
Identification of Serum Peptidome Signatures of Non-Small Cell Lung Cancer

 ,
,
Abstract
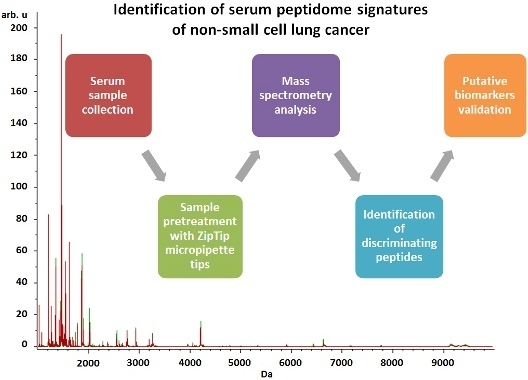
:
1. Introduction
2. Results
2.1. Characteristics of the Study Participants
2.2. Reproducibility Evaluation
2.3. Serum Peptide Profiling
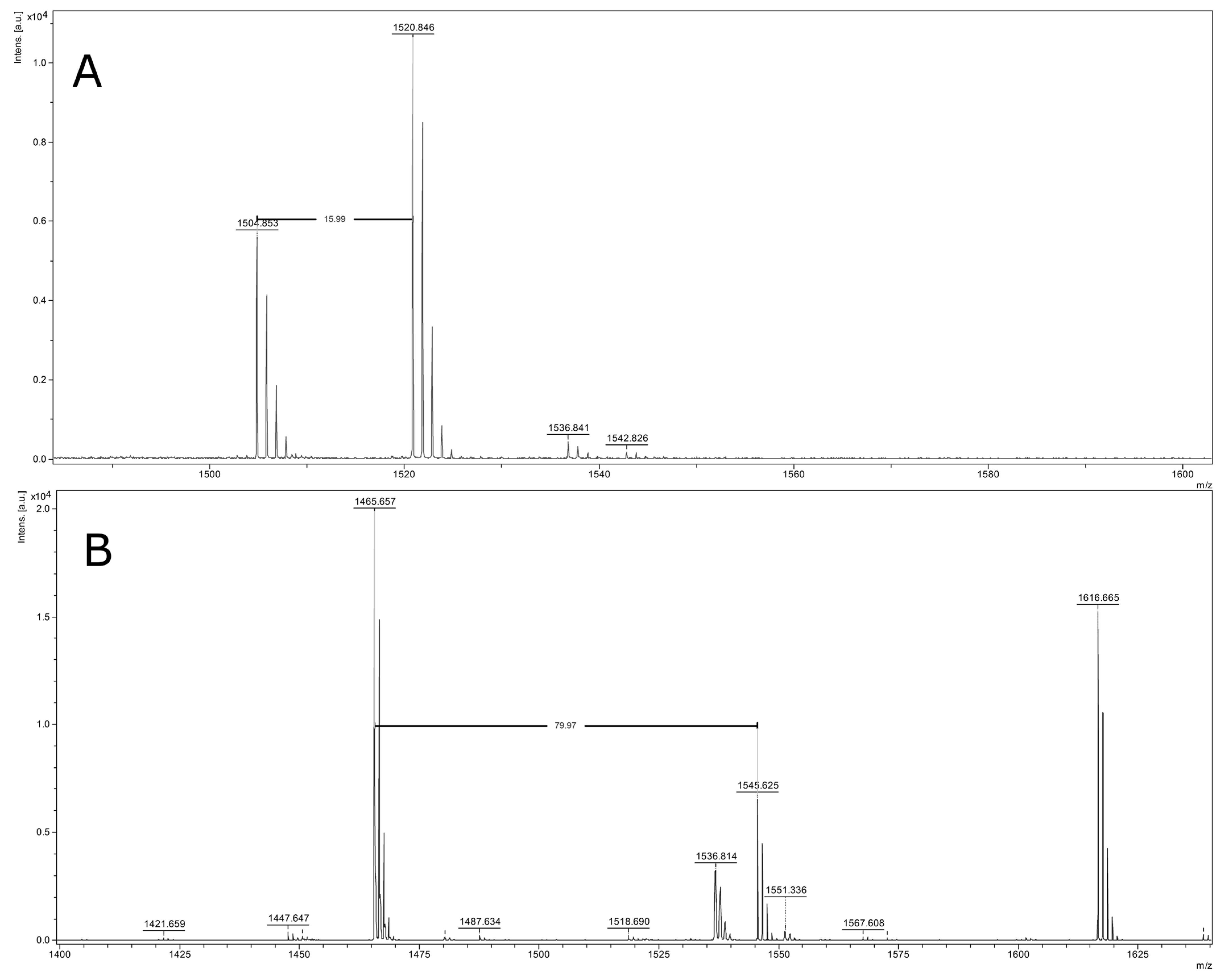
2.4. Identification of Peptide Ions
3. Discussion
4. Materials and Methods
4.1. Characteristics of the Study Participants
4.2. Chemicals and Reagents
4.3. Serum Samples Pretreatment
4.4. MALDI-TOF-MS Analysis
4.5. LC-MS/MS Identification of Selected Ion Peptides
4.6. Determination of Complement C3 by ELISA
4.7. Data Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MALDI-TOF-MS | Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry |
| LC | Lung cancer |
| NSCLC | Non-small cell lung cancer |
| ROC curve | Receiver operating characteristic curve |
| AUC | Area under the ROC curve |
| GA | Genetic algorithms |
| QC | Quick classifier |
| SNN | Supervised neural network |
| TFA | Trifluoroacetic acid |
| ACN | Acetonitrile |
| HCCA | α-Cyano-4-hydroxycinnamic acid |
References and Notes
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics: 2011. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Thakur, R.K.; Yadav, V.K.; Nanda, R.; Chowdhury, S.; Agrawal, A. Lung cancer biomarkers: State of the art. J. Carcinog. 2013, 12, 3. [Google Scholar] [PubMed]
- Patz, E.F.; Campa, M.J.; Gottlin, E.B.; Kusmartseva, I.; Xiang, R.G.; Herndon, J.E. Panel of serum biomarkers for the diagnosis of lung cancer. J. Clin. Oncol. 2007, 25, 5578–5583. [Google Scholar] [CrossRef] [PubMed]
- Patz, E.F.; Pinsky, P.; Gatsonis, C.; Sicks, J.D.; Kramer, B.S.; Tammemägi, M.C.; Chiles, C.; Black, W.C.; Aberle, D.R. Overdiagnosis in low-dose computed tomography screening for lung cancer. JAMA Intern. Med. 2014, 174, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nishiumi, S.; Kobayashi, K.; Shinohara, M.; Hatakeyama, Y.; Kotani, Y.; Hatano, N.; Maniwa, Y.; Nishio, W.; Bamba, T.; et al. A metabolomic approach to lung cancer. Lung Cancer 2011, 74, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, P.J.; Wang, X.F.; Beukemann, M.; Zhang, Q.; Seeley, M.; Mohney, R.; Holt, T.; Pappan, K.L. Metabolite profiles of the serum of patients with non-small cell carcinoma. J. Thorac Oncol. 2016, 11, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Musharraf, S.G.; Mazhar, S.; Choudhary, M.I.; Rizi, N.; Atta-ur-Rahman, A. Plasma metabolite profiling and chemometric analyses of lung cancer along with three controls through gas chromatography-mass spectrometry. Sci. Rep. 2015, 5, 8607. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ma, Z.; Li, A.; Li, H.; Wang, B.; Zhong, J.; Min, L.; Dai, L. Metabolomic profiling of human serum in lung cancer patients using liquid chromatography/hybrid quadrupole time-of-flight mass spectrometry and gas chromatography/mass spectrometry. J. Cancer Res. Clin. Oncol. 