
Nucleotide Excision Repair and Vitamin D—Relevance for Skin Cancer Therapy

Abstract
:
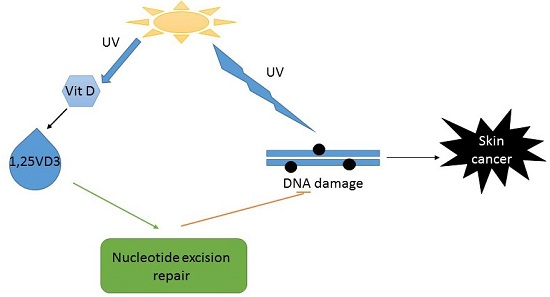{kind=link}
{kind=link}
{kind=link}
{kind=link}
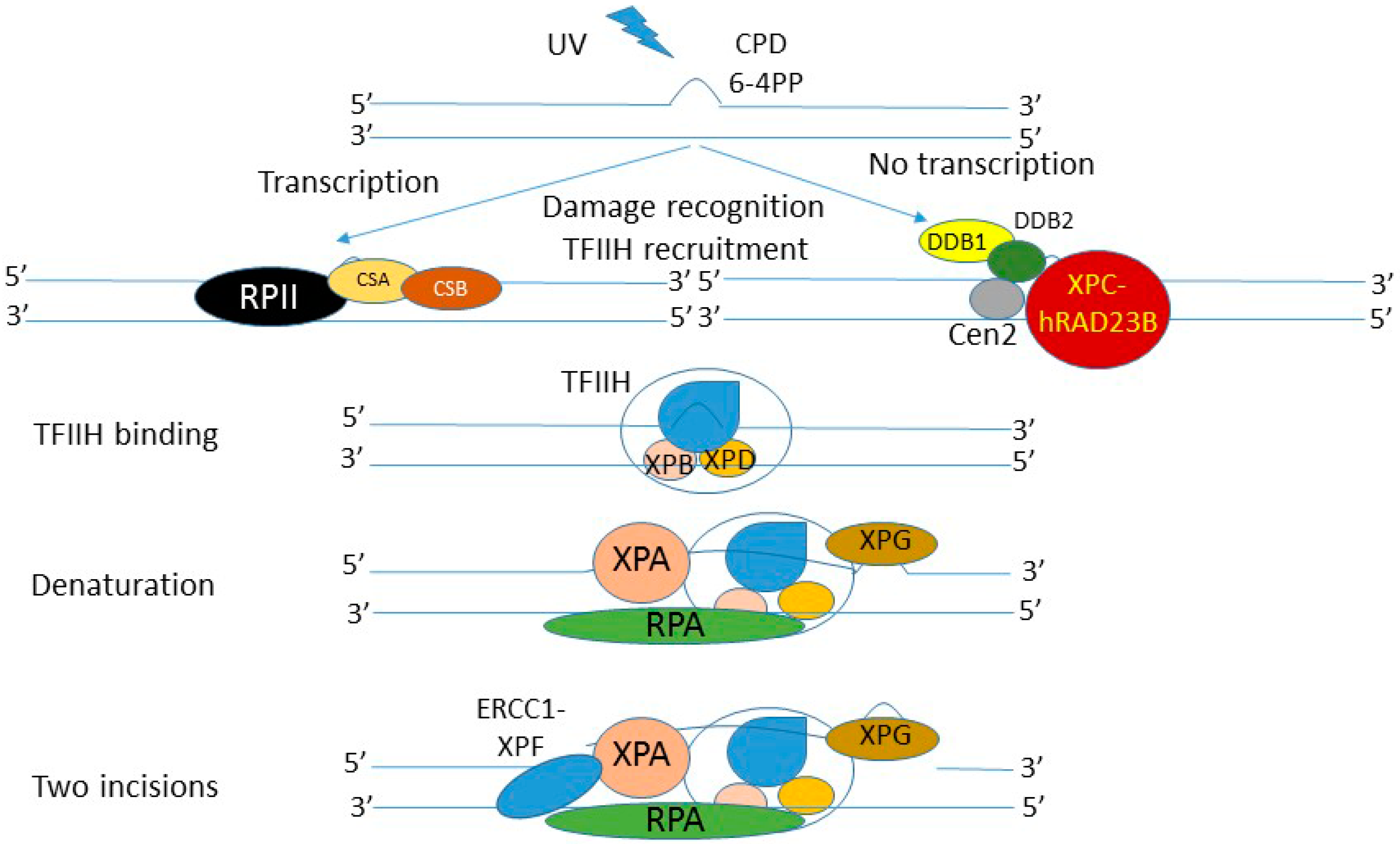{kind=link}
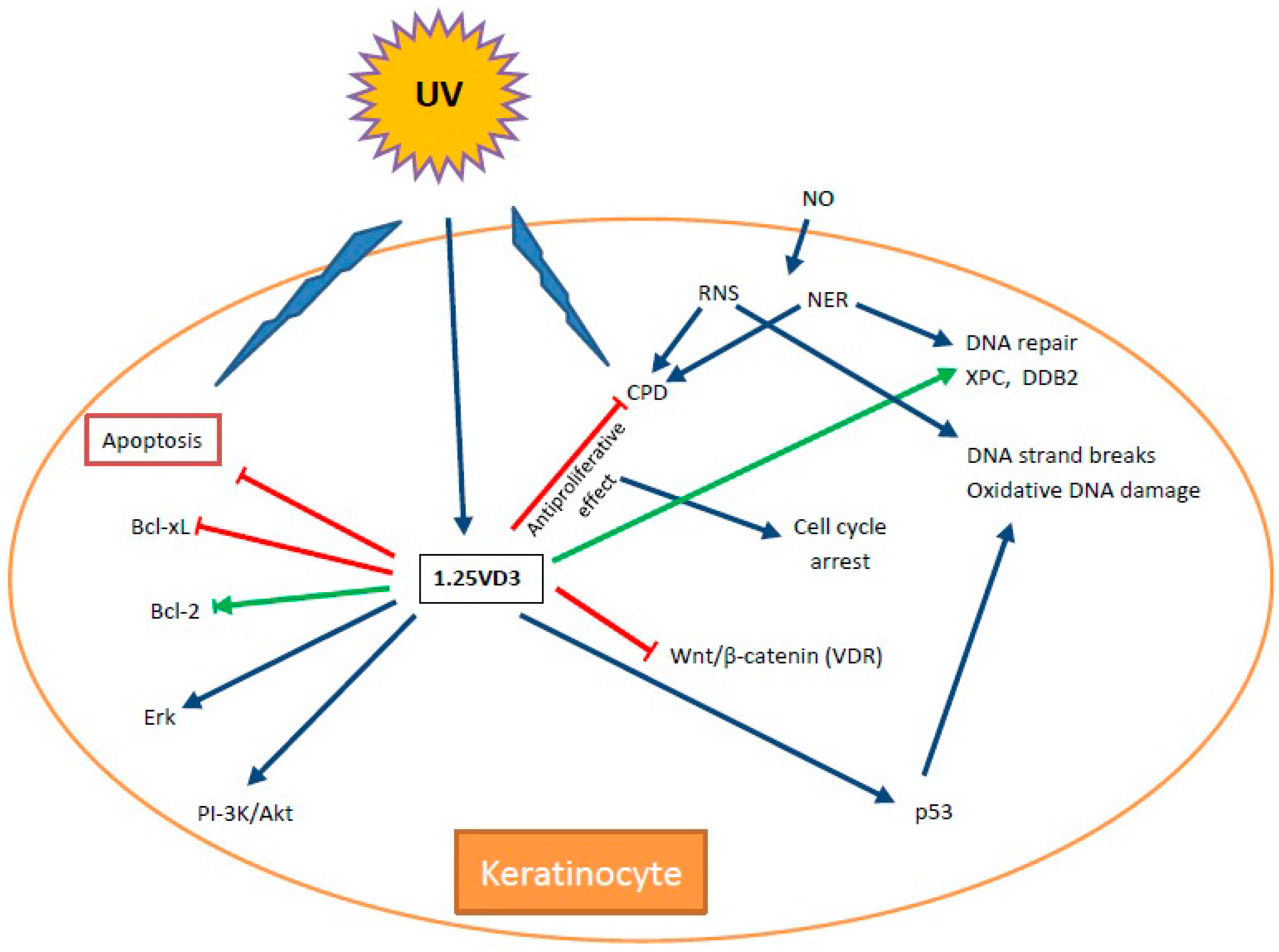{kind=link}
1. Melanoma and Non-Melanoma Skin Cancers
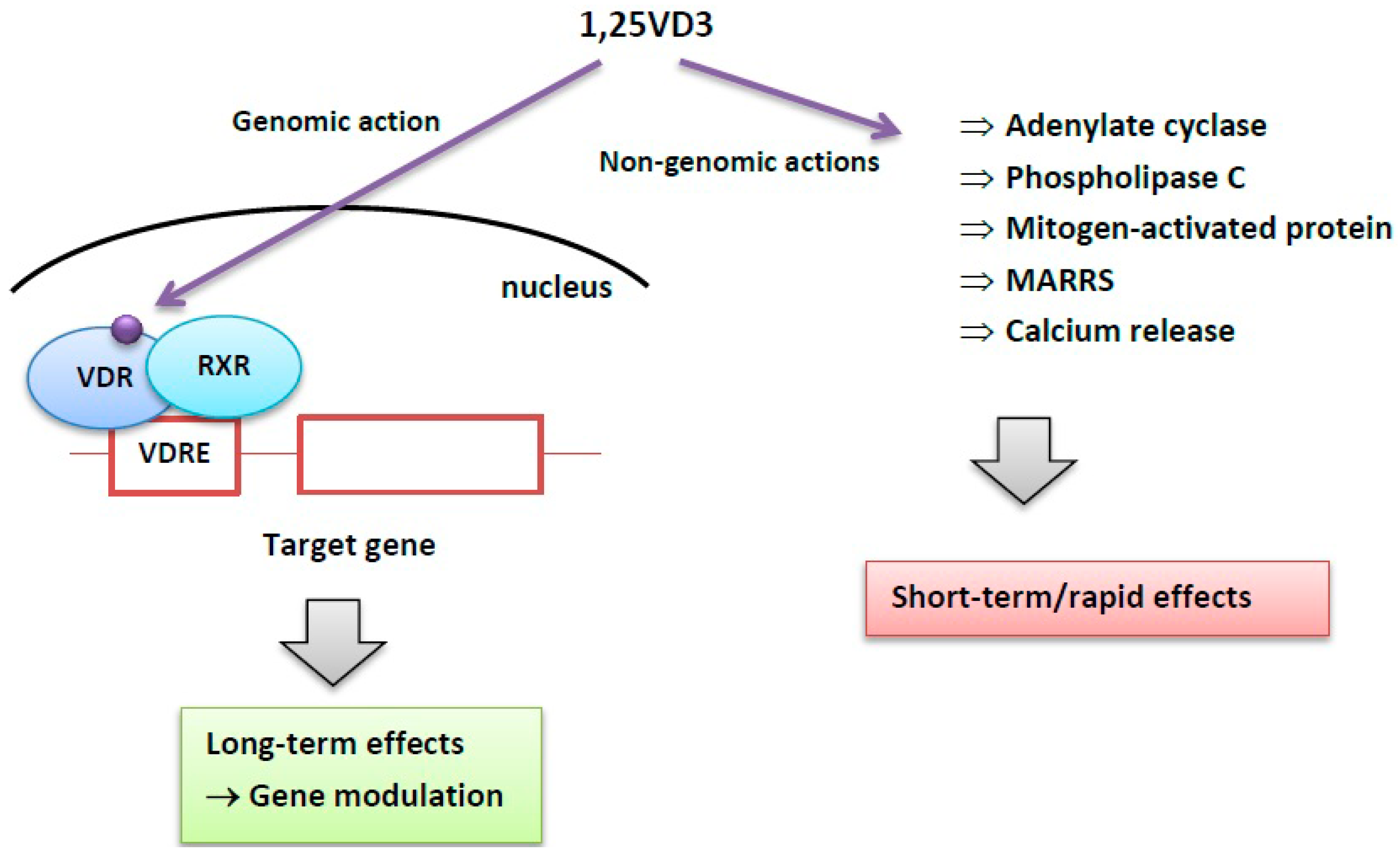
2. Vitamin D and its Role in Physiology and Pathology
3. Vitamin D in Cancer
4. DNA Damage Induced by UV Radiation and Its Role in Skin Carcinogenesis
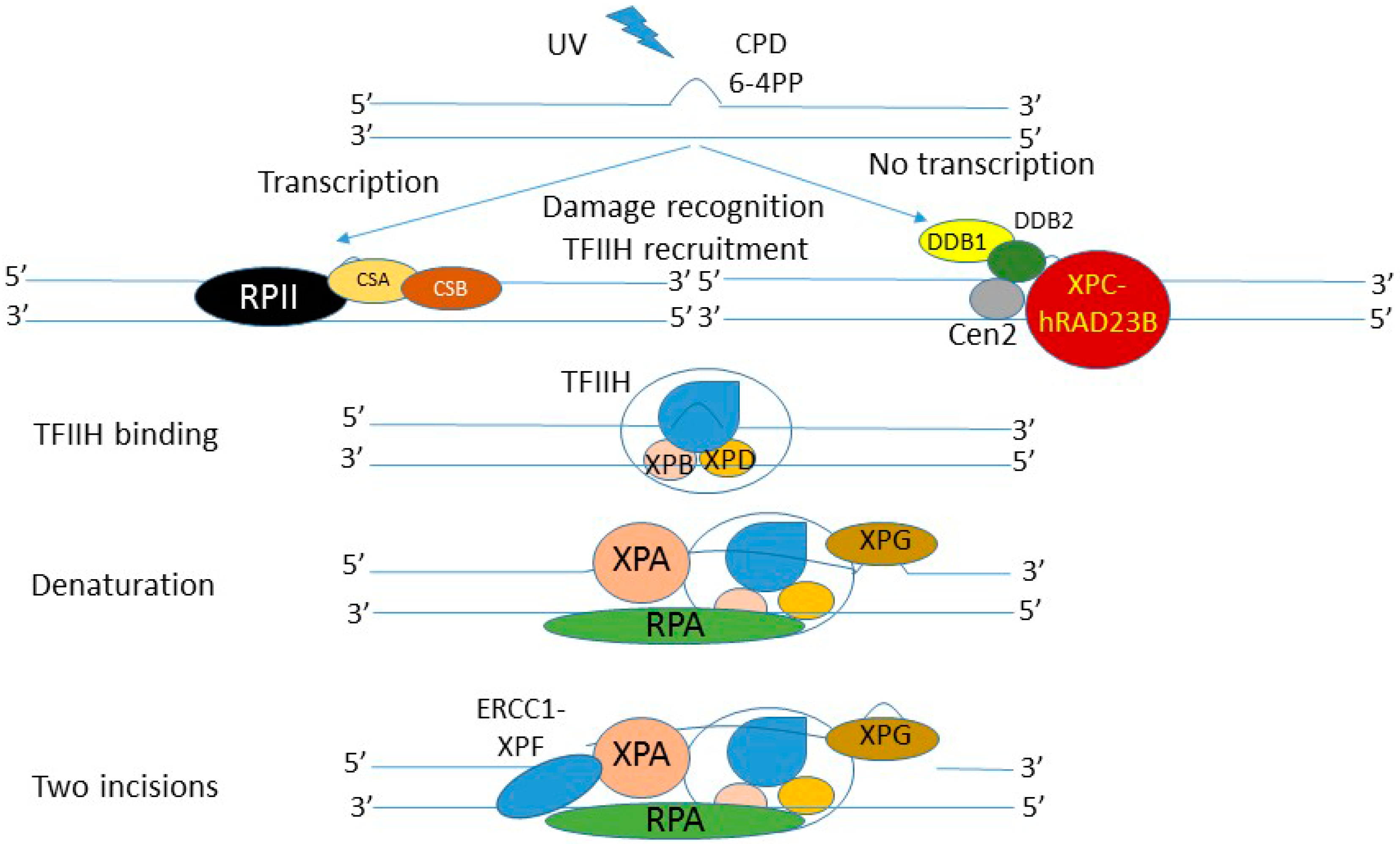
5. Nucleotide Excision Repair—The Most Versatile DNA Repair System
6. Vitamin D and DNA Damage/Repair Induced by UV Radiation
7. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 1,25VD3 | 1,25-Dihydroxyvitamin D3 |
| VDR | Vitamin D receptor |
| NER | Nucleotide excision repair |
| CM | Cutaneous melanoma |
| BCC | Basal cell carcinoma |
| SCC | Squamous cell carcinoma |
| NMSC | Non-melanoma skin cancer |
| BAP1 | BRCA-associated protein 1 |
| CYP2R1, CYP2J2, CYP3A4, CYP27A1, CYP27B1 | cytochrome P-450 enzymes |
| PTH | Parathyroid hormone |
| MS | Multiple sclerosis |
| MHC | Major histocompatibility complex |
| Nrf2 | Nuclear factor (erythroid-derived 2)-like 2 |
| FOS, JUN | Transcription factors |
| RXR | Retinoid X receptor |
| VDRE | Vitamin D response element |
| MARRS | Membrane-associated rapid-response steroid binding protein |
| DAP | Death-associated protein-3 |
| CFKAR | Caspase 8 apoptosis-related cysteine peptidase |
| FADD | Fas-associated death domain |
| Bcl-2 | B-cell lymphoma 2 antiapoptotic factor |
| TERT | Telomerase reverse trancriptase |
| HIF1 | Hypoxia induced factor 1 |
| VEGF | Vascular endothelial growth factor |
| IL-8 | Interleukin 8 |
| COX | Cyclooxygenase |
| MMP | Matrix metalloproteinase |
| Rb | Retinoblastoma protein |
| E2F | Transcription factor, regulator of cell cycle |
| Cdk2/4/6 | Cyclin-dependent kinases |
| IGFBP3 | Insulin-like growth factor binding protein-3 |
| FoxO | Forkhead box O transcription factor |
| ROS | Reactive oxygen species |
| DAP | Death-associated protein-3 |
| CFKAR | Caspase 8 apoptosis-related cysteine peptidase |
| FADD | Fas-associated death domain |
| mTOR, beclin-1 | Proteins involved in autophagy |
| NFκ-B | Nuclear factor kappa-light-chain-enhancer of activated B-cells |
| COX-2 | Cyclooxygenase-2 |
| DDR | DNA damage response |
| CPD | Cyclobutane pyrimidine dimer |
| 6-4PP | Pyrimidine-pyrimidone (6-4) photoproduct |
| TLS | Translesion synthesis |
| EFGR | Epidermal growth factor receptor |
| PTPRK | Protein tyrosine phosphatase kappa |
| MAPK | Mitogen-activated protein kinase |
| AKT/mTOR | Serine/threonine protein kinase B/mammalian target of rapamycin |
| XP | Xeroderma pigmentosum |
| XPA-G | Xeroderma pigmentosum complementation group A-GDDB1 and 2: DNA damage-binding protein 1 and 2 |
| hRAD23B | Protein involved in damage recognition in nucleotide excision repair |
| Cen2 | Centrin-2 |
| RPA | Replication protein A |
| PCNA | Proliferating cell nuclear antigen |
| TFIIH | General transcription factor of RNA polymerase II H |
| CSA, CSB | Proteins of the Cockayne syndrome group: A and B |
| RFC | Replication factor C |
| ERK | MEK/extracellular signal regulated kinase pathway |
| PI-3K/AKT | phosphatidylinositol 3-kinase/Serine/threonine protein kinase B |
| Bcl-2, Bax and Bad | proteins of Bcl-2 family of anti-apoptotic proteins |
| Bcl-xL | B-cell lymphoma-extra large protein |
| RNS | Reactive nitrogen species |
References
- Simões, M.C.; Sousa, J.J.; Pais, A.A. Skin cancer and new treatment perspectives: A review. Cancer Lett. 2015, 357, 8–42. [Google Scholar] [CrossRef] [PubMed]
- D’Orazio, J.; Jarrett, S.; Amaro-Ortiz, A.; Scott, T. UV radiation and the skin. Int. J. Mol. Sci. 2013, 14, 12222–12248. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, D.L.; Saladi, R.N.; Fox, J.L. Ultraviolet radiation and skin cancer. Int. J. Dermatol. 2010, 49, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Boyle, P.; Doré, J.F.; Autier, P.; Ringborg, U. Cancer of the skin: A forgotten problem in Europe. Ann. Oncol. 2004, 15, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Suárez, B.; López-Abente, G.; Martínez, C.; Navarro, C.; Tormo, M.J.; Rosso, S.; Schraub, S.; Gafà, L.; Sancho-Garnier, H.; Wechsler, J.; et al. Occupation and skin cancer: The results of the HELIOS–I multicenter case–control study. BMC Public Health 2007, 7. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, L.; McMorrow, J.; Doolan, P.; McKiernan, E.; Mehta, J.P.; Ryan, E.; Gammell, P.; Joyce, H.; O’Donovan, N.; Walsh, N.; et al. Investigation of the molecular profile of basal cell carcinoma using whole genome microarrays. Mol. Cancer 2006, 5. [Google Scholar] [CrossRef]
- Gordon, R. Skin cancer: An overview of epidemiology and risk factors. Semin. Oncol. Nurs. 2013, 29, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Geller, A.C.; Annas, G.D. Epidemiology of melanoma and nonmelanoma skin cancer. Semin. Oncol. Nurs. 2003, 19, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Lear, J.T.; Szeimies, R.M. Non-melanoma skin cancer. Lancet 2010, 375, 673–685. [Google Scholar] [CrossRef]
