
Caveolae and Caveolin-1 Integrate Reverse Cholesterol Transport and Inflammation in Atherosclerosis

 and
and
Abstract
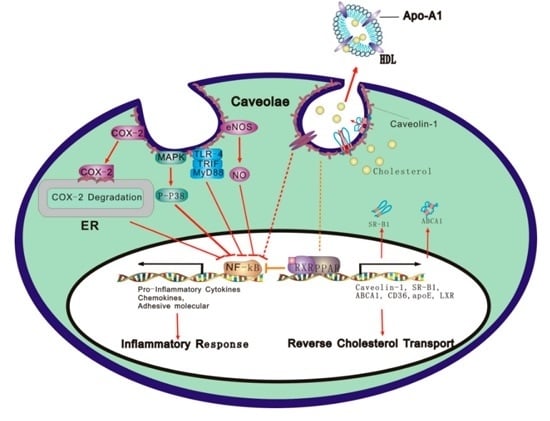
:
{kind=link}
{kind=link}
1. Introduction
2. Relationship between RCT and Inflammatory Response
2.1. Effect of RCT on Inflammatory Response
2.2. Effect of Inflammation on RCT
3. Roles of Caveolae/Caveolins in RCT and Inflammatory Response
3.1. Caveolae and Caveolins
3.2. Caveolae/Cav-1 as a Platform of RCT
3.3. Caveolae/Cav-1 as a Platform for Inflammatory Response
4. Molecular Mechanisms Underlying Caveolae/Cav-1 Integration of RCT and Inflammatory Response
4.1. Caveolae/Cav-1 and the TLR Signaling Pathway
4.2. Caveolae/Cav-1 and the eNOS/NO Signal Pathway
4.3. Caveolae/Cav-1 and the COX Signaling Pathway
4.4. Caveolae/Cav-1 and the MAPK Signal Pathway
4.5. Caveolae/Cav-1 and the Integrin/Adhesion Molecule Signaling Pathway
4.6. Caveolae/Cav-1 and PPARγ and NF-κB Signal Pathways
5. Perspective and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Temel, R.E.; Brown, J.M. Biliary and nonbiliary contributions to reverse cholesterol transport. Curr. Opin. Lipidol. 2012, 23, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.X.; Cao, D.L.; Xiong, Y.; Peng, X.H.; Liao, D.F. A novel model of cholesterol efflux from lipid-loaded cells. Acta Pharmacol. Sin. 2010, 31, 1243–1257. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- McGillicuddy, F.C.; de la Llera Moya, M.; Hinkle, C.C.; Joshi, M.R.; Chiquoine, E.H.; Billheimer, J.T.; Rothblat, G.H.; Reilly, M.P. Inflammation impairs reverse cholesterol transport in vivo. Circulation 2009, 119, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- De Beer, M.C.; Wroblewski, J.M.; Noffsinger, V.P.; Ji, A.; Meyer, J.M.; van der Westhuyzen, D.R.; de Beer, F.C.; Webb, N.R. The impairment of macrophage-to-feces reverse cholesterol transport during inflammation does not depend on serum amyloid A. J. Lipids 2013, 2013, 283486. [Google Scholar] [CrossRef] [PubMed]
- Warrier, M.; Shih, D.M.; Burrows, A.C.; Ferguson, D.; Gromovsky, A.D.; Brown, A.L.; Marshall, S.; McDaniel, A.; Schugar, R.C.; Wang, Z.; et al. The TMAO-generating enzyme flavin monooxygenase 3 is a central regulator of cholesterol balance. Cell Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ishibashi, M.; Seimon, T.; Lee, M.; Sharma, S.M.; Fitzgerald, K.A.; Samokhin, A.O.; Wang, Y.; Sayers, S.; Aikawa, M.; et al. Free cholesterol accumulation in macrophage membranes activates toll-like receptors and p38 mitogen-activated protein kinase and induces cathepsin K. Circ. Res. 2009, 104, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; He, J.; Li, C.; Shyy, J.Y.; Zhu, Y. Cholesterol increases adhesion of monocytes to endothelium by moving adhesion molecules out of caveolae. Biochim. Biophys. Acta 2010, 1801, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Jagielska, J.; Kapopara, P.R.; Salguero, G.; Scherr, M.; Schutt, H.; Grote, K.; Schieffer, B.; Bavendiek, U. Interleukin-1 assembles a proangiogenic signaling module consisting of caveolin-1, tumor necrosis factor receptor-associated factor 6, p38-mitogen-activated protein kinase (MAPK), and MAPK-activated protein kinase 2 in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Kootte, R.S.; Smits, L.P.; van der Valk, F.M.; Dasseux, J.L.; Keyserling, C.H.; Barbaras, R.; Paolini, J.F.; Santos, R.D.; van Dijk, T.H.; Dallinga-van Thie, G.M.; et al. Effect of open-label infusion of an APOA-I-containing particle (CER-001) on RCT and artery wall thickness in patients with FHA. J. Lipid Res. 2015, 56, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Biocca, S.; Iacovelli, F.; Matarazzo, S.; Vindigni, G.; Oteri, F.; Desideri, A.; Falconi, M. Molecular mechanism of statin-mediated lox-1 inhibition. Cell Cycle 2015, 14, 1583–1595. [Google Scholar] [CrossRef] [PubMed]