2015, 141, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Taylor, S.; Barupal, D.; Taguchi, A.; Wohlgemuth, G.; Wikoff, W.; Yoneda, K.; Gandara, D.; Hanash, S.; Kim, K.; et al. Systemic metabolomic changes in blood samples of lung cancer patients identified by gas chromatography time-of-flight mass spectrometry. Metabolites 2015, 5, 192–210. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Song, X.; Zhao, X.; Zou, L.; Xu, G. Serum metabolic profiling study of lung cancer using ultra high performance liquid chromatography/quadrupole time-of-flight mass spectrometry. J. Chromatogr. B. 2014, 966, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ma, Z.; Min, L.; Li, H.; Wang, B.; Zhong, J.; Dai, L. Biomarker identification and pathway analysis by serum metabolomics of lung cancer. Biomed. Res. Int. 2015, 2015, 183624. [Google Scholar] [CrossRef] [PubMed]
- Maeda, J.; Higashiyama, M.; Imaizumi, A.; Nakayama, T.; Yamamoto, H.; Daimon, T.; Yamakado, M.; Imamura, F.; Kodama, K. Possibility of multivariate function composed of plasma amino acid profiles as a novel screening index for non-small cell lung cancer: A case control study. BMC Cancer 2010, 10, 690. [Google Scholar] [CrossRef] [PubMed]
- Shingyoji, M.; Iizasa, T.; Higashiyama, M.; Imamura, F.; Saruki, N.; Imaizumi, A.; Yamamoto, H.; Daimon, T.; Tochikubo, O.; Mitsushima, T.; et al. The significance and robustness of a plasma free amino acid (PFAA) profile-based multiplex function for detecting lung cancer. BMC Cancer 2013, 13, 77. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.A.; Petricoin, E.F. Serum peptidome for cancer detection: Spinning biologic trash into diagnostic gold. J. Clin. Investig. 2006, 116, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.F.; Mikulskis, A.; Kuzdzal, S.; Golenko, E.; Petricoin, E.F.; Liotta, L.A.; Patton, W.F.; Whiteley, G.R.; Rosenblatt, K.; Gurnani, P.; et al. A novel, high-throughput workflow for discovery and identification of serum carrier protein-bound peptide biomarker candidates in ovarian cancer samples. Clin. Chem. 2007, 53, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J. Tumor markers in detection of lung cancer. Adv. Clin. Chem. 2006, 42, 1–41. [Google Scholar] [PubMed]
- Tas, F.; Aydiner, A.; Topuz, E.; Yasasever, V.; Karadeniz, A.; Saip, P. Utility of the serum tumor markers: CYFRA 21.1, carcinoembryonic antigen (CEA), and squamous cell carcinoma antigen (SCC) in squamous cell lung cancer. J. Exp. Clin. Canc. Res. 2000, 19, 477–481. [Google Scholar]
- Kulpa, J.; Wojcik, E.; Reinfuss, M.; Kolodziejski, L. Carcinoembryonic antigen, squamous cell carcinoma antigen, CYFRA 21–1, and neuron-specific enolase in squamous cell lung cancer patients. Clin. Chem. 2002, 48, 1931–1937. [Google Scholar] [PubMed]