- Geller, A.C.; Colditz, G.; Oliveria, S.; Emmons, K.; Jorgensen, C.; Aweh, G.N.; Frazier, A.L. Use of sunscreen, sunburning rates, and tanning bed use among more than 10,000 US children and adolescents. Pediatrics 2002, 109, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Glanz, K.; Buller, D.B.; Saraiya, M. Reducing ultraviolet radiation exposure among outdoor workers: State of the evidence and recommendations. Environ. Health 2007, 6. [Google Scholar] [CrossRef] [PubMed]
- Beissert, S.; Schwarz, T. Mechanisms involved in ultraviolet light-induced immunosuppression. J. Investig. Dermatol. Symp. Proc. 1999, 4, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, S.; Sera, F.; Gandini, S.; Iodice, S.; Caini, S.; Maisonneuve, P.; Fargnoli, M.C. MC1R variants, melanoma and red hair color phenotype: A meta-analysis. Int. J. Cancer 2008, 122, 2753–2760. [Google Scholar] [CrossRef] [PubMed]
- Gandini, S.; Sera, F.; Cattaruzza, M.S.; Pasquini, P.; Zanetti, R.; Masini, C.; Boyle, P.; Melchi, C.F. Meta-analysis of risk factors for cutaneous melanoma: III. Family history, actinic damage and phenotypic factors. Eur. J. Cancer 2005, 41, 2040–2059. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, V.; Stratigos, A.J. Emerging trends in the epidemiology of melanoma. Br. J. Dermatol. 2014, 170, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Udayakumar, D.; Mahato, B.; Gabree, M.; Tsao, H. Genetic determinants of cutaneous melanoma predisposition. Semin. Cutan. Med. Surg. 2010, 29, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; Obenauf, A.C.; Murali, R.; Fried, I.; Griewank, K.G.; Ulz, P.; Windpassinger, C.; Wackernagel, W.; Loy, S.; Wolf, I.; et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat. Genet. 2011, 43, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Valverde, P.; Healy, E.; Jackson, I.; Rees, J.L.; Thody, A.J. Variants of the melanocyte–stimulating hormone receptor gene are associated with red hair and fair skin in humans. Nat. Genet. 1995, 11, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Haussler, M.R.; Whitfield, G.K.; Kaneko, I.; Haussler, C.A.; Hsieh, D.; Hsieh, J.C.; Jurutka, P.W. Molecular mechanisms of vitamin D action. Calcif. Tissue Int. 2013, 92, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Battault, S.; Whiting, S.J.; Peltier, S.L.; Sadrin, S.; Gerber, G.; Maixent, J.M. Vitamin D metabolism, functions and needs: From science to health claims. Eur. J. Nutr. 2013, 52, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Schuster, I. Cytochromes P450 are essential players in the vitamin D signaling system. Biochim. Biophys. Acta 2011, 1814, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Renkema, K.Y.; Alexander, R.T.; Bindels, R.J.; Hoenderop, J.G. Calcium and phosphate homeostasis: Concerted interplay of new regulators. Ann. Med. 2008, 40, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Moreira-Pfrimer, L.D.; Pedrosa, M.A.; Teixeira, L.; Lazaretti-Castro, M. Treatment of vitamin D deficiency increases lower limb muscle strength in institutionalized older people independently of regular physical activity: A randomized double-blind controlled trial. Ann. Nutr. Metab. 2009, 54, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Zittermann, A.; Gummert, J.F. Nonclassical vitamin D action. Nutrients 2010, 2, 408–425. [Google Scholar] [CrossRef] [PubMed]
- Witham, M.D.; Nadir, M.A.; Struthers, A.D. Effect of vitamin D on blood pressure: A systematic review and meta-analysis. J. Hypertens. 2009, 27, 1948–1954. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.H.; Gorman, S.; Finlay-Jones, J.J. Modulation of the immune system by UV radiation: More than just the effects of vitamin D? Nat. Rev. Immunol. 2011, 11, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.L.; Levin, L.I.; Hollis, B.W.; Howard, N.S.; Ascherio, A. Serum 25–hydroxyvitamin D levels and risk of multiple sclerosis. JAMA 2006, 296, 2832–2838. [Google Scholar] [CrossRef] [PubMed]
- Ramagopalan, S.V.; Maugeri, N.J.; Handunnetthi, L.; Lincoln, M.R.; Orton, S.M.; Dyment, D.A.; Deluca, G.C.; Herrera, B.M.; Chao, M.J.; Sadovnick, A.D.; et al. Expression of the multiple sclerosis–associated MHC class II Allele HLA-DRB1*1501 is regulated by vitamin D. PLoS Genet 2009, 5, e1000369. [Google Scholar] [CrossRef] [PubMed]
- Nakai, K.; Fujii, H.; Kono, K.; Goto, S.; Kitazawa, R.; Kitazawa, S.; Hirata, M.; Shinohara, M.; Fukagawa, M.; Nishi, S. Vitamin D activates the Nrf2-Keap1 antioxidant pathway and ameliorates nephropathy in diabetic rats. Am. J. Hypertens. 2014, 27, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Bobilev, I.; Novik, V.; Levi, I.; Shpilberg, O.; Levy, J.; Sharoni, Y.; Studzinski, G.P.; Danilenko, M. The Nrf2 transcription factor is a positive regulator of myeloid differentiation of acute myeloid leukemia cells. Cancer Biol. Ther. 2011, 11, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, G.K.; Hsieh, J.C.; Jurutka, P.W.; Selznick, S.H.; Haussler, C.A.; MacDonald, P.N.; Haussler, M.R. Genomic actions of 1,25-dihydroxyvitamin D3. J. Nutr. 1995, 125, 1690S–1694S. [Google Scholar] [PubMed]