- Doi, H.; Kugiyama, K.; Oka, H.; Sugiyama, S.; Ogata, N.; Koide, S.I.; Nakamura, S.I.; Yasue, H. Remnant lipoproteins induce proatherothrombogenic molecules in endothelial cells through a redox-sensitive mechanism. Circulation 2000, 102, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pendlebury, C.; Dodd, M.M.; Maximova, K.; Vine, D.F.; Jetha, M.M.; Ball, G.D.; Proctor, S.D. Elevated remnant lipoproteins may increase subclinical CVD risk in pre-pubertal children with obesity: A case-control study. Pediatr. Obes. 2013, 8, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Namiri-Kalantari, R.; Gao, F.; Chattopadhyay, A.; Wheeler, A.A.; Navab, K.D.; Farias-Eisner, R.; Reddy, S.T. The dual nature of HDL: Anti-inflammatory and pro-inflammatory. BioFactors 2015, 41, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.; Otvos, J.; Nikolic, D.; Montalto, G.; Toth, P.P.; Banach, M. Subfractions and subpopulations of HDL: An update. Curr. Med. Chem. 2014, 21, 2881–2891. [Google Scholar] [CrossRef] [PubMed]
- Undurti, A.; Huang, Y.; Lupica, J.A.; Smith, J.D.; DiDonato, J.A.; Hazen, S.L. Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J. Biol. Chem. 2009, 284, 30825–30835. [Google Scholar] [CrossRef] [PubMed]
- Cutuli, L.; Pirillo, A.; Uboldi, P.; Kuehn, H.; Catapano, A.L. 15-lipoxygenase-mediated modification of HDL3 impairs ENOS activation in human endothelial cells. Lipids 2014, 49, 317–326. [Google Scholar] [CrossRef] [PubMed]
- De la Llera Moya, M.; McGillicuddy, F.C.; Hinkle, C.C.; Byrne, M.; Joshi, M.R.; Nguyen, V.; Tabita-Martinez, J.; Wolfe, M.L.; Badellino, K.; Pruscino, L.; et al. Inflammation modulates human HDL composition and function in vivo. Atherosclerosis 2012, 222, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Charles-Schoeman, C.; Lee, Y.Y.; Grijalva, V.; Amjadi, S.; FitzGerald, J.; Ranganath, V.K.; Taylor, M.; McMahon, M.; Paulus, H.E.; Reddy, S.T. Cholesterol efflux by high density lipoproteins is impaired in patients with active rheumatoid arthritis. Ann. Rheum. Dis. 2012, 71, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Welch, C.; Pagler, T.A.; Ranalletta, M.; Lamkanfi, M.; Han, S.; Ishibashi, M.; Li, R.; Wang, N.; Tall, A.R. Increased inflammatory gene expression in abc transporter-deficient macrophages: Free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation 2008, 118, 1837–1847. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.; Liao, D.F.; Tang, C.K. Atp-binding membrane cassette transporter A1 (ABCA1): A possible link between inflammation and reverse cholesterol transport. Mol. Med. 2010, 16, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.M.; Handa, P.; Tateya, S.; Schwartz, J.; Tang, C.; Mitra, P.; Oram, J.F.; Chait, A.; Kim, F. Apolipoprotein A-I attenuates palmitate-mediated NF-κB activation by reducing toll-like receptor-4 recruitment into lipid rafts. PLoS ONE 2012, 7, e33917. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Owen, J.S.; Wilson, M.D.; Li, H.; Griffiths, G.L.; Thomas, M.J.; Hiltbold, E.M.; Fessler, M.B.; Parks, J.S. Macrophage ABCA1 reduces MYD88-dependent toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J. Lipid Res. 2010, 51, 3196–3206. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Kim, H.P.; Geng, X.H.; Nakao, A.; Wang, X.; Murase, N.; Drain, P.F.; Wang, X.; Sasidhar, M.; Nabel, E.G.; et al. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of tlrs to lipid rafts. J. Exp. Med. 2006, 203, 2377–2389. [Google Scholar] [CrossRef] [PubMed]