- Indovina, P.; Marcelli, E.; Pentimalli, F.; Tanganelli, P.; Tarro, G.; Giordano, A. Mass spectrometry-based proteomics: The road to lung cancer biomarker discovery. Mass Spectrom. Rev. 2012, 32, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Bajtarevic, A.; Ager, C.; Pienz, M.; Klieber, M.; Schwarz, K.; Ligor, M.; Ligor, T.; Filipiak, W.; Denz, H.; Fiegl, M.; et al. Noninvasive detection of lung cancer by analysis of exhaled breath. BMC Cancer 2009, 9, 348. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Tisch, U.; Adams, O.; Hakim, M.; Shehada, N.; Broza, Y.Y.; Billan, S.; Abdah-Bortnyak, R.; Kuten, A.; Haick, H. Diagnosing lung cancer in exhaled breath using gold nanoparticles. Nat. Nanotechnol. 2009, 4, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.; Altorki, N.; Austin, J.H.M.; Cameron, R.B.; Cataneo, R.N.; Greenberg, J.; Kloss, R.; Maxfield, R.A.; Munawar, M.I.; Pass, H.I.; et al. Prediction of lung cancer using volatile biomarkers in breath. Cancer Biomark. 2007, 3, 95–109. [Google Scholar] [PubMed]
- Poli, D.; Carbognani, P.; Corradi, M.; Goldoni, M.; Acampa, O.; Balbi, B.; Bianchi, L.; Rusca, M.; Mutti, A. Exhaled volatile organic compounds in patients with non-small cell lung cancer: Cross sectional and nested short-term follow-up study. Respir. Res. 2005, 6, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Xiao, X.; Zhang, W.; Zhang, L.; Zhang, W.; Zhou, B.; Chen, G.; He, D. Application of serum SELDI proteomic patterns in diagnosis of lung cancer. BMC Cancer 2005, 5, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hocker, J.R.; Peyton, M.D.; Lerner, M.R.; Mitchell, S.L.; Lightfoot, S.A.; Lander, T.J.; Bates-Albers, L.M.; Vu, N.T.; Hanas, R.J.; Kupiec, T.C.; et al. Serum discrimination of early-stage lung cancer patients using electrospray-ionization mass spectrometry. Lung Cancer 2011, 74, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Han, M.K.; Oh, Y.H.; Kang, J.; Kim, Y.P.; Seo, S.; Kim, J.; Park, K.; Kim, H.S. Protein profiling in human sera for identification of potential lung cancer biomarkers using antibody microarray. Proteomics 2009, 9, 5544–5552. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, V.E.; Arnotskaya, N.E.; Zaridze, D.G. Detection of lung cancer using plasma protein profiling by matrix-assisted laser desorption/ionization mass spectrometry. Eur. J. Mass Spectrom. 2010, 16, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.L.; Yang, S.Y.; Du, J.; Tian, Y.X.; Bu, L.N.; Huo, S.F.; Wang, F.P.; Nan, Y.D. Detection of lung adenocarcinoma using magnetic beads based matrix-assisted laser desorption/ionization time-of-flight mass spectrometry serum protein profiling. Chin. Med. J. 2010, 123, 34–39. [Google Scholar] [PubMed]
- Musharraf, S.G.; Hashmi, N.; Choudhary, M.I.; Rizvi, N.; Usman, A.; Atta-ur-Rahman, A. Comparison of plasma from healthy nonsmokers, smokers, and lung cancer patients: Pattern-based differentiation profiling of low molecular weight proteins and peptides by magnetic bead technology with MALDI-TOF MS. Biomarkers 2012, 17, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Sandanayake, N.S.; Camuzeaux, S.; Sinclair, J.; Blyuss, O.; Andreola, F.; Chapman, M.H.; Webster, G.J.; Smith, R.C.; Timms, J.F.; Pereira, S.P. Identification of potential serum peptide biomarkers of biliary tract cancer using MALDI MS profiling. BMC Clin. Pathol. 