- Norman, A.W. From vitamin D to hormone D: Fundamentals of the vitamin D endocrine system essential for good health. Am. J. Clin Nutr. 2008, 88, 491S–499S. [Google Scholar] [PubMed]
- Ramagopalan, S.V.; Heger, A.; Berlanga, A.J.; Maugeri, N.J.; Lincoln, M.R.; Burrell, A.; Handunnetthi, L.; Handel, A.E.; Disanto, G.; Orton, S.M.; et al. A ChIP–seq defined genome-wide map of vitamin D receptor binding: Associations with disease and evolution. Genome Res. 2010, 20, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Vitamin D cell signalling in health and disease. Biochem. Biophys. Res. Commun. 2015, 460, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Fetahu, I.S.; Höbaus, J.; Kállay, E. Vitamin D and the epigenome. Front. Physiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.; Meurer, M. Vitamin D metabolism. Dermatol. Ther. 2010, 23, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. Vitamin D metabolism, mechanism of action, and clinical applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Saccone, D.; Asani, F.; Bornman, L. Regulation of the vitamin D receptor gene by environment, genetics and epigenetics. Gene 2015, 561, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Mocellin, S. Vitamin D and cancer: Deciphering the truth. Biochim. Biophys. Acta 2011, 1816, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Toner, C.D.; Davis, C.D.; Milner, J.A. The vitamin D and cancer conundrum: Aiming at a moving target. J. Am. Diet. Assoc. 2010, 110, 1492–1500. [Google Scholar] [CrossRef] [PubMed]
- Nürnberg, B.; Gräber, S.; Gärtner, B.; Geisel, J.; Pföhler, C.; Schadendorf, D.; Tilgen, W.; Reichrath, J. Reduced serum 25–hydroxyvitamin D levels in stage IV melanoma patients. Anticancer Res. 2009, 29, 3669–3674. [Google Scholar] [PubMed]
- Moan, J.; Porojnicu, A.C.; Dahlback, A.; Setlow, R.B. Addressing the health benefits and risks, involving vitamin D or skin cancer, of increased sun exposure. Proc. Natl. Acad. Sci. USA 2008, 105, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Ahonen, M.H.; Tenkanen, L.; Teppo, L.; Hakama, M.; Tuohimaa, P. Prostate cancer risk and prediagnostic serum 25-hydroxyvitamin D levels (Finland). Cancer Causes Control 2000, 11, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Tuohimaa, P.; Tenkanen, L.; Ahonen, M.; Lumme, S.; Jellum, E.; Hallmans, G.; Stattin, P.; Harvei, S.; Hakulinen, T.; Luostarinen, T.; et al. Both high and low levels of blood vitamin D are associated with a higher prostate cancer risk: A longitudinal, nested case-control study in the Nordic countries. Int. J. Cancer 2004, 108, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Peters, U.; Albanes, D.; Purdue, M.P.; Abnet, C.C.; Chatterjee, N.; Horst, R.L.; Hollis, B.W.; Huang, W.Y.; Shikany, J.M.; et al. Serum vitamin D concentration and prostate cancer risk: A nested case-control study. J. Natl. Cancer Inst. 2008, 100, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, D.A.; Prindiville, S.; Weiss, D.G.; Willett, W.; VA Cooperative Study Group 380. Risk factors for advanced colonic neoplasia and hyperplastic polyps in asymptomatic individuals. JAMA 2003, 290, 2959–2967. [Google Scholar] [CrossRef] [PubMed]
- Gorham, E.D.; Garland, C.F.; Garland, F.C.; Grant, W.B.; Mohr, S.B.; Lipkin, M.; Newmark, H.L.; Giovannucci, E.; Wei, M.; Holick, M.F. Optimal vitamin D status for colorectal cancer prevention: A quantitative meta analysis. Am. J. Prev. Med. 2007, 32, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Feskanich, D.; Fuchs, C.S.; Willett, W.C.; Hollis, B.W.; Giovannucci, E.L. A nested case control study of plasma 25–hydroxyvitamin D concentrations and risk of colorectal cancer. J. Natl. Cancer Inst. 2007, 99, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Freedman, D.M.; Looker, A.C.; Chang, S.C.; Graubard, B.I. Prospective study of serum vitamin D and cancer mortality in the United States. J. Natl. Cancer Inst. 2007, 99, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.F.; Gorham, E.D.; Mohr, S.B.; Grant, W.B.; Giovannucci, E.L.; Lipkin, M.; Newmark, H.; Holick, M.F.; Garland, F.C. Vitamin D and prevention of breast cancer: Pooled analysis. J. Steroid Biochem. Mol. Biol. 2007, 103, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Robien, K.; Cutler, G.J.; Lazovich, D. Vitamin D intake and breast cancer risk in postmenopausal women: The Iowa Women’s Health Study. Cancer Causes Control 2007, 18, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, P.J.; Ennis, M.; Pritchard, K.I.; Koo, J.; Hood, N. Prognostic effects of 25–hydroxyvitamin D levels in early breast cancer. J. Clin. Oncol. 2009, 27, 3757–3763. [Google Scholar] [CrossRef] [PubMed]
