.Q. Hepatitis C virus infections and genotypes in China. Hepatobiliary Pancreat. Dis. Int. 2002, 1, 194–201. [Google Scholar] [PubMed]
- Apichartpiyakul, C.; Apichartpiyakul, N.; Urwijitaroon, Y.; Gray, J.; Natpratan, C.; Katayama, Y.; Fujii, M.; Hotta, H. Seroprevalence and subtype distribution of hepatitis C virus among blood donors and intravenous drug users in northern/northeastern Thailand. Jpn. J. Infect. Dis. 1999, 52, 121–123. [Google Scholar] [PubMed]
- Narahari, S.; Juwle, A.; Basak, S.; Saranath, D. Prevalence and geographic distribution of Hepatitis C Virus genotypes in Indian patient cohort. Infect. Genet. Evol. 2009, 9, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M.; Tsakiris, L.; Roudot-Thoraval, F.; Pellet, C.; Stuyver, L.; Duval, J.; Dhumeaux, D. Relationship between hepatitis C virus genotypes and sources of infection in patients with chronic hepatitis C. J. Infect. Dis. 1995, 171, 1607–1610. [Google Scholar] [CrossRef] [PubMed]
- Abdulkarim, A.S.; Zein, N.N.; Germer, J.J.; Kolbert, C.P.; Kabbani, L.; Krajnik, K.L.; Hola, A.; Agha, M.N.; Tourogman, M.; Persing, D.H. Hepatitis C virus genotypes and hepatitis G virus in hemodialysis patients from Syria: Identification of two novel hepatitis C virus subtypes. Am. J. Trop. Med. Hyg. 1998, 59, 571–576. [Google Scholar] [PubMed]
- Chamberlain, R.W.; Adams, N.J.; Taylor, L.A.; Simmonds, P.; Elliott, R.M. The complete coding sequence of hepatitis C virus genotype 5a, the predominant genotype in South Africa. Biochem. Biophys. Res. Commun. 1997, 236, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Pybus, O.G.; Barnes, E.; Taggart, R.; Lemey, P.; Markov, P.V.; Rasachak, B.; Syhavong, B.; Phetsouvanah, R.; Sheridan, I.; Humphreys, I.S.; et al. Genetic history of hepatitis C virus in East Asia. J. Virol. 2009, 83, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yao, Y.; Wu, W.; Feng, R.; Wu, Z.; Cun, W.; Dong, S. Hepatitis C virus genotype diversity among intravenous drug users in Yunnan Province, southwestern China. PLoS ONE 2013, 8, e82598. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.L.; Chuang, W.L. Treatment of chronic hepatitis C in Asia: When East meets West. J. Gastroenterol. Hepatol. 2009, 24, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Fried, M.W.; Shiffman, M.L.; Reddy, K.R.; Smith, C.; Marinos, G.; Gonçales, F.L., Jr.; Häussinger, D.; Diago, M.; Carosi, G.; Dhumeaux, D.; et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. New Engl. J. Med. 2002, 347, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Hadziyannis, S.J.; Sette, H., Jr.; Morgan, T.R.; Balan, V.; Diago, M.; Marcellin, P.; Ramadori, G.; Bodenheimer, H., Jr.; Bernstein, D.; Rizzetto, M.; et al. Peginterferon-α2a and ribavirin combination therapy in chronic hepatitis C: A randomized study of treatment duration and ribavirin dose. Ann. Intern. Med. 2004, 140, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P. Genetic diversity and evolution of hepatitis C virus—15 years on. J. Gen. Virol. 2004, 85, 3173–3188. [Google Scholar] [CrossRef] [PubMed]