- Powers, K.A.; Szaszi, K.; Khadaroo, R.G.; Tawadros, P.S.; Marshall, J.C.; Kapus, A.; Rotstein, O.D. Oxidative stress generated by hemorrhagic shock recruits toll-like receptor 4 to the plasma membrane in macrophages. J. Exp. Med. 2006, 203, 1951–1961. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, M.; Morath, S.; Mackie, A.; Hartung, T.; Triantafilou, K. Lateral diffusion of toll-like receptors reveals that they are transiently confined within lipid rafts on the plasma membrane. J. Cell Sci. 2004, 117, 4007–4014. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.-F.; Tang, C.-K. Cholesterol reverse transport from basic to clinical; Beijing Sci. Technol. Press: Beijing, China, 2009. (In Chinese) [Google Scholar]
- Annema, W.; Nijstad, N.; Tolle, M.; de Boer, J.F.; Buijs, R.V.; Heeringa, P.; van der Giet, M.; Tietge, U.J. Myeloperoxidase and serum amyloid a contribute to impaired in vivo reverse cholesterol transport during the acute phase response but not group IIA secretory phospholipase A(2). J. Lipid Res. 2010, 51, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Baranova, I.; Vishnyakova, T.; Bocharov, A.; Chen, Z.; Remaley, A.T.; Stonik, J.; Eggerman, T.L.; Patterson, A.P. Lipopolysaccharide down regulates both scavenger receptor b1 and atp binding cassette transporter A1 in raw cells. Infect. Immun. 2002, 70, 2995–3003. [Google Scholar] [CrossRef] [PubMed]
- Miller, Y.I.; Choi, S.H.; Fang, L.; Harkewicz, R. Toll-like receptor-4 and lipoprotein accumulation in macrophages. Trends Cardiovasc. Med. 2009, 19, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.-L.; Yin, K.; Mo, Z.-C.; Hao, X.-R.; Hu, Y.-W.; Li, X.-X.; Tang, Y.-L.; Tang, C.-K. Lipopolysaccharide down-regulates ABCA1 expression in foam cells in a nucleus factor-κB pathway-dependent manne. Prog. Biochem. Biophys. 2010, 37, 540–548. [Google Scholar] [CrossRef]
- Singla, S.; Predescu, D.; Bardita, C.; Wang, M.; Zhang, J.; Balk, R.A.; Predescu, S. Pro-inflammatory endothelial cell dysfunction is associated with intersectin-1s down-regulation. Respir. Res. 2011, 12, 46. [Google Scholar] [CrossRef] [PubMed]
- Ma, A.Z.; Zhang, Q.; Song, Z.Y. Tnfa alter cholesterol metabolism in human macrophages via PKC-θ-dependent pathway. BMC Biochem. 2013, 14, 20. [Google Scholar] [PubMed]
- Jia, R.; Kurita-Ochiai, T.; Oguchi, S.; Yamamoto, M. Periodontal pathogen accelerates lipid peroxidation and atherosclerosis. J. Dent. Res. 2013, 92, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Malik, P.; Berisha, S.Z.; Santore, J.; Agatisa-Boyle, C.; Brubaker, G.; Smith, J.D. Zymosan-mediated inflammation impairs in vivo reverse cholesterol transport. J. Lipid Res. 2011, 52, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Lucas, M.; Fernandez-Lizarbe, S.; Montesinos, J.; Guerri, C. LPS or ethanol triggers clathrin- and rafts/caveolae-dependent endocytosis of TLR4 in cortical astrocytes. J. Neurochem. 2014, 129, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liao, D.; Bharadwaj, U.; Li, M.; Yao, Q.; Chen, C. C-reactive protein inhibits cholesterol efflux from human macrophage-derived foam cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.R.; Cao, D.L.; Hu, Y.W.; Li, X.X.; Liu, X.H.; Xiao, J.; Liao, D.F.; Xiang, J.; Tang, C.K. IFN-γ down-regulates ABCA1 expression by inhibiting LXRα in a JAK/STAT signaling pathway-dependent manner. Atherosclerosis 2009, 203, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Yamada, E. The fine structure of the gall bladder epithelium of the mouse. J. Biophys. Biochem. Cytol. 1955, 1, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Li, X.A.; Everson, W.V.; Smart, E.J. Caveolae, lipid rafts, and vascular disease. Trends Cardiovasc. Med. 2005, 15, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Fielding, C.J.; Fielding, P.E. Caveolae and intracellular trafficking of cholesterol. Adv. Drug Deliv. Rev. 2001, 49, 251–264. [Google Scholar] [CrossRef]
