
Profilings of MicroRNAs in the Liver of Common Carp (Cyprinus carpio) Infected with Flavobacterium columnare

Abstract
:
1. Introduction
2. Results
2.1. Construction of cDNA Library for Sequencing and Small RNA Discovery
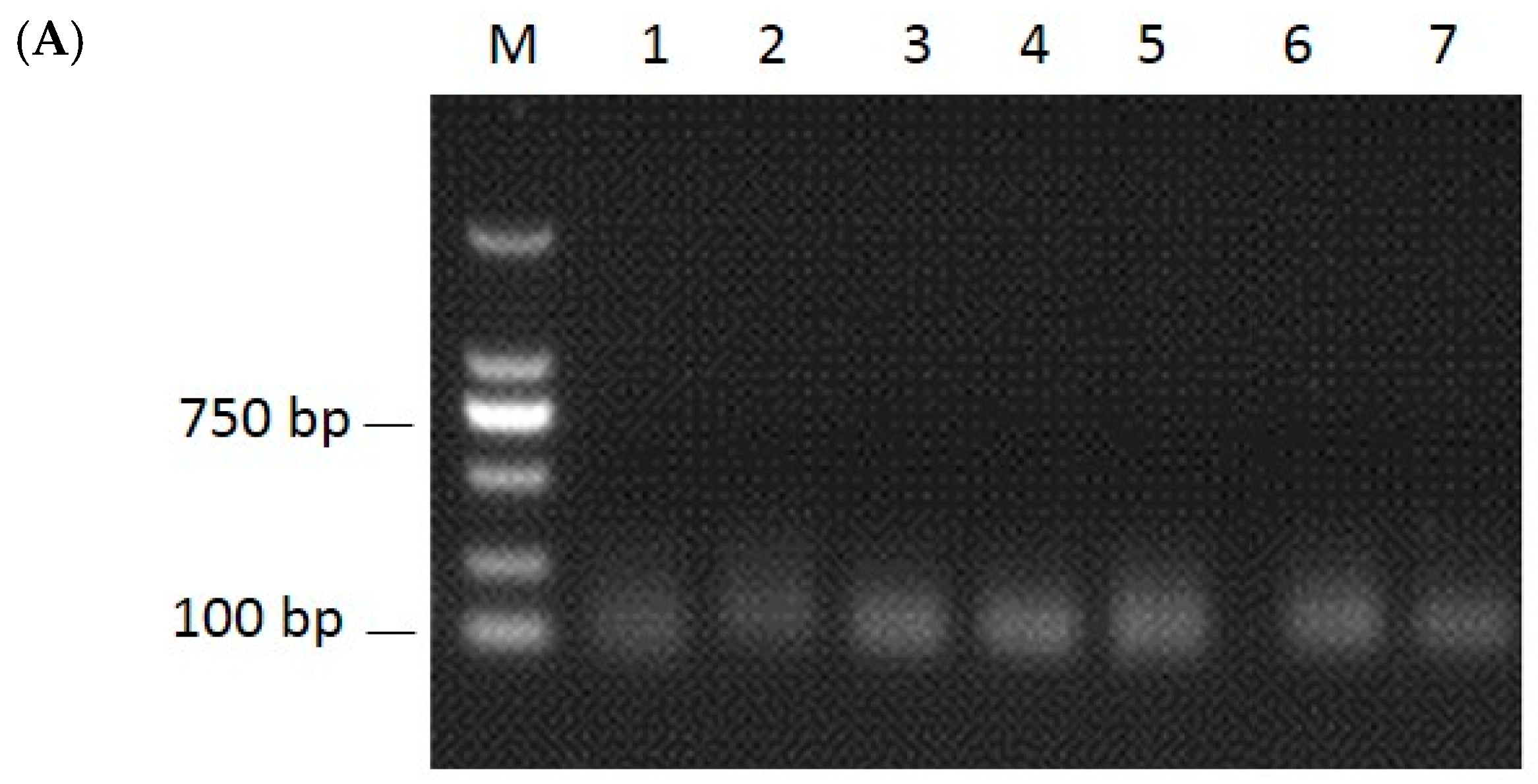
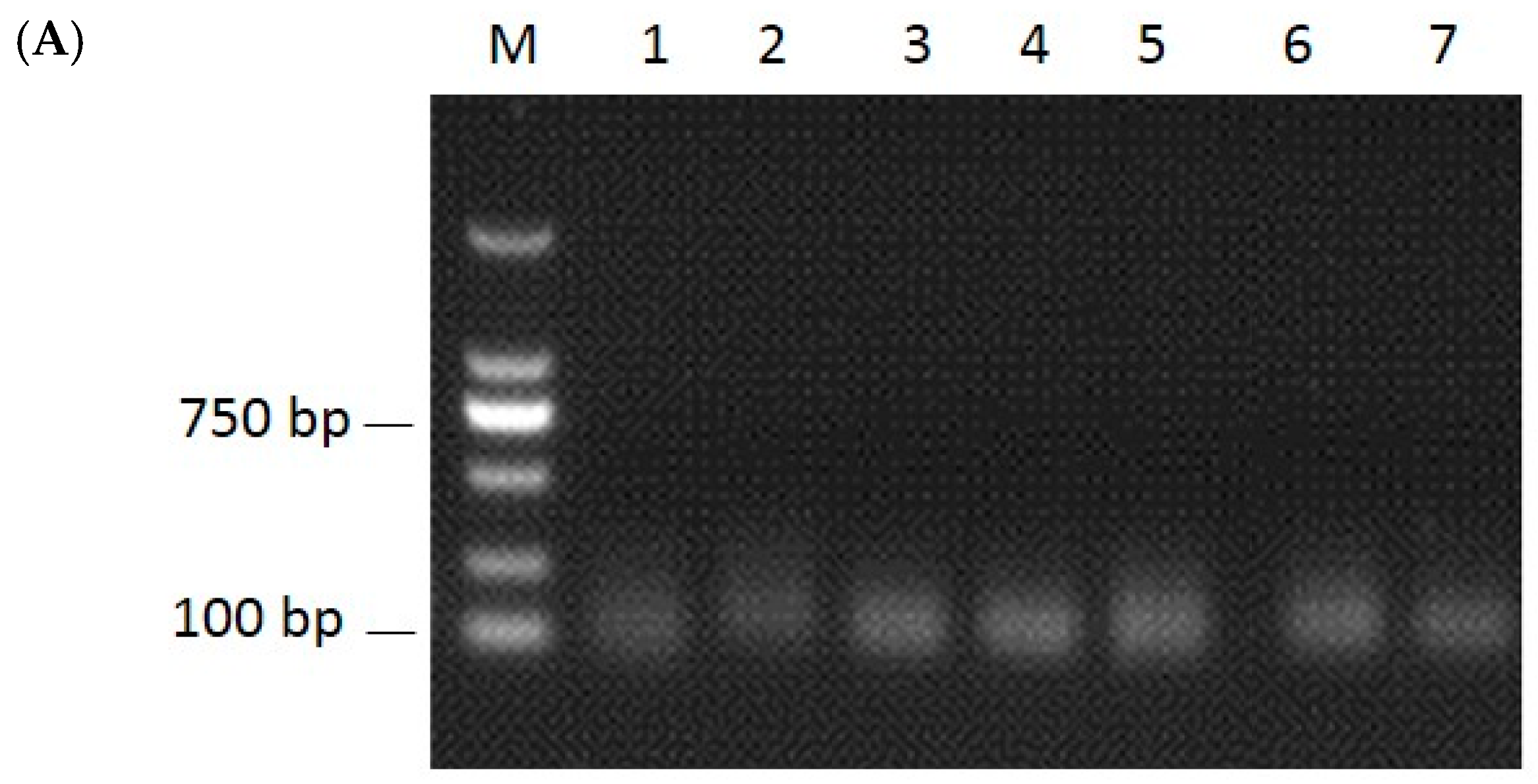
2.2. Identification of miRNAs and Validation by RT-PCR and qRT-PCR
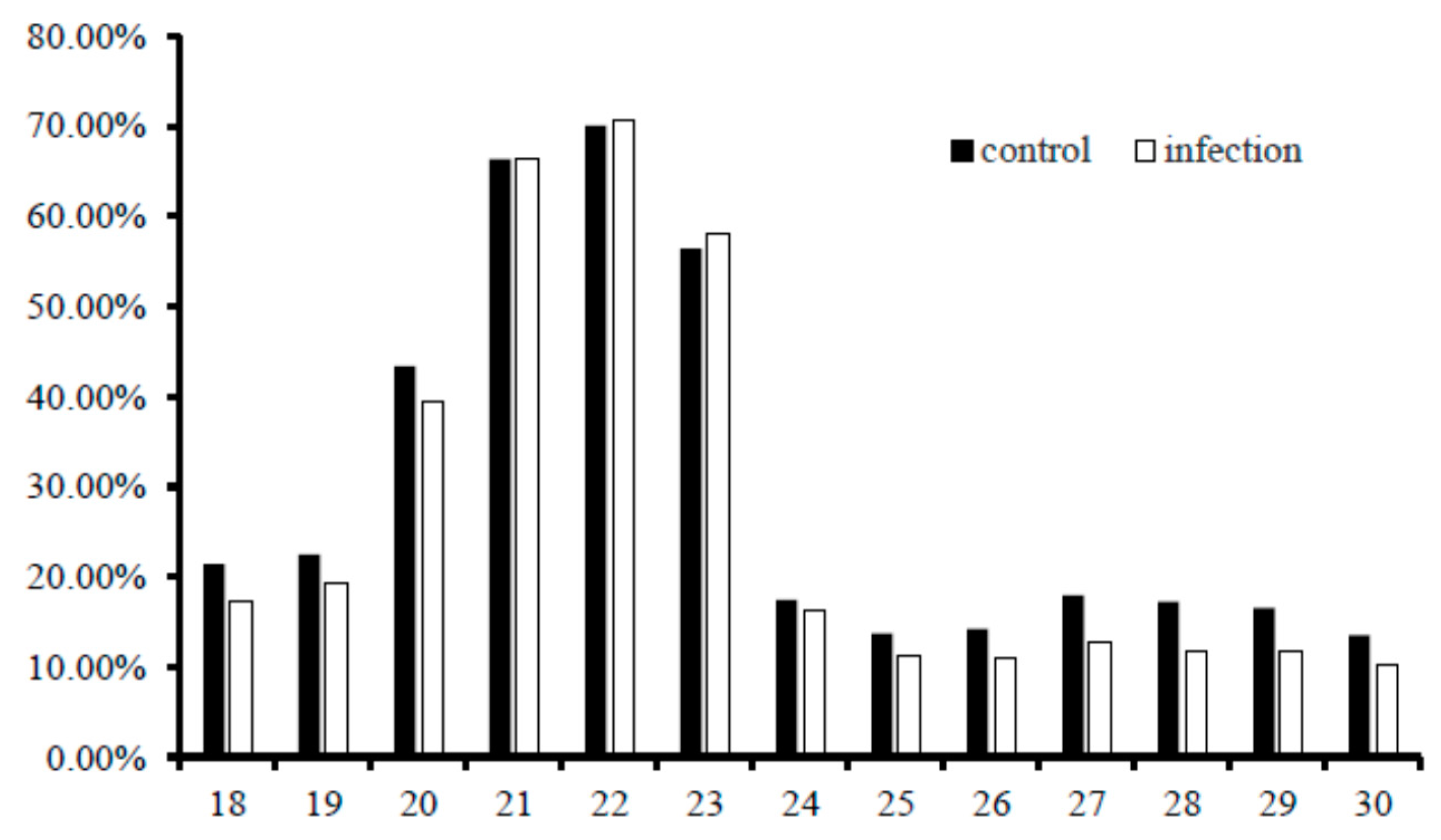
2.3. Conservation Analysis of the Identified Common Carp miRNAs
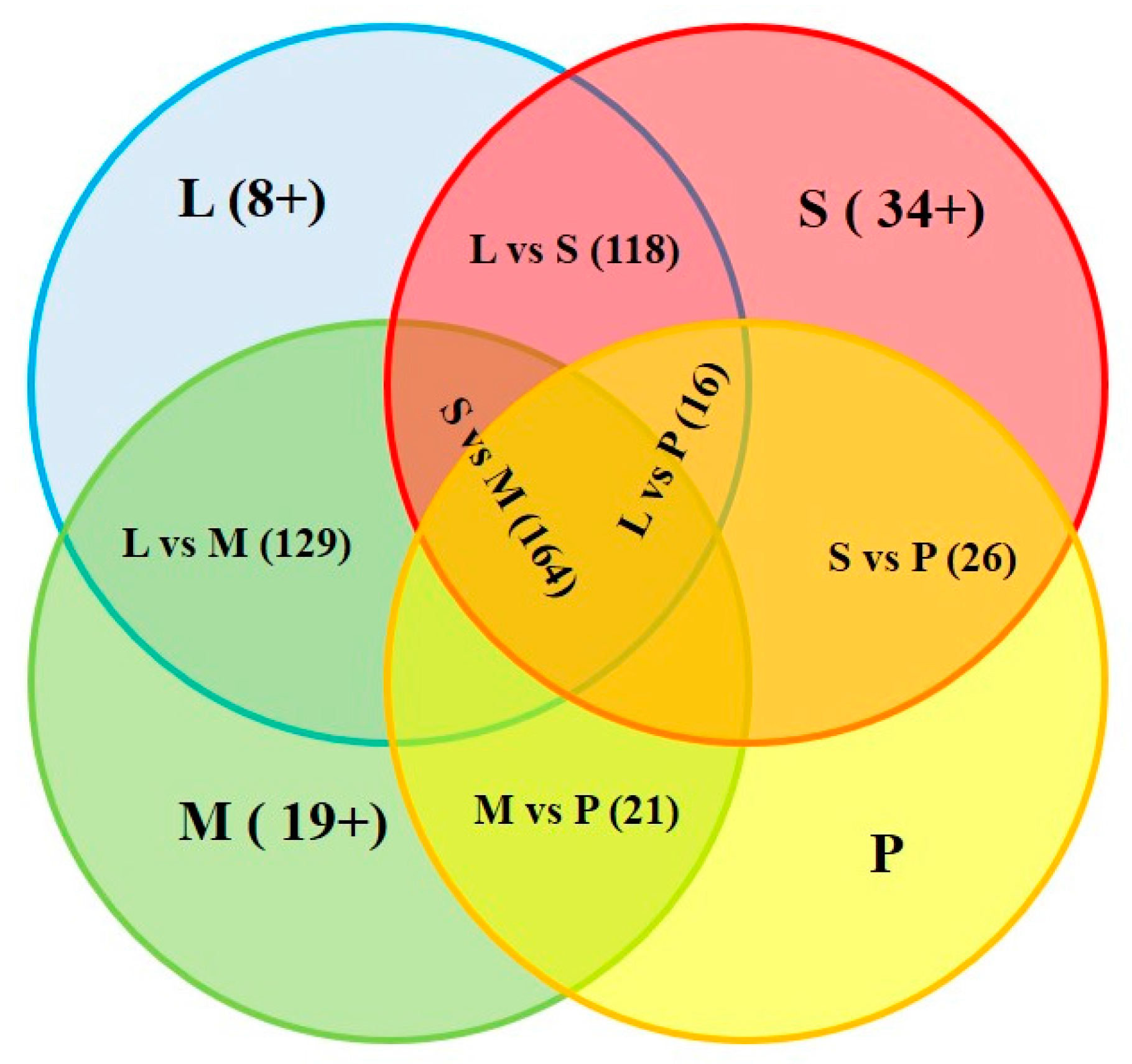
2.4. Differentially Expressed miRNAs (DIE-miRNAs) between Infected and Control Livers of Common Carp
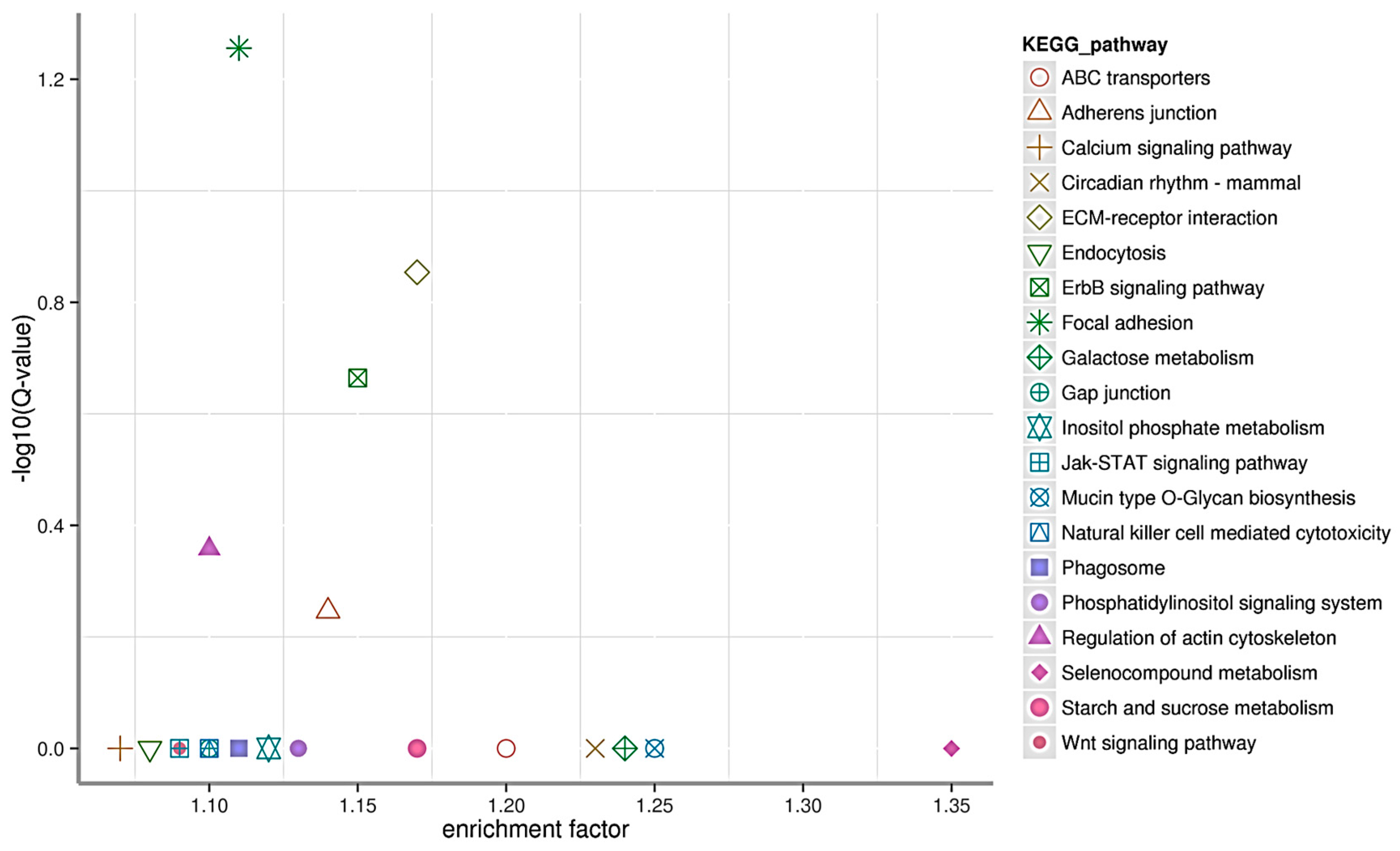
2.5. miRNA Target Genes, Annotation and KEGG Pathway Enrichment Analysis
3. Discussion
4. Materials and Methods
4.1. Fish, Bacteria, and Infection
4.2. Small RNA Isolation and cDNA Library Construction
4.3. Sequence Analysis and Identification of miRNAs
4.4. Target Gene Functional Annotation
4.5. Differential miRNA Expression Analysis
4.6. GO and KEGG Pathway Enrichment Analysis
4.7. RT-PCR and qRT-PCR Analysis of miRNAs
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Declercq, A.M.; Haesebrouck, F.; van den Broeck, W.; Bossier, P.; Decostere, A. Columnaris disease in fish: A review with emphasis on bacterium-host interactions. Vet Res. 2013, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Zhang, Y.; Wen, Y.; Ji, W.; Zhou, Y.; Ji, Y.; Liu, X.; Wang, W.; Asim, M.; Liang, X.; et al. Analysis of the transcriptomic profilings of Mandarin fish (Siniperca chuatsi) infected with Flavobacterium columnare with an emphasis on immune responses. Fish Shellfish Immunol. 2015, 43, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zhang, X.; Wang, X.; Ji, W.; Zhou, Y.; Ji, Y.; Liu, X.; Wang, W.; Asim, M.; Liang, X.; et al. Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat. Genet. 2014, 46, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Declercq, A.M.; Chiers, K.; van den Broeck, W.; Eeckhaut, V.; Cornelissen, M.; Bossier, P.; Haesebrouck, F.; Decostere, A. Interactions of highly and low virulent Flavobacterium columnare isolates with gill tissue in carp and rainbow trout. Vet. Res. 2015, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.R.; Sonenberg, N. The mechanics of miRNA-mediated gene silencing: A look under the hood of miRISC. Nat. Struct. Mol. Biol. 2012, 19, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Jeon, K.; Lee, J.T.; Kim, S.; Kim, V.N. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J. 2002, 21, 4663–4670. [Google Scholar] [CrossRef] [PubMed]
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: An expanding universe. Nat. Rev. Genet. 2009, 10, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhao, Y.; Wen, L.; Liu, Z.; Yan, F.; Gao, C. Identification and characterization of microRNAs in the spleen of common carp immune organ. J. Cell. Biochem. 2014, 115, 1768–1778. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.P.; Xue, W.; Wang, J.T.; Wan, Y.M.; Wang, S.-L.; Xu, P.; Zhang, Y.; Li, J.-T.; Sun, X.-W. Identification of common carp (Cyprinus carpio) microRNAs and microRNA-related SNPs. BMC Genom. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Ding, L.; Li, Y.; Zhang, X.; Liang, Y.; Sun, X.; Teng, C.B. Identification and profiling of microRNAs from skeletal muscle of the common carp. PLoS ONE 2012, 7, e30925. [Google Scholar] [CrossRef] [PubMed]
- Bizuayehu, T.T.; Babiak, I. MicroRNA in teleost fish. Genome Biol. Evol. 2014, 6, 1911–1937. [Google Scholar] [CrossRef] [PubMed]
- Hauck, C.R. Cell adhesion receptors—Signaling capacity and exploitation by bacterial pathogens. Med. Microbiol. Immunol. 2002, 191, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M. An overview of microRNAs. Adv. Drug Deliv. Rev. 2015, 87, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Jiang, F. MicroRNA (miRNA) profiling. Methods Mol. Biol. 2016, 1381, 151–161. [Google Scholar] [PubMed]
- Bartel, D. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.J. Developmental control of cell growth and division in Drosophila. Curr. Opin. Cell Biol. 2010, 22, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Lian, H.; Wang, J.W. Plant developmental transitions: The role of microRNAs and sugars. Curr. Opin. Plant Biol. 2015, 27, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.E. Paxillin and focal adhesion signaling. Nat. Cell Biol. 2000, 2, E231–E236. [Google Scholar] [CrossRef] [PubMed]