- Weck, K. Molecular methods of hepatitis C genotyping. Expert Rev. Mol. Diagn. 2005, 5, 507–520. [Google Scholar] [CrossRef] [PubMed]
- McNaughton, A.L.; Thomson, E.C.; Templeton, K.; Gunson, R.N.; Leitch, E.C. Mixed genotype hepatitis C infections and implications for treatment. Hepatology 2014, 59. [Google Scholar] [CrossRef] [PubMed]
- Pham, S.T.; Bull, R.A.; Bennett, J.M.; Rawlinson, W.D.; Dore, G.J.; Lloyd, A.R.; White, P.A. Frequent multiple hepatitis C virus infections among injection drug users in a prison setting. Hepatology 2010, 52, 1564–1572. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, T.; Nguyen, H.C.; Ishizaki, A.; Chung, P.T.; Hoang, T.T.; Nguyen, V.T.; Kageyama, S.; Oka, S.; Pham, V.T.; Ichimura, H. Multiple routes of hepatitis C virus transmission among injection drug users in Hai Phong, Northern Vietnam. J. Med. Virol. 2010, 82, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Lu, L.; Tee, K.K.; Zhao, W.; Wu, J.; Yu, J.; Li, X.; Lin, Y.; Mukhtar, M.M.; Hagedorn, C.H.; Takebe, Y. The unique HCV genotype distribution and the discovery of a novel subtype 6u among IDUs co-infected with HIV-1 in Yunnan, China. J. Med. Virol. 2008, 80, 1142–1152. [Google Scholar] [CrossRef] [PubMed]
- Dunford, L.; Carr, M.J.; Dean, J.; Waters, A.; Nguyen, L.T.; Ta Thi, T.H.; Thi, L.A.; Do, H.D.; Thi, T.T.; Nguyen, H.T.; et al. Hepatitis C virus in Vietnam: High prevalence of infection in dialysis and multi-transfused patients involving diverse and novel virus variants. PLoS ONE 2012, 7, e41266. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.P.; Jeffers, T.; Younoszai, Z.; Fazel, Y.; Younossi, Z.M. The changing landscape of hepatitis C virus therapy: Focus on interferon-free treatment. Therap. Adv. Gastroenterol. 2015, 8, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Smith, M.K.; Wang, L.; Su, Y.; Wang, L.; Guo, W.; Wang, L.; Cui, Y.; Wang, N. Hepatitis C virus infection in China: An emerging public health issue. J. Viral Hepat. 2015, 22, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.X.; Zhou, H.J.; Wang, J.H.; Xiang, X.G.; Zhuang, Y.; Guo, S.M.; Gui, H.L.; Zhao, G.D.; Tang, W.L.; Wang, H.; et al. Distribution of hepatitis C virus genotypes in Chinese patients with chronic hepatitis C: Correlation with patients’ characteristics and clinical parameters. J. Dig. Dis. 2012, 13, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.; Yang, S.; Feng, S.; Wang, Q.; Liu, S.; Xing, H.; Xie, W.; Zhu, L.; Cheng, J. Hepatitis C virus genotype and subtype distribution in Chinese chronic hepatitis C patients: Nationwide spread of HCV genotypes 3 and 6. Virol. J. 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Wang, M.; Xia, W.; Tian, L.; Xu, R.; Li, C.; Wang, J.; Rong, X.; Xiong, H.; Huang, K.; et al. Migration patterns of hepatitis C virus in China characterized for five major subtypes based on samples from 411 volunteer blood donors from 17 provinces and municipalities. J. Virol 2014, 88, 7120–7129. [Google Scholar] [CrossRef] [PubMed]