- Maceckova, M.; Martiskova, H.; Koudelka, A.; Kubala, L.; Lojek, A.; Pekarova, M. Bone marrow-derived macrophages exclusively expressed caveolin-2: The role of inflammatory activators and hypoxia. Immunobiology 2015, 220, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Gutierrez-Pajares, J.L.; Iturrieta, J.; Lisanti, M.P.; Frank, P.G. Endothelial caveolin-1 plays a major role in the development of atherosclerosis. Cell Tissue Res. 2014, 356, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Chen, S.J.; Tsao, C.M.; Wu, C.C. Perivascular adipose tissue inhibits endothelial function of rat aortas via caveolin-1. PLoS ONE 2014, 9, e99947. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.Y.; Lee, M.J.; Summer, R.; Liu, L.; Fried, S.K.; Pilch, P.F. Pleiotropic effects of cavin-1 deficiency on lipid metabolism. J. Biol. Chem. 2014, 289, 8473–8483. [Google Scholar] [CrossRef] [PubMed]
- Sanon, V.P.; Sawaki, D.; Mjaatvedt, C.H.; Jourdan-Le Saux, C. Myocardial tissue caveolae. Compr. Physiol. 2015, 5, 871–886. [Google Scholar] [PubMed]
- Murata, M.; Peranen, J.; Schreiner, R.; Wieland, F.; Kurzchalia, T.V.; Simons, K. Vip21/caveolin is a cholesterol-binding protein. Proc. Natl. Acad. Sci. USA 1995, 92, 10339–10343. [Google Scholar] [CrossRef] [PubMed]
- Fielding, P.E.; Fielding, C.J. Intracellular transport of low density lipoprotein derived free cholesterol begins at clathrin-coated pits and terminates at cell surface caveolae. Biochemistry 1996, 35, 14932–14938. [Google Scholar] [CrossRef] [PubMed]
- Schelgel, A.; Lisanti, M.P. A molecular dissection of caveolin-1 membrane attachment and oligomerization. Two separate regions of the caveolin-1 c-terminal domain mediate membrane binding and oligomer/oligomer interactions in vivo. J. Biol. Chem. 2000, 275, 21605–21617. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Woodman, S.E.; Lisanti, M.P. Caveolae: From cell biology to animal physiology. Pharmacol. Rev. 2002, 54, 431–467. [Google Scholar] [CrossRef] [PubMed]
- Fork, C.; Hitzel, J.; Nichols, B.J.; Tikkanen, R.; Brandes, R.P. Flotillin-1 facilitates toll-like receptor 3 signaling in human endothelial cells. Basic Res. Cardiol. 2014, 109, 439. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Higashimoto, Y.; Taira, J.; Yamagishi, S. Pigment epithelium-derived factor (PEDF) binds to caveolin-1 and inhibits the pro-inflammatory effects of caveolin-1 in endothelial cells. Biochem. Biophys. Res. Commun. 2013, 441, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, R.; Mu, H.; Wang, X.; Yao, Q.; Chen, C. Reverse cholesterol transport and cholesterol efflux in atherosclerosis. QJM Mon. J. Assoc. Phys. 2005, 98, 845–856. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosi, N.; Volonte, C. Metabotropic purinergic receptors in lipid membrane microdomains. Curr. Med. Chem. 2013, 20, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Zhang, Y.; Yan, Z.; Wang, Z.G.; Liu, G.; Minshall, R.D.; Malik, A.B.; Hu, G. Caveolin-1 TYR14 phosphorylation induces interaction with TLR4 in endothelial cells and mediates myd88-dependent signaling and sepsis-induced lung inflammation. J. Immunol. 2013, 191, 6191–6199. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Nadeau, P.E.; Mergia, A. HIV inhibits endothelial reverse cholesterol transport through impacting subcellular caveolin-1 trafficking. Retrovirology 2015, 12, 62. [Google Scholar] [CrossRef] [PubMed]