- Kuper, H.; Yang, L.; Sandin, S.; Lof, M.; Adami, H.O.; Weiderpass, E. Prospective study of solar exposure, dietary vitamin D intake, and risk of breast cancer among middle-aged women. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2558–2561. [Google Scholar] [CrossRef] [PubMed]
- McCullough, M.L.; Stevens, V.L.; Patel, R.; Jacobs, E.J.; Bain, E.B.; Horst, R.L.; Gapstur, S.M.; Thun, M.J.; Calle, E.E. Serum 25–hydroxyvitamin D concentrations and postmenopausal breast cancer risk: A nested case control study in the Cancer Prevention Study–II Nutrition Cohort. Breast Cancer Res. 2009, 11. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.; Kost, S.B.; Ennis, B.; Stumpf, W.; Kumar, R. Effect of 1,25-dihydroxyvitamin D3 on mouse mammary tumor (GR) cells: Evidence for receptors, cellular uptake, inhibition of growth and alteration in morphology at physiologic concentrations of hormone. J. Bone Miner Res. 1986, 1, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Skowronski, R.J.; Peehl, D.M.; Feldman, D. Vitamin D and prostate cancer: 1,25 dihydroxyvitamin D3 receptors and actions in human prostate cancer cell lines. Endocrinology 1993, 132, 1952–1960. [Google Scholar] [PubMed]
- Fleet, J.C. Molecular actions of vitamin D contributing to cancer prevention. Mol. Aspects Med. 2008, 29, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Díaz, G.D.; Paraskeva, C.; Thomas, M.G.; Binderup, L.; Hague, A. Apoptosis is induced by the active metabolite of vitamin D3 and its analogue EB1089 in colorectal adenoma and carcinoma cells: Possible implications for prevention and therapy. Cancer Res. 2000, 60, 2304–2312. [Google Scholar] [PubMed]
- Zhuang, S.H.; Burnstein, K.L. Antiproliferative effect of 1α,25-dihydroxyvitamin D3 in human prostate cancer cell line LNCaP involves reduction of cyclin-dependent kinase 2 activity and persistent G1 accumulation. Endocrinology 1998, 139, 1197–1207. [Google Scholar] [PubMed]
- Hill, R.; Song, Y.; Cardiff, R.D.; Van Dyke, T. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell 2005, 123, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Vieth, R.; McCarten, K.; Norwich, KH. Role of 25-hydroxyvitamin D3 dose in determining rat 1,25-dihydroxyvitamin D3 production. Am. J. Physiol. 1990, 258, E780–E789. [Google Scholar] [PubMed]
- Liu, M.; Lee, M.H.; Cohen, M.; Bommakanti, M.; Freedman, L.P. Transcriptional activation of the Cdk inhibitor p21 by vitamin D3 leads to the induced differentiation of the myelomonocytic cell line U937. Genes Dev. 1996, 10, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Holt, P.R.; Arber, N.; Halmos, B.; Forde, K.; Kissileff, H.; McGlynn, K.A.; Moss, S.F.; Kurihara, N.; Fan, K.; Yang, K.; et al. Colonic epithelial cell proliferation decreases with increasing levels of serum 25-hydroxy vitamin D. Cancer Epidemiol. Biomark. Prev. 2002, 11, 113–119. [Google Scholar]
- Hager, G.; Formanek, M.; Gedlicka, C.; Thurnher, D.; Knerer, B.; Kornfehl, J. 1,25(OH)2 vitamin D3 induces elevated expression of the cell cycle-regulating genes P21 and P27 in squamous carcinoma cell lines of the head and neck. Acta Otolaryngol. 2001, 121, 103–109. [Google Scholar] [PubMed]
- Bouillon, R.; Eelen, G.; Verlinden, L.; Mathieu, C.; Carmeliet, G.; Verstuyf, A. Vitamin D and cancer. J. Steroid Biochem. Mol. Biol. 2006, 102, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.; Krishnan, A.V.; Feldman, D. Molecular mechanisms mediating the anti–proliferative effects of Vitamin D in prostate cancer. J. Steroid Biochem. Mol. Biol. 2005, 97, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Boyle, B.J.; Zhao, X.Y.; Cohen, P.; Feldman, D. Insulin-like growth factor binding protein-3 mediates 1α,25-dihydroxyvitamin D3 growth inhibition in the LNCaP prostate cancer cell line through p21/WAF1. J. Urol. 2001, 165, 1319–1324. [Google Scholar] [CrossRef]
- Peng, L.; Malloy, P.J.; Feldman, D. Identification of a functional vitamin D response element in the human insulin–like growth factor binding protein-3 promoter. Mol. Endocrinol. 2004, 18, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- An, B.S.; Tavera–Mendoza, L.E.; Dimitrov, V.; Wang, X.; Calderon, M.R.; Wang, H.J.; White, J.H. Stimulation of Sirt1–regulated FoxO protein function by the ligand–bound vitamin D receptor. Mol. Cell Biol. 2010, 30, 4890–4900. [Google Scholar] [CrossRef] [PubMed]
- Moukayed, M.; Grant, W.B. Molecular link between vitamin D and cancer prevention. Nutritients 2013, 5, 3993–4021. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yu, W.D.; Hershberger, P.A.; Flynn, G.; Kong, R.X.; Trump, D.L.; Johnson, C.S. 1α,25-Dihydroxyvitamin D3 potentiates cisplatin antitumor activity by p73 induction in a squamous cell carcinoma model. Mol. Cancer Ther. 2008, 7, 3047–3055. [Google Scholar] [CrossRef] [PubMed]
- Blutt, S.E.; McDonnell, T.J.; Polek, T.C.; Weigel, N.L. Calcitriol-induced apoptosis in LNCaP cells is blocked by overexpression of Bcl-2. Endocrinology 2000, 141, 10–17. [Google Scholar] [PubMed]