2014, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Abramowicz, A.; Wojakowska, A.; Gdowicz-Klosok, A.; Polanska, J.; Rodziewicz, P.; Polanowski, P.; Namysl-Kaletka, A.; Pietrowska, M.; Wydmanski, J.; Widlak, P. Identification of serum proteome signatures of locally advanced and metastatic gastric cancer: A pilot study. J. Transl. Med. 2015, 13, 304. [Google Scholar] [CrossRef] [PubMed]
- Fania, C.; Sogno, I.; Vasso, M.; Torretta, E.; Leone, R.; Bruno, A.; Consonni, P.; Albini, A.; Gelfi, C. A PSA-guided approach for a better diagnosis of prostatic adenocarcinoma based on MALDI profiling and peptide identification. Clin. Chim. Acta 2015, 439, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, J.T.; Somers, K.D.; Cazares, L.H.; Malik, G.; Adam, B.L.; Stack, B.C.; Wright, G.L.; Semmes, O.J. Serum protein profiles to identify head and neck cancer. Clin. Cancer Res. 2004, 10, 1625–1632. [Google Scholar] [CrossRef] [PubMed]
- Pietrowska, M.; Polanska, J.; Marczak, L.; Behrendt, K.; Nowicka, E.; Stobiecki, M.; Polanski, A.; Tarnawski, R.; Widlak, P. Mass spectrometry-based analysis of therapy-related changes in serum proteome patterns of patients with early-stage breast cancer. J. Transl. Med. 2010, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.A.; Petricoin, E.F. Mass spectrometry-based protein biomarker discovery: Solving the remaining challenges to reach the promise of clinical benefit. Clin. Chem. 2010, 56, 1641–1642. [Google Scholar] [CrossRef] [PubMed]
- Tiss, A.; Smith, C.; Camuzeaux, S.; Kabir, M.; Gayther, S.; Menon, U.; Waterfield, M.; Timms, J.; Jacobs, I.; Cramer, R. Serum peptide profiling using MALDI mass spectrometry: Avoiding the pitfalls of coated magnetic beads using well-established ZipTip technology. Proteomics 2007, 1, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, J.; Matysiak, J.; Kokot, P.; Nowicki, P.; Dereziński, P.; Kokot, Z.J. The application of fuzzy statistics and linear discriminant analysis as criteria for optimizing the preparation of plasma for matrix-assisted laser desorption/ionization mass spectrometry peptide profiling. Clin. Chim. Acta 2015, 448, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Callesen, A.K.; Madsen, J.S.; Vach, W.; Kruse, T.A.; Mogensen, O.; Jensen, O.N. Serum protein profiling by solid phase extraction and mass spectrometry: A future diagnostics tool? Proteomics 2009, 9, 1428–1441. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, J.; Klupczynska, A.; Dereziński, P.; Matysiak, J.; Kokot, P.; Nowak, D.; Gajęcka, M.; Nowak-Markwitz, E.; Kokot, Z.J. A combined metabolomic and proteomic analysis of gestational diabetes mellitus. Int. J. Mol. Sci. 2015, 16, 30034–30045. [Google Scholar] [CrossRef] [PubMed]
- Pio, R.; Ajona, D.; Lambris, J.D. Complement inhibition in cancer therapy. Semin. Immunol. 2013, 25, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, M.J.; Sughrue, M.E.; Kane, A.J.; Mills, S.A.; Parsa, A.T. Cancer and the complement cascade. Mol. Cancer Res. 2010, 8, 1453–1465. [Google Scholar] [CrossRef] [PubMed]