- Kuiken, C.; Mizokami, M.; Deleage, G.; Yusim, K.; Penin, F.; Shin-I, T.; Charavay, C.; Tao, N.; Crisan, D.; Grando, D.; et al. Hepatitis C databases, principles and utility to researchers. Hepatology 2006, 43, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Li, C.; Fu, Y.; Thaikruea, L.; Thongswat, S.; Maneekarn, N.; Apichartpiyakul, C.; Hotta, H.; Okamoto, H.; Netski, D.; et al. Complete genomes for hepatitis C virus subtypes 6f, 6i, 6j and 6m: Viral genetic diversity among Thai blood donors and infected spouses. J. Gen. Virol. 2007, 88, 1505–1518. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Njouom, R.; Pepin, J.; Nakano, T.; Bennett, P.; Pybus, O.G.; Lu, L. Characterization of full-length hepatitis C virus sequences for subtypes 1e, 1h and 1l, and a novel variant revealed Cameroon as an area in origin for genotype 1. J. Gen. Virol 2013, 94, 1780–1790. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) Single HCV Infection Group | ||||||||
|---|---|---|---|---|---|---|---|---|
| Sample | NS5B | C/E1 | Sample | NS5B | C/E1 | Sample | NS5B | C/E1 |
| SC1 | 3b | 3b | SC18 | nd | 3b | SC35 | 1b | 1b |
| SC2 | 3a | 3a | SC19 | 3b | 3b | SC36 | 1b | 1b |
| SC3 | 3a | 3a | SC20 | 3b | 3b | SC37 | 6n | 6n |
| SC4 | 6n | 6n | SC21 | 6n | 6n | SC38 | 3a | 3a |
| SC5 | 3b | 3b | SC22 | 3b | 3b | SC39 | 1b | 1b |
| SC6 | 3a | 3a | SC23 | 3b | 3b | SC40 | 6n | 6n |
| SC7 | 3b | 3b | SC24 | 6a | nd | SC41 | 3b | 3b |
| SC8 | 3a | 3a | SC25 | 3b | 3b | SC42 | 1b | 1b |
| SC9 | 3b | 3b | SC26 | 6n | 6n | SC43 | 6n | 6n |
| SC10 | 6n | 6n | SC27 | 6n | 6n | SC44 | 6n | 6n |
| SC11 | 6a | 6a | SC28 | 3b | 3b | SC45 | U | 6v |
| SC12 | 3b | 3b | SC29 | 3b | 3b | SC46 | 3b | 3b |
| * SC13 | 3b | 6a | SC30 | 3b | 3b | SC47 | 3b | 3b |
| SC14 | 3b | 3b | SC31 | 1b | 1b | SC48 | 6n | 6n |
| SC15 | 3b | 3b | SC32 | 1b | 1b | SC49 | 6n | 6n |
| SC16 | 6n | 6n | SC33 | 6n | 6n | SC50 | 6a | 6a |
| SC17 | 3b | 3b | SC34 | U | U | – | – | – |
| Total | 50 | |||||||
| (B) HCV and HIV Co-Infection Group | ||||||||
| Sample | NS5B | C/E1 | Sample | NS5B | C/E1 | Sample | NS5B | C/E1 |
| CC1 | 6a | 6a | * CC18 | 3b | 6a | CC35 | 3b | 3b |
| CC2 | 6a | 6a | CC19 | 6n | 6n | CC36 | 6a | 6a |
| CC3 | 3b | 3b | CC20 | 3b | 3b | CC37 | 3a | 3a |
| CC4 | 3b | 3b | CC21 | 3a | 3a | CC38 | 3b | 3b |
| CC5 | nd | 6a | CC22 | 3b | nd | CC39 | 3a | 3a |
| CC6 | 3a | 3a | CC23 | 3a | 3a | CC40 | 3b | 3b |
| CC7 | 6n | 6n | CC24 | 3b | nd | CC41 | 3a | 3a |
| CC8 | 3a | 3a | CC25 | 3a | 3a | CC42 | 6a | 6a |
| CC9 | 6n | 6n | CC26 | nd | 1b | CC43 | 3a | 3a |
| CC10 | 6n | 6n | CC27 | 6a | 6a | CC44 | 6n | 6n |
| CC11 | 3a | nd | CC28 | 3b | 3b | CC45 | 6a | 6a |
| CC12 | 3b | 3b | CC29 | 3a | 3a | CC46 | 1b | 1b |
| CC13 | 3b | 3b | CC30 | 6a | 6a | CC47 | 3b | 3b |
| CC14 | 6n | 6n | CC31 | 1b | 1b | CC48 | 3b | 3b |