- Swami, S.; Raghavachari, N.; Muller, U.R.; Bao, Y.P.; Feldman, D. Vitamin D growth inhibition of breast cancer cells: Gene expression patterns assessed by cDNA microarray. Breast Cancer Res. Treat 2003, 80, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Sergeev, I.N. Vitamin D and cellular Ca2+ signaling in breast cancer. Anticancer Res. 2012, 32, 299–302. [Google Scholar] [PubMed]
- Jiang, F.; Bao, J.; Li, P.; Nicosia, S.V.; Bai, W. Induction of ovarian cancer cell apoptosis by 1,25-dihydroxyvitamin D3 through the down–regulation of telomerase. J. Biol. Chem. 2004, 279, 53213–53221. [Google Scholar] [CrossRef] [PubMed]
- Høyer–Hansen, M.; Bastholm, L.; Mathiasen, I.S.; Elling, F.; Jäättelä, M. Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death. Cell Death Differ. 2005, 12, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Mantell, D.J.; Owens, P.E.; Bundred, N.J.; Mawer, E.B.; Canfield, A.E. 1α,25-dihydroxyvitamin D(3) inhibits angiogenesis in vitro and in vivo. Circ. Res. 2000, 87, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shoshan, M.; Amir, S.; Dang, D.T.; Dang, L.H.; Weisman, Y.; Mabjeesh, N.J. 1α,25-dihydroxyvitamin D3 (Calcitriol) inhibits hypoxia-inducible factor-1/vascular endothelial growth factor pathway in human cancer cells. Mol. Cancer Ther. 2007, 6, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.; Han, G.; Seshadri, M.; Gillard, B.M.; Yu, W.D.; Foster, B.A.; Trump, D.L.; Johnson, C.S. Role of vitamin D receptor in the antiproliferative effects of calcitriol in tumor-derived endothelial cells and tumor angiogenesis in vivo. Cancer Res. 2009, 69, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.Y.; Yao, J.; Lee, Y.F. 1α,25-Dihydroxyvitamin D3 suppresses interleukin-8-mediated prostate cancer cell angiogenesis. Carcinogenesis 2006, 27, 1883–1893. [Google Scholar] [CrossRef] [PubMed]
- Aparna, R.; Subhashini, J.; Roy, K.R.; Reddy, G.S.; Robinson, M.; Uskokovic, M.R.; Venkateswara Reddy, G.; Reddanna, P. Selective inhibition of cyclooxygenase-2 (COX-2) by 1α,25-dihydroxy-16-ene-23-yne-vitamin D3, a less calcemic vitamin D analog. J. Cell Biochem. 2008, 104, 1832–1842. [Google Scholar] [CrossRef] [PubMed]
- Sung, V.; Feldman, D. 1,25-Dihydroxyvitamin D3 decreases human prostate cancer cell adhesion and migration. Mol. Cell Endocrinol. 2000, 164, 133–143. [Google Scholar] [CrossRef]
- Liu, W.; Guo, M.; Ezzat, S.; Asa, S.L. Vitamin D inhibits CEACAM1 to promote insulin/IGF-I receptor signaling without compromising anti-proliferative action. Lab Investig. 2011, 91, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.Y.; Yeh, S.D.; Lee, Y.F. 1α,25-dihydroxyvitamin D3 inhibits prostate cancer cell invasion via modulation of selective proteases. Carcinogenesis 2006, 27, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Baek, M.S.; Yoon, D.S.; Park, J.S.; Yoon, B.W.; Oh, B.S.; Park, J.; Kim, H.J. Vitamin D inhibits expression and activity of matrix metalloproteinase in human lung fibroblasts (HFL-1) cells. Tuberc Respir Dis. 2014, 77, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Díaz, L.; Díaz-Muñoz, M.; García-Gaytán, A.C.; Méndez, I. Mechanistic effects of calcitriol in cancer biology. Nutrients 2015, 7, 5020–5050. [Google Scholar] [CrossRef] [PubMed]
- Saunders, D.E.; Christensen, C.; Wappler, N.L.; Schultz, J.F.; Lawrence, W.D.; Malviya, V.K.; Malone, J.M.; Deppe, G. Inhibition of c-myc in breast and ovarian carcinoma cells by 1,25-dihydroxyvitamin D3, retinoic acid and dexamethasone. Anticancer Drugs 1993, 4, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Kasiappan, R.; Shen, Z.; Tse, A.K.; Jinwal, U.; Tang, J.; Lungchukiet, P.; Sun, Y.; Kruk, P.; Nicosia, S.V.; Zhang, X.; et al. 1,25-Dihydroxyvitamin D3 suppresses telomerase expression and human cancer growth through microRNA-498. J. Biol. Chem. 2012, 287, 41297–41309. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.B.; Goetsch, P.D.; Pike, J.W. VDR/RXR and TCF4/β-catenin cistromes in colonic cells of colorectal tumor origin: Impact on c-FOS and c-MYC gene expression. Mol. Endocrinol. 2012, 26, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Candeliere, G.A.; Jurutka, P.W.; Haussler, M.R.; St-Arnaud, R. A composite element binding the vitamin D receptor, retinoid X receptor α, and a member of the CTF/NF-1 family of transcription factors mediates the vitamin D responsiveness of the c-FOS promoter. Mol. Cell Biol. 1996, 16, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Okano, K.; Usa, T.; Ohtsuru, A.; Tsukazaki, T.; Miyazaki, Y.; Yonekura, A.; Namba, H.; Shindoh, H.; Yamashita, S. Effect of 22-oxa-1,25-dihydroxyvitamin D3 on human thyroid cancer cell growth. Endocr. J. 1999, 46, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Camacho, J. Ether à go-go potassium channels and cancer. Cancer Lett. 2006, 233, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cázares-Ordoñez, V.; González-Duarte, R.J.; Díaz, L.; Ishizawa, M.; Uno, S.; Ortíz, V.; Ordoñez-Sánchez, M.L.; Makishima, M.; Larrea, F.; Avila, E. A cis-acting element in the promoter of human ether à go-go 1 potassium channel gene mediates repression by calcitriol in human cervical cancer cells. Biochem. Cell Biol. 2015, 93, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. Vitamin D receptor, a tumor suppressor in skin. Can J. Physiol. Pharmacol. 