- Danen, E.H.; Yamada, K.M. Fibronectin, integrins, and growth control. J. Cell. Physiol. 2001, 189, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.D.; Skaria, K.B.; Yarden, Y. Signal transduction and oncogenesis by ErbB/HER receptors. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Sakamoto, H.; Sugiura, S.; Ogawa, M. Endothelial cell motility is compatible with junctional integrity. J. Cell. Physiol. 2007, 211, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Li, W.; He, W.; Jiang, N.; Zhang, G.X.; Chen, W.; Yang, H.F.; Liu, Q.L.; Huang, Y.N.; Zhang, L.; et al. miR-184 post-transcriptionally regulates SOX7 expression and promotes cell proliferation in human hepatocellular carcinoma. PLoS ONE 2014, 9, e88796. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Gao, W.; Liu, B.; Ye, W. Upregulation of miR-184 enhances the malignant biological behavior of human glioma cell line A172 by targeting FIH-1. Cell. Physiol. Biochem. 2014, 34, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.R.; Wang, Q.; Zhang, X.B.; Wang, S.J. Regulatory role of microRNA184 in osteosarcoma cells. Genet. Mol. Res. 2015, 14, 14246–14252. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Mai, C.; Yang, H.; Zhen, Y.; Yu, X.; Hua, S.; Wu, Q.; Jiang, Q.; Zhang, Y.; Song, X.; et al. Candidate tumour suppressor CCDC19 regulates miR-184 direct targeting of C-Myc thereby suppressing cell growth in non-small cell lung cancers. J. Cell. Mol. Med. 2014, 18, 1667–1679. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Chen, D.; Li, Y.; Zhang, E.; Yu, Z.; Chen, T.; Jiang, Z.; Ni, L.; Yang, S.; Gui, Y. MicroRNA-184 functions as tumor suppressor in renal cell carcinoma. Exp. Ther. Med. 2015, 9, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Grimaldo, S.; Yuen, D.; Theis, J.; Ng, M.; Ecoiffier, T.; Chen, L. MicroRNA-184 regulates corneal lymphangiogenesis. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7209–7213. [Google Scholar] [CrossRef] [PubMed]
- Maru, D.M.; Singh, R.R.; Hannah, C.; Albarracin, C.T.; Li, Y.X.; Abraham, R.; Romans, A.M.; Yao, H.; Luthra, M.G.; Anandasabapathy, S. MicroRNA-196a is a potential marker of progression during Barrett’s metaplasiadysplasia-invasive adenocarcinoma sequence in esophagus. Am. J. Pathol. 2009, 174, 1940–1948. [Google Scholar] [CrossRef] [PubMed]