- Maness, P.F.; Orengo, A. Serum complement levels in patients with digestive tract carcinomas and other neoplastic diseases. Oncology 1977, 34, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Ytting, H.; Jensenius, J.C.; Christensen, I.J.; Thiel, S.; Nielsen, H.J. Increased activity of the mannan-binding lectin complement activation pathway in patients with colorectal cancer. Scand. J. Gastroenterol. 2004, 39, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Markiewski, M.M.; DeAngelis, R.A.; Benencia, F.; Ricklin-Lichtsteiner, S.K.; Koutoulaki, A.; Gerard, C.; Coukos, G.; Lambris, J.D. Modulation of the antitumor immune response by complement. Nat. Immunol. 2008, 9, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Gmiński, J.; Mykała-Cieśla, J.; Machalski, M.; Drózdz, M.; Najda, J. Immunoglobulins and complement components in patients with lung cancer. Tuberk. Toraks 2004, 30, 39–44. [Google Scholar]
- Corrales, L.; Ajona, D.; Rafail, S.; Lasarte, J.J.; Riezu-Boj, J.I.; Lambris, J.D.; Rouzaut, A.; Pajares, M.J.; Montuenga, L.M.; Pio, R. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J. Immunol. 2012, 189, 4674–4683. [Google Scholar] [CrossRef] [PubMed]
- Watson, N.F.; Durrant, L.G.; Madjd, Z.; Ellis, I.O.; Scholefield, J.H.; Spendlove, I. Expression of the membrane complement regulatory protein CD59 (protectin) is associated with reduced survival in colorectal cancer patients. Cancer Immunol. Immunother. 2006, 55, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Jung, M.; Burkhardt, M.; Stephan, C.; Schnorr, D.; Loening, S.; Jung, K.; Dietel, M.; Kristiansen, G. Increased CD59 protein expression predicts a PSA relapse in patients after radical prostatectomy. Prostate 2005, 62, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Farrell, D.H.; Thiagarajan, P.; Chung, D.W.; Davie, E.W. Role of fibrinogen α and γ chain sites in platelet aggregation. Proc. Natl. Acad. Sci. USA 1992, 89, 10729–10732. [Google Scholar] [CrossRef] [PubMed]
- Theodorescu, D.; Wittke, S.; Ross, M.M.; Walden, M.; Conaway, M.; Just, I.; Mischak, H.; Frierson, H.F. Discovery and validation of new protein biomarkers for urothelial cancer: A prospective analysis. Lancet Oncol. 2006, 7, 230–240. [Google Scholar] [CrossRef]
- Bergen, H.R.; Vasmatzis, G.; Cliby, W.A.; Johnson, K.L.; Oberg, A.L.; Muddiman, D.C. Discovery of ovarian cancer biomarkers in serum using NanoLC electrospray ionization TOF and FT-ICR mass spectrometry. Dis. Markers 2004, 19, 239–249. [Google Scholar] [CrossRef]