- Maniatis, N.A.; Brovkovych, V.; Allen, S.E.; John, T.A.; Shajahan, A.N.; Tiruppathi, C.; Vogel, S.M.; Skidgel, R.A.; Malik, A.B.; Minshall, R.D. Novel mechanism of endothelial nitric oxide synthase activation mediated by caveolae internalization in endothelial cells. Circ. Res. 2006, 99, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Smart, E.J.; Ying, Y.; Donzell, W.C.; Anderson, R.G. A role for caveolin in transport of cholesterol from endoplasmic reticulum to plasma membrane. J. Biol. Chem. 1996, 271, 29427–29435. [Google Scholar] [PubMed]
- Uittenbogaard, A.; Ying, Y.; Smart, E.J. Characterization of a cytosolic heat-shock protein-caveolin chaperone complex. Involvement in cholesterol trafficking. J. Biol. Chem. 1998, 273, 6525–6532. [Google Scholar] [CrossRef] [PubMed]
- Kurzchalia, T.V.; Dupree, P.; Monier, S. VIP21-caveolin, a protein of the trans-Golgi network and caveolae. FEBS Lett. 1994, 346, 88–91. [Google Scholar] [CrossRef]
- Zhou, M.; Parr, R.D.; Petrescu, A.D.; Payne, H.R.; Atshaves, B.P.; Kier, A.B.; Ball, J.M.; Schroeder, F. Sterol carrier protein-2 directly interacts with caveolin-1 in vitro and in vivo. Biochemistry 2004, 43, 7288–7306. [Google Scholar] [CrossRef] [PubMed]
- Chao, W.T.; Tsai, S.H.; Lin, Y.C.; Lin, W.W.; Yang, V.C. Cellular localization and interaction of ABCA1 and caveolin-1 in aortic endothelial cells after HDL incubation. Biochem. Biophys. Res. Commun. 2005, 332, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Ma, C.; Hsu, W.C.; Lo, H.F.; Yang, V.C. Molecular interaction between caveolin-1 and ABCA1 on high-density lipoprotein-mediated cholesterol efflux in aortic endothelial cells. Cardiovasc. Res. 2007, 75, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Li, A.C.; Binder, C.J.; Gutierrez, A.; Brown, K.K.; Plotkin, C.R.; Pattison, J.W.; Valledor, A.F.; Davis, R.A.; Willson, T.M.; Witztum, J.L.; et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARα, β/δ, and γ. J. Clin. Investig. 2004, 114, 1564–1576. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Oh, G.S.; Yoon, J.; Lee, G.G.; Lee, K.U.; Kim, S.W. Hepatic TRAP80 selectively regulates lipogenic activity of liver x receptor. J. Clin. Investig. 2015, 125, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Zhang, X.J.; Liu, C.X.; Wang, X.P.; Zhang, Y. PPARγ1-induced caveolin-1 enhances cholesterol efflux and attenuates atherosclerosis in apolipoprotein E-deficient mice. J. Vasc. Res. 2010, 47, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Chinetti, G.; Lestavel, S.; Bocher, V.; Remaley, A.T.; Neve, B.; Torra, I.P.; Teissier, E.; Minnich, A.; Jaye, M.; Duverger, N.; et al. PPAR-α and PPAR-γ activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat. Med. 2001, 7, 53–58. [Google Scholar] [PubMed]
- Chinetti, G.; Gbaguidi, F.G.; Griglio, S.; Mallat, Z.; Antonucci, M.; Poulain, P.; Chapman, J.; Fruchart, J.C.; Tedgui, A.; Najib-Fruchart, J.; et al. CLA-1/SR-BI is expressed in atherosclerotic lesion macrophages and regulated by activators of peroxisome proliferator-activated receptors. Circulation 2000, 101, 2411–2417. [Google Scholar] [CrossRef] [PubMed]
- Argmann, C.A.; Sawyez, C.G.; McNeil, C.J.; Hegele, R.A.; Huff, M.W. Activation of peroxisome proliferator-activated receptor γ and retinoid x receptor results in net depletion of cellular cholesteryl esters in macrophages exposed to oxidized lipoproteins. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Galetto, R.; Albajar, M.; Polanco, J.I.; Zakin, M.M.; Rodriguez-Rey, J.C. Identification of a peroxisome-proliferator-activated-receptor response element in the apolipoprotein E gene control region. Biochem. J. 2001, 357, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Llaverias, G.; Vazquez-Carrera, M.; Sanchez, R.M.; Noe, V.; Ciudad, C.J.; Laguna, J.C.; Alegret, M. Rosiglitazone upregulates caveolin-1 expression in THP-1 cells through a PPAR-dependent mechanism. J. Lipid Res. 2004, 45, 2015–2024. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, E.; Chinetti-Gbaguidi, G.; Staels, B. Regulation of macrophage functions by PPAR-α, PPAR-γ, and LXRS in mice and men. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Chidlow, J.H., Jr.; Sessa, W.C. Caveolae, caveolins, and cavins: Complex control of cellular signalling and inflammation. Cardiovasc. Res. 2010, 86, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Medina, F.A.; de Almeida, C.J.; Dew, E.; Li, J.; Bonuccelli, G.; Williams, T.M.; Cohen, A.W.; Pestell, R.G.; Frank, P.G.; Tanowitz, H.B.; et al. Caveolin-1-deficient mice show defects in innate immunity and inflammatory immune response during salmonella enterica serovar typhimurium infection. Infect. Immun. 2006, 74, 6665–6674. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Arzuaga, X.; Kluemper, C.T.; Caraballo, A.; Toborek, M.; Hennig, B. Quercetin blocks caveolae-dependent pro-inflammatory responses induced by co-planar PCBS. Environ. Int. 2010, 36, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Petriello, M.C.; Newsome, B.; Hennig, B. Influence of nutrition in PCB-induced vascular inflammation. Environ. Sci. Pollut. Res. Int. 2014, 21, 6410–6418. [Google Scholar] [CrossRef] [PubMed]
- Engel, D.; Beckers, L.; Wijnands, E.; Seijkens, T.; Lievens, D.; Drechsler, M.; Gerdes, N.; Soehnlein, O.; Daemen, M.J.; Stan, R.V.; et al. Caveolin-1 deficiency decreases atherosclerosis by hampering leukocyte influx into the arterial wall and generating a regulatory T-cell response. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 3838–3848. [Google Scholar] [CrossRef] [PubMed]
- Layne, J.; Majkova, Z.; Smart, E.J.; Toborek, M.; Hennig, B. Caveolae: A regulatory platform for nutritional modulation of inflammatory diseases. J. Nutr. Biochem. 2011, 22, 807–811. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, C.J.; Witkiewicz, A.K.; Jasmin, J.F.; Tanowitz, H.B.; Sotgia, F.; Frank, P.G.; Lisanti, M.P. Caveolin-2-deficient mice show increased sensitivity to endotoxemia. Cell Cycle 2011, 10, 2151–2161. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Kim, H.P.; Nakahira, K.; Ryter, S.W.; Choi, A.M. The heme oxygenase-1/carbon monoxide pathway suppresses TLR4 signaling by regulating the interaction of TLR4 with caveolin-1. J. Immunol. 2009, 182, 3809–3818. [Google Scholar] [CrossRef] [PubMed]
- Garrean, S.; Gao, X.P.; Brovkovych, V.; Shimizu, J.; Zhao, Y.Y.; Vogel, S.M.; Malik, A.B. Caveolin-1 regulates NF-κB activation and lung inflammatory response to sepsis induced by lipopolysaccharide. J. Immunol. 2006, 177, 4853–4860. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J. Nitric oxide as a unique signaling molecule in the vascular system: A historical overview. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2002, 53, 503–514. [Google Scholar]
- Bucci, M.; Gratton, J.P.; Rudic, R.D.; Acevedo, L.; Roviezzo, F.; Cirino, G.; Sessa, W.C. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat. Med. 2000, 6, 1362–1367. [Google Scholar] [PubMed]
- Zhu, L.; Schwegler-Berry, D.; Castranova, V.; He, P. Internalization of caveolin-1 scaffolding domain facilitated by antennapedia homeodomain attenuates PAF-induced increase in microvessel permeability. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H195–H201. [Google Scholar] [CrossRef] [PubMed]
- Gratton, J.P.; Fontana, J.; O’Connor, D.S.; Garcia-Cardena, G.; McCabe, T.J.; Sessa, W.C. Reconstitution of an endothelial nitric-oxide synthase (ENOS), HSP90, and caveolin-1 complex in vitro. Evidence that HSP90 facilitates calmodulin stimulated displacement of enos from caveolin-1. J. Biol. Chem. 2000, 275, 22268–22272. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cardena, G.; Martasek, P.; Masters, B.S.; Skidd, P.M.; Couet, J.; Li, S.; Lisanti, M.P.; Sessa, W.C. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J. Biol. Chem. 1997, 272, 25437–25440. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Chu, C.; Lin, A.; Jo, H.; Ikezu, T.; Okamoto, T.; Kohtz, D.S.; Lisanti, M.P. Caveolin-mediated regulation of signaling along the P42/44 map kinase cascade in vivo. A role for the caveolin-scaffolding domain. FEBS Lett. 1998, 428, 205–211. [Google Scholar] [CrossRef]