- Bloomston, M.; Frankel, W.L.; Petrocca, F.; Volinia, S.; Alder, H.; Hagan, J.P.; Liu, C.G.; Bhatt, D.; Taccioli, C.; Croce, C.M. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic ancreatitis. JAMA 2007, 297, 1901–1908. [Google Scholar] [CrossRef] [PubMed]
- Szafranska, A.E.; Doleshal, M.; Edmunds, H.S.; Gordon, S.; Luttges, J.; Munding, J.B.; Barth, R.J., Jr.; Gutmann, E.J.; Suriawinata, A.A.; Marc Pipas, J. Analysis of microRNAs in pancreatic fine-needle aspirates can classify benign and malignant tissues. Clin. Chem. 2008, 54, 1716–1724. [Google Scholar] [CrossRef] [PubMed]
- Hui, A.B.; Shi, W.; Boutros, P.C.; Miller, N.; Pintilie, M.; Fyles, T.; McCready, D.; Wong, D.; Gerster, K.; Waldron, L.; et al. Robust global micro-RNA profiling with formalin-fixed paraffin-embedded breast cancer tissues. Lab. Investig. 2009, 89, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Debernardi, S.; Skoulakis, S.; Molloy, G.; Chaplin, T.; Dixon-McIver, A.; Young, B.D. MicroRNA miR-181a correlates with morphological sub-class of acute myeloid leukaemia and the expression of its target genes in global genome-wide analysis. Leukemia 2007, 21, 912–916. [Google Scholar] [CrossRef] [PubMed]
- Thorsteinsdottir, U.; Sauvageau, G.; Hough, M.R.; Dragowska, W.; Lansdorp, P.M.; Lawrence, H.J.; Largman, C.; Humphries, R.K. Overexpression of HOXA10 in murine hematopoietic cells perturbs both myeloid and lymphoid differentiation and leads to acute myeloid leukemia. Mol. Cell. Biol. 1997, 17, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Thorsteinsdottir, U.; Mamo, A.; Kroon, E.; Jerome, L.; Bijl, J.; Lawrence, H.J.; Humphries, K.; Sauvageau, G. Overexpression of the myeloid leukemia-associated Hoxa9 gene in bone marrow cells induces stem cell expansion. Blood 2002, 99, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Schimanski, C.C.; Frerichs, K.; Rahman, F.; Berger, M.; Lang, H.; Galle, P.R.; Moehler, M.; Gockel, I. High miR-196a levels promote the oncogenic phenotype of colorectal cancer cells. World J. Gastroenterol. 2009, 15, 2089–2096. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Zhang, X.Y.; Zhang, B.F.; Yang, C.Q.; Chen, X.M.; Gao, H.J. Initial study of microRNA expression profiles of colonic cancer without lymph node metastasis. J. Dig. Dis. 2010, 11, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Braig, S.; Mueller, D.W.; Rothhammer, T.; Bosserhoff, A.K. MicroRNA miR-196a is a central regulator of HOX-B7 and BMP4 expression in malignant melanoma. Cell. Mol. Life Sci. 2010, 67, 3535–3548. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Ye, H.; Teng, Y.; Wang, Y.L.; Yang, G.; Li, X.B.; Zhang, C.; Yang, X.; Yang, Z.Z.; Yang, X. Akt-p53-miR-365-cyclin D1/cdc25A axis contributes to gastric tumorigenesis induced by PTEN deficiency. Nat. Commun. 2013, 4, 2544. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Liu, W.; Ma, S.; Cao, H.; Peng, X.; Guo, L.; Zhou, X.; Zheng, L.; Guo, L.; Wan, M. A novel onco-miR-365 induces cutaneous squamous cell carcinoma. Carcinogenesis 2013, 34, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Rice, S.; Salzberg, A.; Runkle, E.A.; Liao, J.; Zander, D.S.; Mu, D. miR-365 regulates lung cancer and developmental gene thyroid transcription factor 1. Cell Cycle 2012, 11, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Liu, L.; Zheng, W.; Chen, L.; Wu, X.; Xu, Y.; Du, X.; Han, W. microRNA-365, down-regulated in colon cancer, inhibits cell cycle progression and promotes apoptosis of colon cancer cells by probably targeting Cyclin D1 and Bcl-2. Carcinogenesis 2012, 33, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Zhang, Z.; Li, X.; Liu, H. MicroRNA-365 inhibits growth, invasion and metastasis of malignant melanoma by targeting NRP1 expression. Int. J. Clin. Exp. Pathol. 2015, 8, 4913–4922. [Google Scholar] [CrossRef] [PubMed]
- Cavodeassi, F.; Carreira-Barbosa, F.; Young, R.M.; Concha, M.L.; Allende, M.L.; Houart, C.; Tada, M.; Wilson, S.W. Early stages of zebrafish eye formation require the coordinated activity of Wnt11, Fz5, and the Wnt/b-catenin pathway. Neuron 2005, 47, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Kapsimali, M.; Kloosterman, W.P.; de Bruijn, E.; Rosa, F.; Plasterk, R.H.; Wilson, S.W. MicroRNAs show a wide diversity of expression profiles in the developing and mature central nervous system. Genome Biol. 2007, 8. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, D.R.; Cittelly, D.M.; Richer, J.K. Steroid receptors and microRNAs: Relationships revealed. Steroids 2011, 76, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.W.; de França, L.R.; Lareyre, J.J.; LeGac, F. Spermatogenesis in fish. Gen. Comp. Endocrinol. 2010, 165, 390–411. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Tu, J.; Yuan, J.; Liu, X.; Zhao, L.; Dawar, F.U.; Khattak, M.N.; Hegazy, A.M.; Chen, N.; Vakharia, V.N.; et al. Identification and characterization of microRNAs in snakehead fish cell line upon snakehead fish vesiculovirus infection. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Clean Reads | Annotated Small RNA | Unannotated Small RNA | Mapped siRNA Reads | Mapped Rate (%) |
|---|---|---|---|---|---|
| Control | 16,907,877 | 3,210,400 | 13,697,477 | 9,181,153 | 54.3 |
| Infection | 13,599,310 | 3,758,445 | 9,840,865 | 6,479,172 | 47.6 |
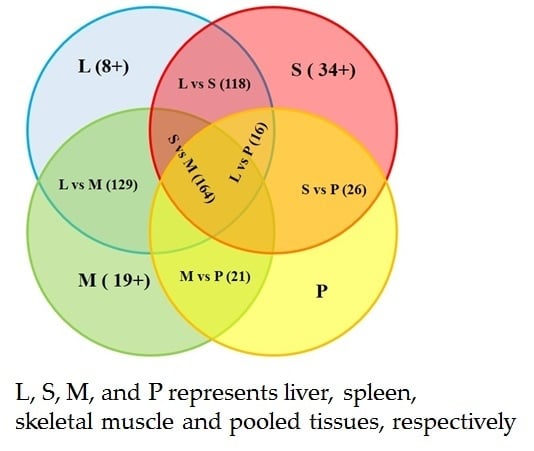
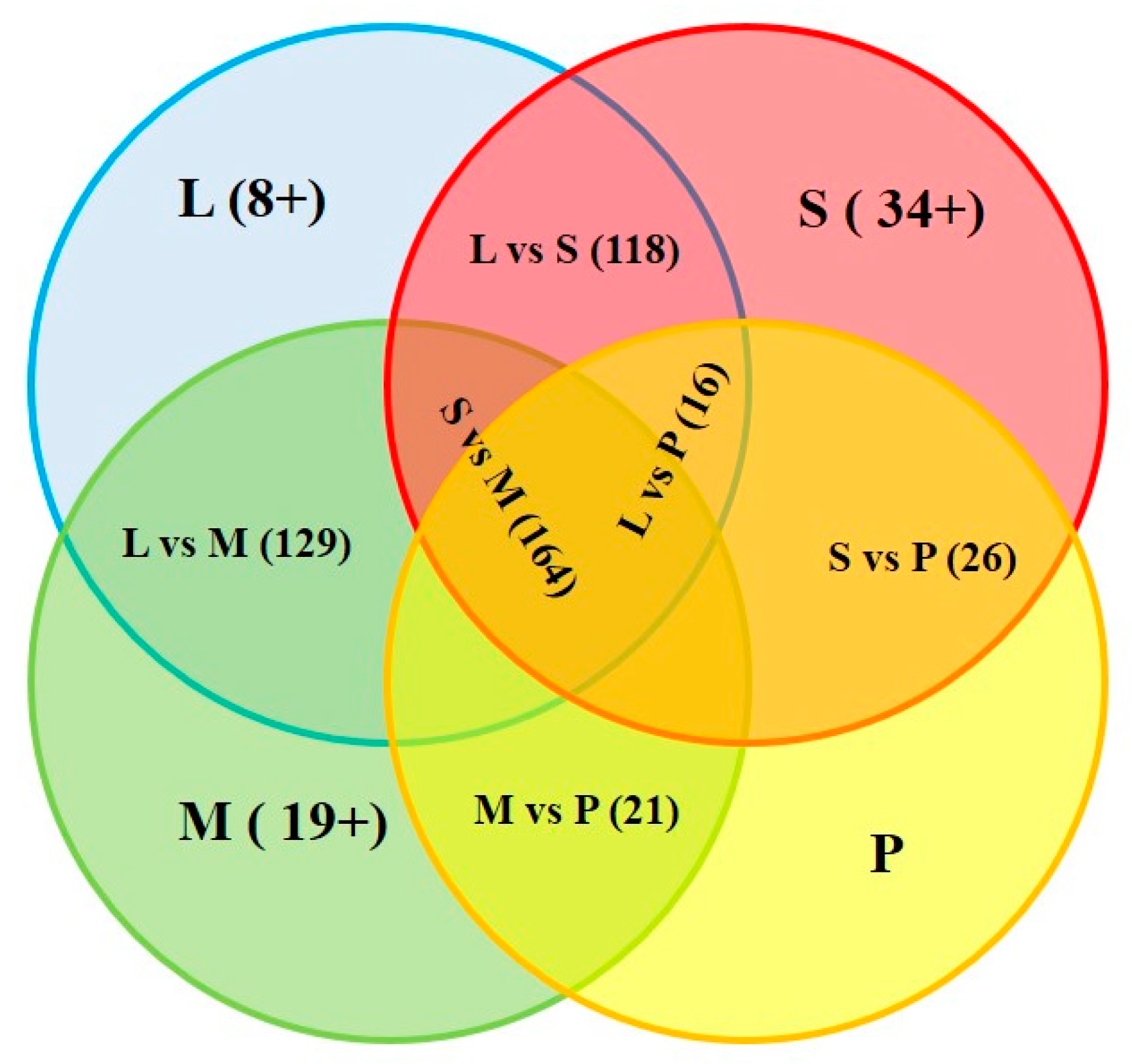
| Tissue | Liver (142) | Spleen (193) | Skeletal Muscle (188) | Pooled Tissues (80) |
|---|---|---|---|---|
| liver | – | 118 (L: 24; S: 75) | 129 (L: 13; M: 59) | 16 (L: 126; P: 64) |
| spleen | – | – | 164 (S: 29; M: 24) | 26 (S: 167; P: 54) |
| skeletal muscle | – | – | – | 21 (M: 167; P: 59) |
| pooled tissues | – | – | – | – |
| No. | miRNA Family | cel | dme | ccr | dre | ipu | xtr | gga | mmu | hsa | ccr Read |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | miR-let_7 | 1 | 1 | 5 | 18 | 21 | 9 | 11 | 12 | 12 | 185,356 |
| 2 | miR-8 | 1 | 1 | 3 | 6 | 6 | 3 | 3 | 5 | 5 | 41,287 |
| 3 | miR-1 | 1 | 1 | 2 | 4 | 3 | 4 | 4 | 4 | 3 | 943 |
| 4 | miR-34 | 1 | 1 | 1 | 3 | 2 | 5 | 3 | 3 | 3 | 305 |
| 5 | miR-9 | 1 | 4 | 1 | 7 | 7 | 4 | 2 | 3 | 3 | 77 |
| 6 | miR-124 | 1 | 1 | 2 | 6 | 5 | 1 | 4 | 3 | 3 | 12 |
| 7 | miR-2 | 1 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 8 | miR-63 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 9 | miR-392 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 10 | miR-10 | 0 | 4 | 7 | 15 | 15 | 8 | 5 | 8 | 8 | 313,126 |
| 11 | miR-25 | 0 | 2 | 3 | 4 | 4 | 5 | 1 | 4 | 4 | 49,817 |