- Ebert, M.P.; Niemeyer, D.; Deininger, S.O.; Wex, T.; Knippig, C.; Hoffmann, J.; Sauer, J.; Albrecht, W.; Malfertheiner, P.; Röcken, C. Identification and confirmation of increased fibrinopeptide a serum protein levels in gastric cancer sera by magnet bead assisted MALDI-TOF mass spectrometry. J. Proteome Res. 2006, 5, 2152–2158. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, J.; Shaffer, D.R.; Philip, J.; Chaparro, C.A.; Erdjument-Bromage, H.; Olshen, A.B.; Fleisher, M.; Lilja, H.; Brogi, E.; Boyd, J.; et al. Differential exoprotease activities confer tumor-specific serum peptidome patterns. J. Clin. Investig. 2006, 116, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, J.; Martorella, A.J.; Lawlor, K.; Philip, J.; Fleisher, M.; Robbins, R.J.; Tempst, P. Serum peptidome patterns that distinguish metastatic thyroid carcinoma from cancer-free controls are unbiased by gender and age. Mol. Cell. Proteomics 2006, 5, 1840–1852. [Google Scholar] [CrossRef] [PubMed]
- Maes, E.; Mertens, I.; Valkenborg, D.; Pauwels, P.; Rolfo, C.; Baggerman, G. Proteomics in cancer research: Are we ready for clinical practice? Crit. Rev. Oncol. Hematol. 2015, 96, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Calderaro, A.; Arcangeletti, M.C.; Rodighiero, I.; Buttrini, M.; Gorrini, C.; Motta, F.; Germini, D.; Medici, M.C.; Chezzi, C.; de Conto, F. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry applied to virus identification. Sci. Rep. 2014, 4, 6803. [Google Scholar] [CrossRef] [PubMed]
- Basics on data preparation, model generation and spectra classification. In ClinProTools 3.0 User Manual; Bruker Daltonics: Bremen, Germany, 2011; pp. 51–96.
- Villanueva, J.; Lawlor, K.; Toledo-Crow, R.; Tempst, P. Automated serum peptide profiling. Nat. Protoc. 2006, 1, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Matysiak, J.; Światły, A.; Hajduk, J.; Matysiak, J.; Kokot, Z.J. Influence of honeybee sting on peptidome profile in human serum. Toxins 2015, 7, 1808–1820. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Zhao, F.; Zhang, H.; Meng, F.; Zhang, J. Novel strategy for typing Mycoplasma pneumoniae isolates by use of matrix-assisted laser desorption ionization-time of flight mass spectrometry coupled with ClinProTools. J. Clin. Microbiol. 2014, 52, 3038–3043. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Characteristic | Training Set | Test Set | Total | |||
|---|---|---|---|---|---|---|
| NSCLC Patients | Controls | NSCLC Patients | Controls | NSCLC Patients | Controls | |
| No. of Subjects | 67 | 47 | 23 | 16 | 90 | 63 |
| Sex | ||||||
| Male | 40 (59.7%) | 31 (66.0%) | 18 (78.3%) | 10 (62.5%) | 58 (64.4%) | 41 (65.1%) |
| Female | 27 (40.3%) | 16 (34.0%) | 5 (21.7) | 6 (37.5%) | 32 (35.6%) | 22 (34.9%) |
| Age | ||||||
| Mean (SD) | 63 (6.5) | 62 (8.5) | 66 (8.0) | 62 (10.0) | 64 (6.9) | 62 (8.7) |
| Range | 48–86 | 43–78 | 53–81 | 45–77 | 48–86 | 43–78 |
| Histological type | ||||||
| Squamous cell carcinoma | 39 (58.2%) | 12 (52.2%) | 50 (55.6%) | |||
| Adenocarcinoma | 28 (41.8%) | 11 (47.8%) | 40 (44.4%) | |||
| Grade of cancer differentiation | ||||||
| G1 | 1 | 1 | 2 | |||
| G2 | 31 | 12 | 43 | |||
| G2/3 | 6 | 2 | 8 | |||
| G3 | 23 | 6 | 29 | |||
| Unknown | 6 | 2 | 8 | |||
| TNM stage | ||||||
| IA | 14 | 3 | 17 | |||
| IB | 19 | 6 | 25 | |||
| IIA | 14 | 5 | 19 | |||
| IIB | 8 | 2 | 10 | |||
