
Mechanisms of Cell Killing Response from Low Linear Energy Transfer (LET) Radiation Originating from 177Lu Radioimmunotherapy Targeting Disseminated Intraperitoneal Tumor Xenografts

Abstract
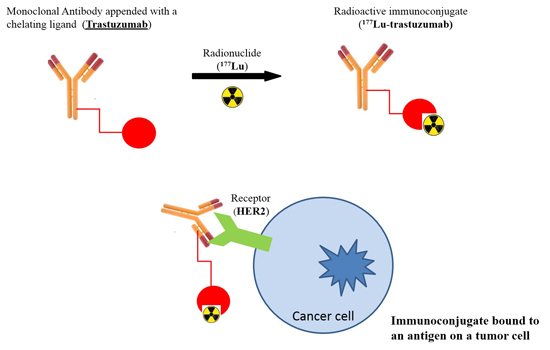
:
1. Introduction
2. Results
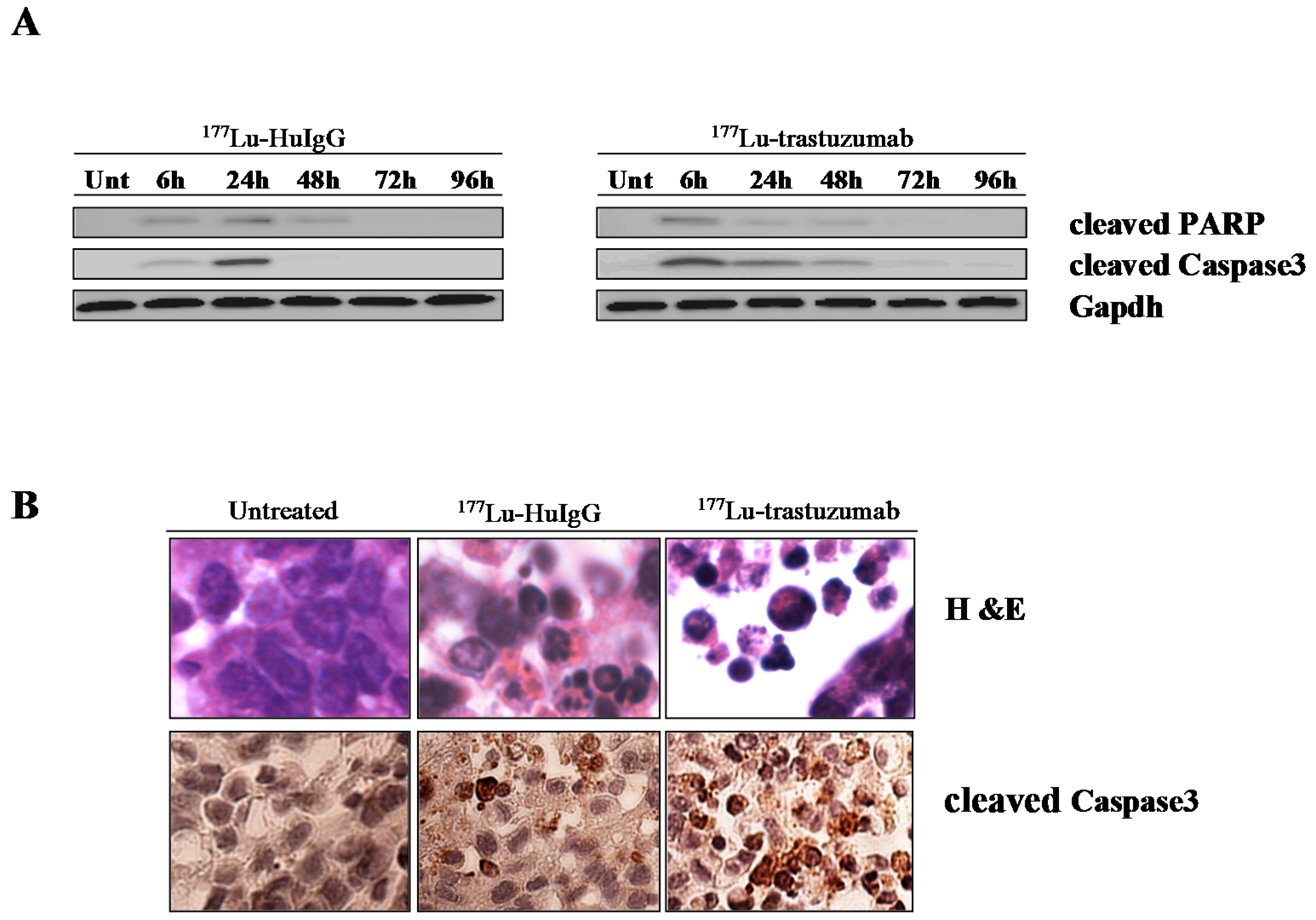