2015, 93, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Mancebo, S.E.; Wang, S.Q. Skin cancer: Role of ultraviolet radiation in carcinogenesis. Rev. Environ. Health 2014, 29, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.P.; Häder, D.P. UV-induced DNA damage and repair: A review. Photochem. Photobiol. Sci. 2002, 1, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.G.; Tsaalbi-Shtylik, A.; Hendriks, G.; Verspuy, J.; Gali, H.; Haracska, L.; de Wind, N. Mammalian polymerase zeta is essential for post-replication repair of UV-induced DNA lesions. DNA Repair (Amst) 2009, 8, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- Tommasi, S.; Swiderski, P.M.; Tu, Y.; Kaplan, B.E.; Pfeifer, G.P. Inhibition of transcription factor binding by ultraviolet-induced pyrimidine dimers. Biochemistry 1996, 35, 15693–15703. [Google Scholar] [CrossRef] [PubMed]
- Donahue, B.A.; Yin, S.; Taylor, J.S.; Reines, D.; Hanawalt, P.C. Transcript cleavage by RNA polymerase II arrested by a cyclobutane pyrimidine dimer in the DNA template. Proc. Natl. Acad. Sci. USA 1994, 91, 8502–8506. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Traganos, F.; Darzynkiewicz, Z. Kinetics of the UV-induced DNA damage response in relation to cell cycle phase. Correlation with DNA replication. Cytometry A 2010, 77, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Soehnge, H.; Ouhtit, A.; Ananthaswamy, H.N. mechanisms of induction of skin cancer by UV radiation. Front. Biosci. 1997, 2, 538–551. [Google Scholar]
- Bossi, O.; Gartsbein, M.; Leitges, M.; Kuroki, T.; Grossman, S.; Tennenbaum, T. UV irradiation increases ROS production via PKCdelta signaling in primary murine fibroblasts. J. Cell Biochem. 2008, 105, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shao, Y.; Zhou, J.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation-induces epidermal growth factor receptor (EGFR) nuclear translocation in human keratinocytes. J. Cell Biochem. 2009, 107, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, S.; Tsai, K.Y. Roles of the immune system in skin cancer. Br. J. Dermatol. 2011, 165, 953–965. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.R.; McGibbon, D.; Stefanini, M. Xeroderma pigmentosum. Orphanet J. Rare Dis. 2011, 70. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, A.; Sarasin, A. DNA damage and gene therapy of xeroderma pigmentosum, a human DNA repair-deficient disease. Mutat. Res. 2015, 776. [Google Scholar] [CrossRef] [PubMed]
- Bradford, P.T.; Goldstein, A.M.; Tamura, D.; Khan, S.G.; Ueda, T.; Boyle, J.; Oh, K.S.; Imoto, K.; Inui, H.; Moriwaki, S.; et al. Cancer and neurologic degeneration in xeroderma pigmentosum: Long term follow-up characterises the role of DNA repair. J. Med. Genet. 2011, 48, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Young, L.C.; Listgarten, J.; Trotter, M.J.; Andrew, S.E.; Tron, V.A. Evidence that dysregulated DNA mismatch repair characterizes human nonmelanoma skin cancer. Br. J. Dermatol. 2008, 158, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Yu, D.; Cho, Y.Y.; Bode, A.M.; Ma, W.; Yao, K.; Li, S.; Li, J.; Bowden, G.T.; Dong, Z. Sunlight UV-induced skin cancer relies upon activation of the p38α signaling pathway. Cancer Res. 2013, 73, 2181–2188. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, Y.; Stratton, S.P.; Curiel-Lewandrowski, C.; Warneke, J.; Hu, C.; Bowden, G.T.; Dickinson, S.E.; Dong, Z.; Bode, A.M.; Saboda, K.; et al. Activation of the PI3K/Akt/mTOR and MAPK signaling pathways in response to acute solar-simulated light exposure of human skin. Cancer Prev. Res. 2015, 8, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Yeung, P.L.; Chiang, A.K. Induction of MAPK- and ROS-dependent autophagy and apoptosis in gastric carcinoma by combination of romidepsin and bortezomib. Oncotarget 2015. [Google Scholar] [CrossRef]
- Strozyk, E.; Kulms, D. The role of AKT/mTOR pathway in stress response to UV-irradiation: Implication in skin carcinogenesis by regulation of apoptosis, autophagy and senescence. Int. J. Mol. Sci. 2013, 14, 15260–15285. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, V.; Piva, T.J. A comparative study of UV-induced cell signalling pathways in human keratinocyte-derived cell lines. Arch. Dermatol. Res. 2013, 305, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Hess, M.T.; Schwitter, U.; Petretta, M.; Giese, B.; Naegeli, H. Bipartite substrate discrimination by human nucleotide excision repair. Proc. Natl. Acad. Sci. USA 1997, 94, 6664–6669. [Google Scholar] [CrossRef] [PubMed]