| CC15 | nd | 3b | CC32 | 3b | 3b | CC49 | 6a | 6a |
| CC16 | 3b | 3b | CC33 | 3b | 3b | – | – | – |
| CC17 | 6n | 6n | CC34 | 3b | 3b | – | – | – |
| Total | 49 | |||||||
| Characteristics/Subtypes | Sample Numbers (%) | Total | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 1b | 3a | 3b | 6n | 6a | 6v | Unassigned | Dual-Infection | ||
| Gender | |||||||||
| Male | 6 (9.0) | 13 (19.4) | 27 (40.3) | 10 (14.9) | 9 (13.4) | 1 (1.5) | 1 (1.5) | – | 67 |
| Female | 3 (9.4) | 3 (9.4) | 11 (34.4) | 10 (31.2) | 3 (9.4) | – | – | 2 (6.2) | 32 |
| Total | 9 (9.1) | 16 (16.2) | 38 (38.4) | 20 (20.2) | 12 (12.1) | 1 (1.0) | 1 (1.0) | 2 (2.0) | 99 |
| Races | |||||||||
| Han | 7 (8.5) | 15 (18.3) | 32 (39.0) | 16 (19.5) | 10 (12.2) | 1 (1.2) | – | 1 (1.2) | 82 |
| Hani | 1 (10.0) | 1 (10.0) | 3 (30.0) | 4 (40.0) | – | – | – | 1 (10.0) | 10 |
| Yi | – | – | 2 (50.0) | – | 2 (50.0) | – | – | – | 4 |
| Others | 1 (33.3) | – | 1 (33.3) | – | – | – | 1 (33.3) | – | 3 |
| Route of Transmission | |||||||||
| Blood Contact | 5 (10.4) | 5 (10.4) | 20 (41.7) | 13 (27.1) | 2 (4.2) * | 1 (2.1) | 1 (2.1) | 1 (2.1) | 48 |
| IDU | 2 (5.1) | 7 (17.9) | 15 (38.5) | 6 (15.4) | 8 (20.5) * | – | – | 1 (2.6) | 39 |
| Sex | 2 (18.2) | 4 (36.4) | 3 (27.3) | 1 (9.1) | 1 (9.1) | – | – | – | 11 |
| Vertical | – | – | – | – | 1 (100) | – | – | – | 1 |
| HIV Infection | |||||||||
| Yes | 3 (6.1) | 11 (22.4) | 18 (36.7) | 7 (14.3) | 9 (18.4) | – | – | 1 (2.0) | 49 |
| No | 6 (12.0) | 5 (10.0) | 20 (40.0) | 13 (26.0) | 3 (6.0) | 1 (2.0) | 1 (2.0) | 1 (2.0) | 50 |
| HCV Subtypes | Yunnan (%) n = 80 | Yunnan (%) n = 100 | Honghe (%) n = 39 | Vietnam (%) n = 145 |
|---|---|---|---|---|
| Genotype 1 | 21.2 | 12.0 | 5.1 | 70.0 |
| 1a | 1.2 | 2.0 | 0 | 42.0 |
| 1b | 20.0 | 10.0 | 5.1 | 28.0 |
| Genotype 3 | 53.8 | 41.0 | 56.4 | 2.0 |
| 3a | 23.8 | 12.0 | 17.9 | 1.0 |
| 3b | 30.0 | 29.0 | 38.5 | 1.0 |
| Genotype 6 | 25.0 | 47.0 | 35.9 | 28.0 |
| 6a | 5.0 | 15.0 | 20.5 | 22.8 |
| 6n | 11.2 | 30.0 | 15.4 | 0 |
| 6 others | 8.8 | 2.0 | 0 | 5.5 |
| Year of Study | 2000–2003 | 2013 | 2014 | 2008–2009 |
| References | [35] | [25] | This study | [36] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Jiang, C.; Hu, S.; Diao, Q.; Li, J.; Si, W.; Chen, M.; Zhao, R.Y. Evolving Diversity of Hepatitis C Viruses in Yunnan Honghe, China. Int. J. Mol. Sci. 2016, 17, 403. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030403
Yang L, Jiang C, Hu S, Diao Q, Li J, Si W, Chen M, Zhao RY. Evolving Diversity of Hepatitis C Viruses in Yunnan Honghe, China. International Journal of Molecular Sciences. 2016; 17(3):403. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030403
Chicago/Turabian StyleYang, Lanhui, Chenyan Jiang, Song Hu, Qiongni Diao, Jia Li, Wei Si, Mei Chen, and Richard Y. Zhao. 2016. "Evolving Diversity of Hepatitis C Viruses in Yunnan Honghe, China" International Journal of Molecular Sciences 17, no. 3: 403. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030403