| 12 | miR-7 | 0 | 1 | 2 | 4 | 6 | 3 | 4 | 3 | 3 | 5689 |
| 13 | miR-375 | 0 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 1 | 1846 |
| 14 | miR-29 | 0 | 1 | 2 | 3 | 3 | 4 | 4 | 4 | 4 | 1215 |
| 15 | miR-184 | 0 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 743 |
| 16 | miR-133 | 0 | 1 | 2 | 4 | 3 | 4 | 4 | 3 | 3 | 98 |
| 17 | miR-190 | 0 | 1 | 1 | 2 | 2 | 1 | 1 | 2 | 2 | 13 |
| 18 | miR-210 | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 455 |
| 19 | miR-193 | 0 | 1 | 1 | 3 | 0 | 1 | 2 | 2 | 2 | 21 |
| 20 | miR-33 | 0 | 1 | 0 | 0 | 0 | 2 | 1 | 1 | 2 | 0 |
| 21 | miR-314 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 22 | miR-31 | 0 | 2 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 0 |
| 23 | miR-263 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 24 | miR-219 | 0 | 1 | 0 | 3 | 3 | 1 | 2 | 3 | 3 | 0 |
| 25 | miR-216 | 0 | 1 | 0 | 2 | 2 | 1 | 3 | 2 | 2 | 0 |
| No. | Name of miRNA | Control TPM | Infected TPM | Folds |
|---|---|---|---|---|
| 1 | miR-365 | 30.03 | 77.28 | +2.57 |
| 2 | miR-196b | 3.64 | 10.03 | +2.75 |
| 3 | miR-153b | 2.12 | 4.80 | +2.26 |
| 4 | miR-301a | 0.76 | 2.72 | +3.58 |
| 5 | miR-133a-5p | 0.30 | 0.84 | +2.75 |
| 6 | miR-132b | 0.61 | 1.67 | +2.75 |
| 7 | miR-184 | 112.68 | 45.53 | −2.47 |
| 8 | miR-124a | 1.21 | 0.21 | −5.76 |
| 9 | miR-124b | 0.61 | 0.21 | −2.91 |
| 10 | ID_LG1_121431 | 0.15 | 2.09 | +13.77 |
| 11 | ID_LG10_120287 | 2.73 | 13.16 | +4.82 |
| 12 | ID_000000701_311347 | 6988.20 | 26,568.39 | +3.80 |
| 13 | ID_000000130_358988 | 6988.20 | 26,568.39 | +3.80 |
| 14 | ID_LG9_186104 | 1.36 | 4.59 | +3.37 |
| 15 | ID_LG32_105872 | 33.37 | 73.52 | +2.20 |
| 16 | ID_LG42_11512 | 437.08 | 924.83 | +2.12 |
| 17 | ID_000001225_235345 | 3.94 | 8.35 | +2.12 |
| 18 | ID_LG17_113850 | 5.76 | 2.51 | −2.30 |
| 19 | ID_LG40_164682 | 12.13 | 5.22 | −2.32 |
| 20 | ID_000000552_212634 | 11.98 | 5.01 | −2.39 |
| 21 | ID_LG20_173934 | 13.95 | 5.01 | −2.78 |
| 22 | ID_LG11_136853 | 5.46 | 1.88 | −2.90 |
| 23 | ID_000000219_202880 | 3.79 | 1.25 | −3.03 |
| 24 | ID_000028861_247413 | 5.16 | 1.67 | −3.09 |
| 25 | ID_LG30_147265 | 40.19 | 9.61 | −4.18 |
| 26 | ID_000001954_374705 | 40.19 | 9.61 | −4.18 |