| IIIA | 12 | 7 | 19 | |||
| Mass (m/z) | NSCLC | Control Group | p-Value 1 | AUC 2 |
|---|---|---|---|---|
| 1568.45 | 6.33 ± 4.27 | 2.75 ± 0.91 | <0.000001 | 0.85 |
| 1546.72 | 54.19 ± 41.34 | 24.09 ± 14.03 | 0.00000386 | 0.78 |
| 1617.88 | 66.99 ± 48.77 | 29.24 ± 23.84 | 0.00000386 | 0.78 |
| 1520.16 | 13.56 ± 4.36 | 9.49 ± 4.28 | 0.00003860 | 0.78 |
| 4466.98 | 0.31 ± 0.19 | 0.51 ± 0.23 | 0.00002280 | 0.75 |
| 4803.17 | 0.29 ± 0.18 | 0.56 ± 0.34 | 0.00000975 | 0.77 |
| 4787.36 | 0.52 ± 0.48 | 1.21 ± 1.12 | 0.00001960 | 0.79 |
| 2083.30 | 1.07 ± 1.42 | 2.24 ± 2.57 | 0.00001960 | 0.76 |
| Non-Small Cell Lung Cancer 1 | Lung Adenocarcinoma | Lung Squamous Cell Carcinoma | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Quick Classifier | Supervised Neural Network | Genetic Algorithm | Quick Classifier | Supervised Neural Network | Genetic Algorithm | Quick Classifier | Supervised Neural Network | Genetic Algorithm | |
| Mass [Da] | 1520.16 | 1546.72 | 1466.90 | 1568.45 | 1568.45 | 1450.94 | 1520.16 | 1546.72 | 2884.63 |
| 1538.14 | 1617.88 | 1568.45 | 7923.94 | 1305.41 | 4626.52 | 1617.88 | 1527.52 | ||
| 1546.72 | 1505.34 | 1450.94 | 5004.12 | 1546.72 | 1741.57 | 1520.16 | |||
| 1568.45 | 1880.97 | 2755.6 | 1505.42 | 2884.63 | 1564.1 | ||||
| 1617.88 | 1617.88 | 2604.28 | 6630.86 | 4787.36 | 1546.72 | ||||
| 4466.98 | 1520.16 | 1450.94 | 1466.85 | 5808.48 | |||||
| 4680.61 | 1546.72 | 1628.01 | 9289.98 | 3882.95 | |||||
| 4803.17 | 5904.39 | 1741.49 | 1510.24 | 6589.21 | |||||
| 6528.67 | 6330.86 | 2673.89 | 6304.32 | 1078.16 | |||||
| 6432.69 | 2555.04 | 1021.00 | 5919.08 | ||||||
| 1905.19 | |||||||||
| 4787.36 | |||||||||
| 2575.89 | |||||||||
| 1888.66 | |||||||||
| 1546.72 | |||||||||
| Cross validation (%) 2 | 75.54 | 47.87 | 71.89 | 84.31 | 86.35 | 74.71 | 69.51 | 60.64 | 74.66 |
| Recognition capability (%) 2 | 76.33 | 56.47 | 96.22 | 87.46 | 95.79 | 97.87 | 75.80 | 50.86 | 96.51 |
| Specificity (%) 3 | 64.40 | 66.70 | - | - | 86.70 | ||||
| Sensitivity (%) 3 | 94.40 | 72.70 | - | - | 66.70 | ||||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klupczynska, A.; Swiatly, A.; Hajduk, J.; Matysiak, J.; Dyszkiewicz, W.; Pawlak, K.; Kokot, Z.J. Identification of Serum Peptidome Signatures of Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2016, 17, 410. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17040410
Klupczynska A, Swiatly A, Hajduk J, Matysiak J, Dyszkiewicz W, Pawlak K, Kokot ZJ. Identification of Serum Peptidome Signatures of Non-Small Cell Lung Cancer. International Journal of Molecular Sciences. 2016; 17(4):410. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17040410
Chicago/Turabian StyleKlupczynska, Agnieszka, Agata Swiatly, Joanna Hajduk, Jan Matysiak, Wojciech Dyszkiewicz, Krystian Pawlak, and Zenon J. Kokot. 2016. "Identification of Serum Peptidome Signatures of Non-Small Cell Lung Cancer" International Journal of Molecular Sciences 17, no. 4: 410. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17040410