- Taylor, S.Y.; Dixon, H.M.; Yoganayagam, S.; Price, N.; Lang, D. Folic acid modulates enos activity via effects on posttranslational modifications and protein-protein interactions. Eur. J. Pharmacol. 2013, 714, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Terasaka, N.; Westerterp, M.; Koetsveld, J.; Fernandez-Hernando, C.; Yvan-Charvet, L.; Wang, N.; Sessa, W.C.; Tall, A.R. ATP-binding cassette transporter G1 and high-density lipoprotein promote endothelial no synthesis through a decrease in the interaction of caveolin-1 and endothelial no synthase. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2219–2225. [Google Scholar] [CrossRef] [PubMed]
- Feron, O.; Dessy, C.; Moniotte, S.; Desager, J.P.; Balligand, J.L. Hypercholesterolemia decreases nitric oxide production by promoting the interaction of caveolin and endothelial nitric oxide synthase. J. Clin. Investig. 1999, 103, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.K.; Yuan, J.; Gao, X.P.; Garrean, S.; Brovkovych, V.; Malik, A.B.; Tiruppathi, C.; Zhao, Y.Y. Caveolin-1 deficiency dampens toll-like receptor 4 signaling through enos activation. Am. J. Pathol. 2010, 176, 2344–2351. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat. Immunol. 2001, 2, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Suzuki, S.; Duncan, G.S.; Millar, D.G.; Wada, T.; Mirtsos, C.; Takada, H.; Wakeham, A.; Itie, A.; Li, S.; et al. Severe impairment of interleukin-1 and toll-like receptor signalling in mice lacking irak-4. Nature 2002, 416, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Henzel, W.J.; Gao, X. Irak: A kinase associated with the interleukin-1 receptor. Science (New York, N.Y.) 1996, 271, 1128–1131. [Google Scholar] [CrossRef]
- Ninomiya-Tsuji, J.; Kishimoto, K.; Hiyama, A.; Inoue, J.; Cao, Z.; Matsumoto, K. The kinase TAK1 can activate the NIK-I κB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 1999, 398, 252–256. [Google Scholar] [PubMed]
- Sun, J.; Kohr, M.J.; Nguyen, T.; Aponte, A.M.; Connelly, P.S.; Esfahani, S.G.; Gucek, M.; Daniels, M.P.; Steenbergen, C.; Murphy, E. Disruption of caveolae blocks ischemic preconditioning-mediated s-nitrosylation of mitochondrial proteins. Antioxid. Redox Signal. 2012, 16, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Nguyen, T.; Aponte, A.M.; Menazza, S.; Kohr, M.J.; Roth, D.M.; Patel, H.H.; Murphy, E.; Steenbergen, C. Ischaemic preconditioning preferentially increases protein S-nitrosylation in subsarcolemmal mitochondria. Cardiovasc. Res. 2015, 106, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.H.; Qiu, J.; Wang, Y.; Ji, X.; Liu, X.J.; You, B.A.; Sheng, Y.P.; Li, X.; Gao, H.Q. Profilin-1 promotes the development of hypertension-induced cardiac hypertrophy. J. Hypertens. 2013, 31, 576–586; discussion 586. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.Y.; Deng, W.G.; Gilroy, D.W.; Shyue, S.K.; Wu, K.K. Colocalization and interaction of cyclooxygenase-2 with caveolin-1 in human fibroblasts. J. Biol. Chem. 2001, 276, 34975–34982. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.F.; Liou, J.Y.; Huang, T.Y.; Lin, Y.S.; Yeh, A.L.; Tam, K.; Tsai, T.H.; Wu, K.K.; Shyue, S.K. Caveolin-1 facilitates cyclooxygenase-2 protein degradation. J. Cell. Biochem. 2010, 109, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Santizo, R.A.; Xu, H.L.; Galea, E.; Muyskens, S.; Baughman, V.L.; Pelligrino, D.A. Combined endothelial nitric oxide synthase upregulation and caveolin-1 downregulation decrease leukocyte adhesion in pial venules of ovariectomized female rats. Stroke J. Cereb. Circ. 2002, 33, 613–616. [Google Scholar] [CrossRef]