| 27 | ID_000000316_205927 | 40.19 | 9.61 | −4.18 |
| 28 | ID_LG27_175722 | 3.49 | 0.63 | −5.57 |
| 29 | ID_000028889_299350 | 1.52 | 0.00 | – |
| 30 | ID_000008345_310753 | 1.52 | 0.00 | – |
| No. | miRNA Species | miRNA Family | Control TPM | Infected TPM | Infected/Control TPM (miRNA Species) | Infected/Control TPM (miRNA Family) |
|---|---|---|---|---|---|---|
| 1 | miR-365 | miR-365 | 30.03 | 77.28 | 2.57 * | 2.57 * |
| 2 | miR-301a | miR-130 | 0.76 | 2.72 | 3.58 * | 0.90 |
| 3 | miR-130b | 27.75 | 26.94 | 0.97 | ||
| 4 | miR-130c | 44.59 | 37.18 | 0.83 | ||
| 5 | miR-130a | 7.13 | 5.64 | 0.79 | ||
| 6 | miR-153b | miR-153 | 2.12 | 4.80 | 2.26 * | 1.94 |
| 7 | miR-153c | 8.19 | 15.25 | 1.86 | ||
| 8 | miR-196b | miR-196 | 3.64 | 10.03 | 2.75 * | 1.77 |
| 9 | miR-196a | 4.25 | 3.97 | 0.93 | ||
| 10 | miR-133a-5p | miR-133 | 0.30 | 0.84 | 2.75 * | 1.80 |
| 11 | miR-133a-3p | 14.56 | 25.90 | 1.78 | ||
| 12 | miR-132b | miR-132 | 0.61 | 1.67 | 2.75 * | 1.78 |
| 13 | miR-132a | 3.49 | 5.64 | 1.62 | ||
| 14 | miR-9-5p | miR-9 | 0.46 | 0.21 | 0.46 | 1.13 |
| 15 | miR-9-3p | 11.22 | 12.95 | 1.15 | ||
| 16 | miR-184 | miR-184 | 112.68 | 45.53 | 0.40 * | 0.40 * |
| 17 | miR-124b | miR-124 | 0.61 | 0.21 | 0.34 * | 0.23 * |
| 18 | miR-124a | 1.21 | 0.21 | 0.17 * |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, L.; Lu, H.; Meng, Q.; Wang, J.; Wang, W.; Yang, L.; Lin, L. Profilings of MicroRNAs in the Liver of Common Carp (Cyprinus carpio) Infected with Flavobacterium columnare. Int. J. Mol. Sci. 2016, 17, 566. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17040566
Zhao L, Lu H, Meng Q, Wang J, Wang W, Yang L, Lin L. Profilings of MicroRNAs in the Liver of Common Carp (Cyprinus carpio) Infected with Flavobacterium columnare. International Journal of Molecular Sciences. 2016; 17(4):566. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17040566
Chicago/Turabian StyleZhao, Lijuan, Hong Lu, Qinglei Meng, Jinfu Wang, Weimin Wang, Ling Yang, and Li Lin. 2016. "Profilings of MicroRNAs in the Liver of Common Carp (Cyprinus carpio) Infected with Flavobacterium columnare" International Journal of Molecular Sciences 17, no. 4: 566. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17040566