- Noll, D.M.; Mason, T.M.; Miller, P.S. Formation and repair of interstrand cross-links in DNA. Chem. Rev. 2006, 106, 277–301. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, K.; Okamoto, T.; Shimizu, Y.; Masutani, C.; Iwai, C.; Hanaoka, F. A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 2001, 15, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Fitch, M.E.; Nakajima, S.; Yasui, A.; Ford, J.M. In vivo recruitment of XPC to UV-induced cyclobutane pyrimidine dimers by the DDB2 gene product. J. Biol. Chem. 2003, 278, 46906–46910. [Google Scholar] [CrossRef] [PubMed]
- Bunick, C.G.; Miller, M.R.; Fuller, B.E.; Fanning, E.; Chazin, W.J. Biochemical and structural domain analysis of xeroderma pigmentosum complementation group C protein. Biochemistry 2006, 45, 14965–14979. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, J.E. Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat. Rev. Cancer 2005, 5, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, Y. Reversing DNA methylation: Mechanism, genomic and biological functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Farrell, A.W.; Halliday, G.M.; Lyons, J.G. Chromatin structure following UV-induced DNA damage— Repair or death? Int. J. Mol. Sci. 2011, 12, 8063–8085. [Google Scholar] [CrossRef] [PubMed]
- De Haes, P.; Garmyn, M.; Verstuyf, A.; De Clercq, P.; Vandewalle, M.; Degreef, H.; Vantieghem, K.; Bouillon, R.; Segaert, S. 1,25-Dihydroxyvitamin D3 and analogues protect primary human keratinocytes against UVB-induced DNA damage. J. Photochem. Photobiol. B 2005, 78, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Al Mahroos, M.; Yaar, M.; Phillips, T.J.; Bhawan, J.; Gilchrest, B.A. Effect of sunscreen application on UV-induced thymine dimers. Arch. Dermatol. 2002, 138, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, S.K. Skin photoprotection by green tea: Antioxidant and immunomodulatory effects. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2003, 3, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Antille, C.; Tran, C.; Sorg, O.; Carraux, P.; Didierjean, L.; Saurat, J.H. Vitamin A exerts a photoprotective action in skin by absorbing ultraviolet B radiation. J. Investig. Dermatol. 2003, 121, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Saladi, R.; Lu, Y.; Wang, Y.; Palep, S.R.; Moore, J.; Phelps, R.; Shyong, E.; Lebwohl, M.G. Isoflavone genistein: Photoprotection and clinical implications in dermatology. J. Nutr. 2003, 133, 3811S–3819S. [Google Scholar] [PubMed]
- Lee, C.H.; Wu, S.B.; Hong, C.H.; Yu, H.S.; Wei, Y.H. Molecular Mechanisms of UV-Induced Apoptosis and Its Effects on Skin Residential Cells: The Implication in UV-Based Phototherapy. Int. J. Mol. Sci. 2013, 14, 6414–6435. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.S.; Reichrath, J. Sunlight vitamin D and skin cancer. Anticancer Agents Med. Chem. 2013, 13, 83–97. [Google Scholar] [CrossRef] [PubMed]
- De Haes, P.; Garmyn, M.; Carmeliet, G.; Degreef, H.; Vantieghem, K.; Bouillon, R.; Segaert, S. Molecular pathways involved in the anti-apoptotic effect of 1,25-dihydroxyvitamin D3 in primary human keratinocytes. J. Cell Biochem. 2004, 93, 951–967. [Google Scholar] [CrossRef] [PubMed]
- De Haes, P.; Garmyn, M.; Degreef, H.; Vantieghem, K.; Bouillon, R.; Segaert, S. 1,25-Dihydroxyvitamin D3 inhibits ultraviolet B-induced apoptosis, Jun kinase activation, and interleukin-6 production in primary human keratinocytes. J. Cell Biochem. 2003, 89, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Fukuya, Y.; Higaki, M.; Higaki, Y.; Kawashima, M. Effect of vitamin D3 on the increased expression of Bcl-xL in psoriasis. Arch. Dermatol. Res. 2002, 293, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Webster, M.R.; Kugel, C.H., 3rd; Weeraratna, A.T. The Wnts of change: How Wnts regulate phenotype switching in melanoma. Biochim. Biophys. Acta 2015, 1856, 244–251. [Google Scholar] [PubMed]
- Gordon-Thomson, C.; Gupta, R.; Tongkao-on, W.; Ryan, A.; Halliday, G.M.; Mason, R.S. 1α,25 dihydroxyvitamin D3 enhances cellular defences against UV-induced oxidative and other forms of DNA damage in skin. Photochem. Photobiol. Sci. 2012, 11, 1837–1847. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, H.; Halliwell, B. Damage to DNA by reactive oxygen and nitrogen species: Role in inflammatory disease and progression to cancer. Biochem. J. 1996, 313, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.; LaRusso, N.F.; Burgart, L.J.; Gores, G.J. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. 2000, 60, 184–190. [Google Scholar] [PubMed]