- Anderson, R.G. Caveolae: Where incoming and outgoing messengers meet. Proc. Natl. Acad. Sci. USA 1993, 90, 10909–10913. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Kim, H.P.; Song, R.; Choi, A.M. Caveolin-1 confers antiinflammatory effects in murine macrophages via the MKK3/p38 MAPK pathway. Am. J. Respir. Cell Mol. Biol. 2006, 34, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Yao, S.T.; Zhou, X.; Si, Y.H.; Sang, H.; Wang, J.F.; Shang, Z.P. Inhibitory effect of caveolin-1 on endoplasmic reticulum stress-induced apoptosis in macrophages via p38 MAPK pathway. Sheng Li Xue Bao 2012, 64, 149–154. (In Chinese) [Google Scholar] [PubMed]
- Gaus, K.; Le Lay, S.; Balasubramanian, N.; Schwartz, M.A. Integrin-mediated adhesion regulates membrane order. J. Cell Biol. 2006, 174, 725–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salanueva, I.J.; Cerezo, A.; Guadamillas, M.C.; del Pozo, M.A. Integrin regulation of caveolin function. J. Cell. Mol. Med. 2007, 11, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Malan, D.; Elischer, A.; Hesse, M.; Wickstrom, S.A.; Fleischmann, B.K.; Bloch, W. Deletion of integrin linked kinase in endothelial cells results in defective RTK signaling caused by caveolin 1 mislocalization. Development 2013, 140, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Shu, H.; Wong, B.; Zhou, G.; Li, Y.; Berger, J.; Woods, J.W.; Wright, S.D.; Cai, T.Q. Activation of PPARα or γ reduces secretion of matrix metalloproteinase 9 but not interleukin 8 from human monocytic THP-1 cells. Biochem. Biophys. Res. Commun. 2000, 267, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Meier, C.A.; Chicheportiche, R.; Juge-Aubry, C.E.; Dreyer, M.G.; Dayer, J.M. Regulation of the interleukin-1 receptor antagonist in THP-1 cells by ligands of the peroxisome proliferator-activated receptor γ. Cytokine 2002, 18, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Ricote, M.; Akiyama, T.E.; Gonzalez, F.J.; Glass, C.K. PPARγ and PPARdelta negatively regulate specific subsets of lipopolysaccharide and IFN-γ target genes in macrophages. Proc. Natl. Acad. Sci. USA 2003, 100, 6712–6717. [Google Scholar] [CrossRef] [PubMed]
- Zelcer, N.; Tontonoz, P. Sumoylation and PPARγ: Wrestling with inflammatory signaling. Cell Metab. 2005, 2, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Glass, C.K. PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta 2007, 1771, 926–935. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, L.; Zhu, N.; Ao, B.-X.; Liu, C.; Shi, Y.-N.; Du, K.; Chen, J.-X.; Zheng, X.-L.; Liao, D.-F. Caveolae and Caveolin-1 Integrate Reverse Cholesterol Transport and Inflammation in Atherosclerosis. Int. J. Mol. Sci. 2016, 17, 429. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030429
Qin L, Zhu N, Ao B-X, Liu C, Shi Y-N, Du K, Chen J-X, Zheng X-L, Liao D-F. Caveolae and Caveolin-1 Integrate Reverse Cholesterol Transport and Inflammation in Atherosclerosis. International Journal of Molecular Sciences. 2016; 17(3):429. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030429
Chicago/Turabian StyleQin, Li, Neng Zhu, Bao-Xue Ao, Chan Liu, Ya-Ning Shi, Ke Du, Jian-Xiong Chen, Xi-Long Zheng, and Duan-Fang Liao. 2016. "Caveolae and Caveolin-1 Integrate Reverse Cholesterol Transport and Inflammation in Atherosclerosis" International Journal of Molecular Sciences 17, no. 3: 429. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030429