2.1. 177Lu-Trastzumab Induces Apoptosis in Intraperitoneal (i.p.) Human Colon Carcinoma Treated Xenografts
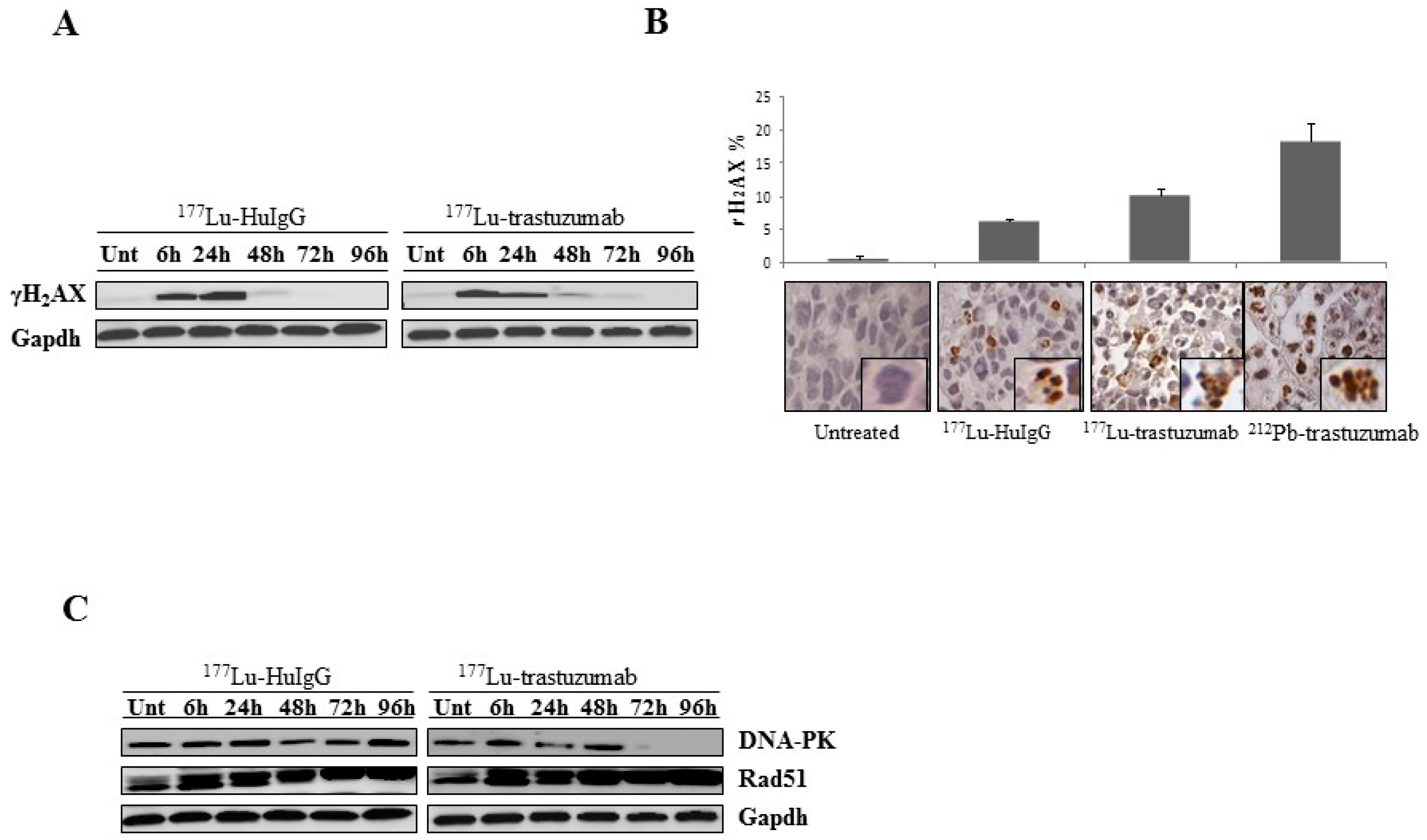