- Bau, D.T.; Gurr, J.R.; Jan, K.Y. Nitric oxide is involved in arsenite inhibition of pyrimidine dimer excision. Carcinogenesis 2001, 22, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.H.; Wei, W.; Liu, L. Regulation of DNA repair by S-nitrosylation. Biochim. Biophys. Acta 2012, 1820, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Dixon, K.M.; Deo, S.S.; Holliday, C.J.; Slater, M.; Halliday, G.M.; Reeve, V.E.; Mason, R.S. Photoprotection by 1,25 dihydroxyvitamin D3 is associated with an increase in p53 and a decrease in nitric oxide products. J. Investig. Dermatol. 2007, 127, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Demetriou, S.K.; Ona-Vu, K.; Teichert, A.E.; Cleaver, J.E.; Bikle, D.D.; Oh, D.H. Vitamin D receptor mediates DNA repair and is UV inducible in intact epidermis but not in cultured keratinocytes. J. Investig. Dermatol. 2012, 132, 2097–2100. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.; Oh, D. Efficient repair of UV-induced DNA damage in terminally differentiated keratinocytes. J. Investig. Dermatol. 2002, 119. [Google Scholar] [CrossRef]
- Bohr, V.A. Preferential DNA repair in active genes. Dan. Med. Bull. 1987, 34, 309–320. [Google Scholar] [PubMed]
- Hanawalt, P.C. Revisiting the rodent repairadox. Environ. Mol. Mutagen. 2001, 38, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.P.; Kim, M.J.; Jung, M.Y.; Jeon, H.; Goo, J.; Ahn, S.K.; Lee, S.H.; Elias, P.M.; Choi, E.H. Biopositive effects of low-dose UVB on epidermis: Coordinate upregulation of antimicrobial peptides and permeability barrier reinforcement. J. Investig. Dermatol. 2008, 128, 2880–2887. [Google Scholar] [CrossRef] [PubMed]
- Lesiak, A.; Narbutt, J.; Wodz, K.; Pawliczak, R.; Rogowski-Tylman, M.; Sysa-Jedrzejowska, A.; Young, A.R. Repeated suberythemal UVB preexposure protects against high-dose UVB-induced expression of vitamin D receptor protein in human Skin. J. Investig. Dermatol. 2011, 131, 2332–2335. [Google Scholar] [CrossRef] [PubMed]
- Mallbris, L.; Edström, D.W.; Sundblad, L.; Granath, F.; Stahle, M. UVB upregulates the antimicrobial protein hCAP18 mRNA in human skin. J. Investig. Dermatol. 2005, 125, 1072–1074. [Google Scholar] [CrossRef] [PubMed]
- Courtois, S.J.; Segaert, S.; Degreef, H.; Bouillon, R.; Garmyn, M. Ultraviolet B suppresses vitamin D receptor gene expression in keratinocytes. Biochem. Biophys. Res. Commun. 1998, 246, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Segaert, S.; Garmyn, M.; Degreef, H.; Bouillon, R. Retinoic acid modulates the anti-proliferative effect of 1,25-dihydroxyvitamin D3 in cultured human epidermal keratinocytes. J. Investig Dermatol. 1997, 109, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Reichrath, J.; Rass, K. Ultraviolet damage, DNA repair and vitamin D in nonmelanoma skin cancer and in malignant melanoma: An update. Adv. Exp. Med. Biol. 2014, 810, 208–233. [Google Scholar] [PubMed]
- Lehmann, A.R. The xeroderma pigmentosum group D (XPD) gene: One gene, two functions, three diseases. Genes Dev. 2001, 15, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Drané, P.; Compe, E.; Catez, P.; Chymkowitch, P.; Egly, J.M. Selective regulation of vitamin D receptor-responsive genes by TFIIH. Mol. Cell 2004, 16, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Moll, P.R.; Sander, V.; Frischauf, A.M.; Richter, K. Expression profiling of vitamin D treated primary human keratinocytes. J. Cell. Biochem. 2007, 100, 574–592. [Google Scholar] [CrossRef] [PubMed]
- Le May, N.; Egly, J.M.; Coin, F. True lies: The double life of the nucleotide excision repair factors in transcription and DNA repair. J. Nucleic Acids 2010. [Google Scholar] [CrossRef] [PubMed]
- Bienertová-Vašků, J.; Drábová, K.; Zlámal, F.; Tomandl, J.; Kýr, M.; Šplíchal, Z.; Štěrba, J. Pre-treatment VD levels and VDR receptors as potential predictors of occurrence and overall survival in paediatric patients with solid tumours—A single institution pilot study. Tumour Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- So, J.Y.; Suh, N. Targeting cancer stem cells in solid tumors by vitamin D. J. Steroid Biochem. Mol. Biol. 2015, 148, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Charehbili, A.; Hamdy, N.A.; Smit, V.T.; Kessels, L.; van Bochove, A.; van Laarhoven, H.W.; Putter, H.; Meershoek-Klein Kranenbarg, E.; van Leeuwen-Stok, A.E.; van der Hoeven, J.J.; et al. Dutch Breast Cancer Research Group (BOOG). Vitamin D (25-0H D3) status and pathological response to neoadjuvant chemotherapy in stage II/III breast cancer: Data from the NEOZOTAC trial (BOOG 10–01). Breast 2015. [Google Scholar] [CrossRef]
- Leiter, U.; Eigentler, T.; Garbe, C. Epidemiology of skin cancer. Adv. Exp. Med. Biol. 2014, 810. [Google Scholar] [CrossRef]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawlowska, E.; Wysokinski, D.; Blasiak, J. Nucleotide Excision Repair and Vitamin D—Relevance for Skin Cancer Therapy. Int. J. Mol. Sci. 2016, 17, 372. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17040372
Pawlowska E, Wysokinski D, Blasiak J. Nucleotide Excision Repair and Vitamin D—Relevance for Skin Cancer Therapy. International Journal of Molecular Sciences. 2016; 17(4):372. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17040372
Chicago/Turabian StylePawlowska, Elzbieta, Daniel Wysokinski, and Janusz Blasiak. 2016. "Nucleotide Excision Repair and Vitamin D—Relevance for Skin Cancer Therapy" International Journal of Molecular Sciences 17, no. 4: 372. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17040372