2.2. 177Lu-Trastuzumab Induces DNA Damage and Interferes with DNA Repair
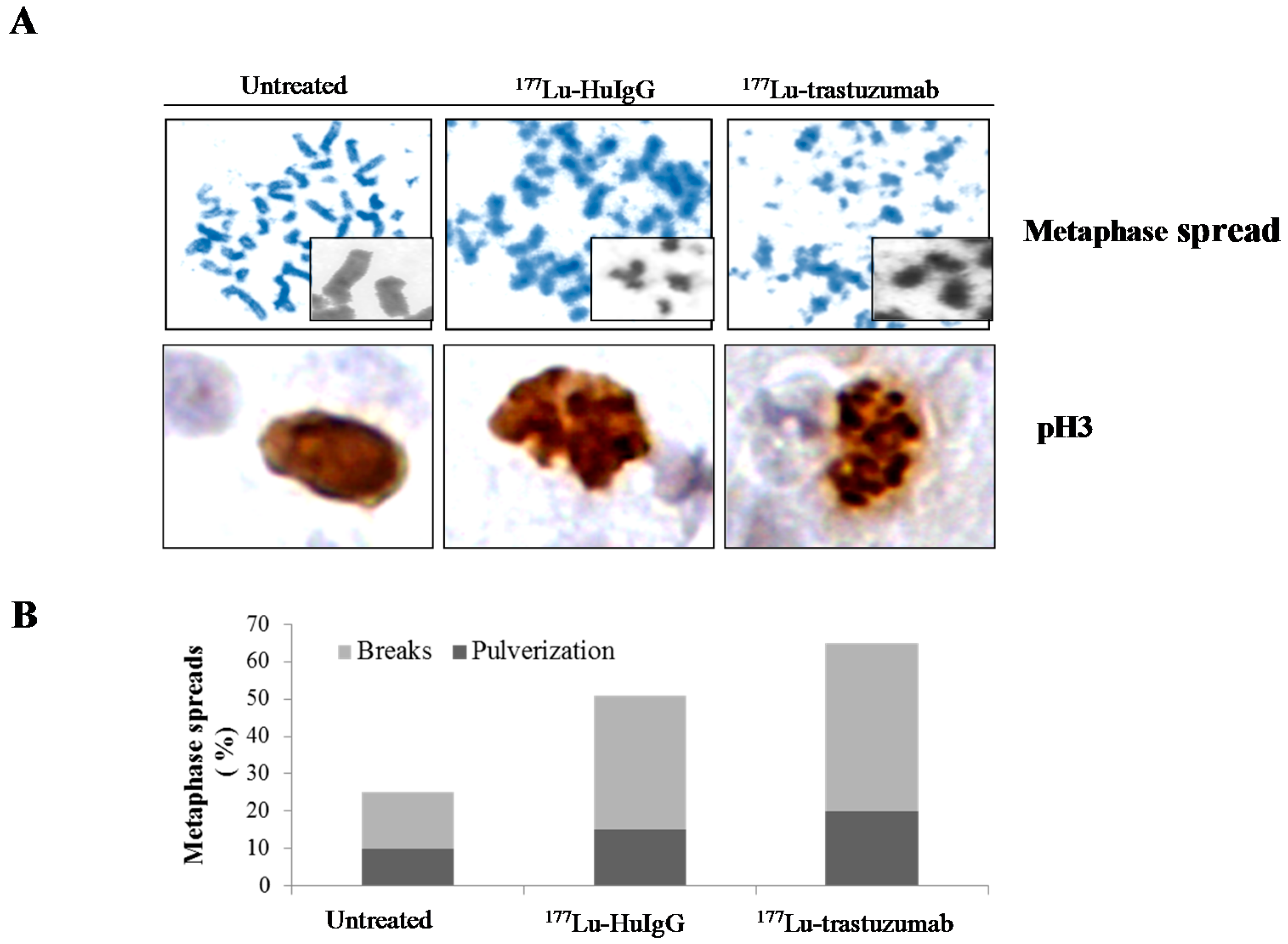
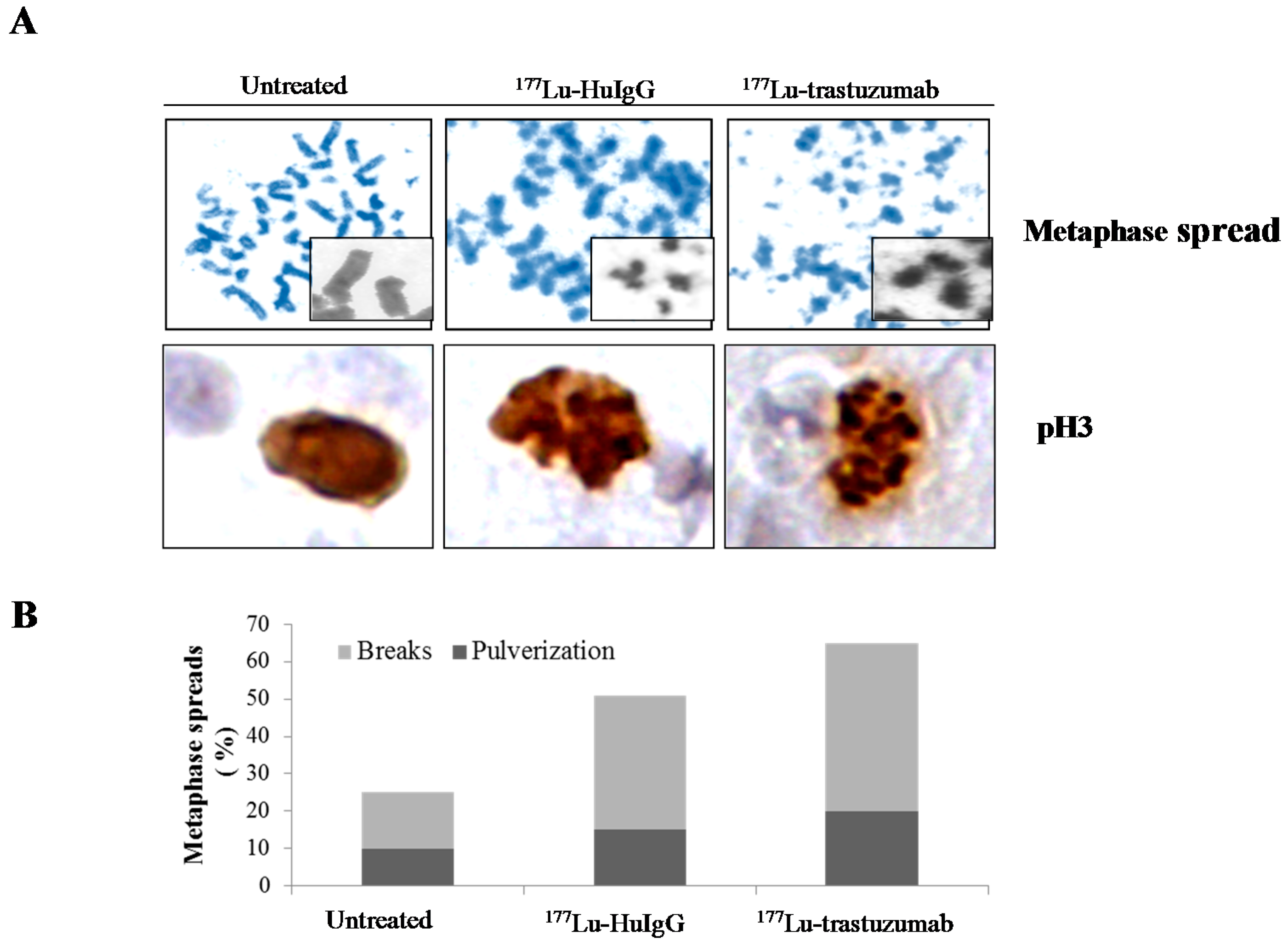
2.3. Metaphase Spread
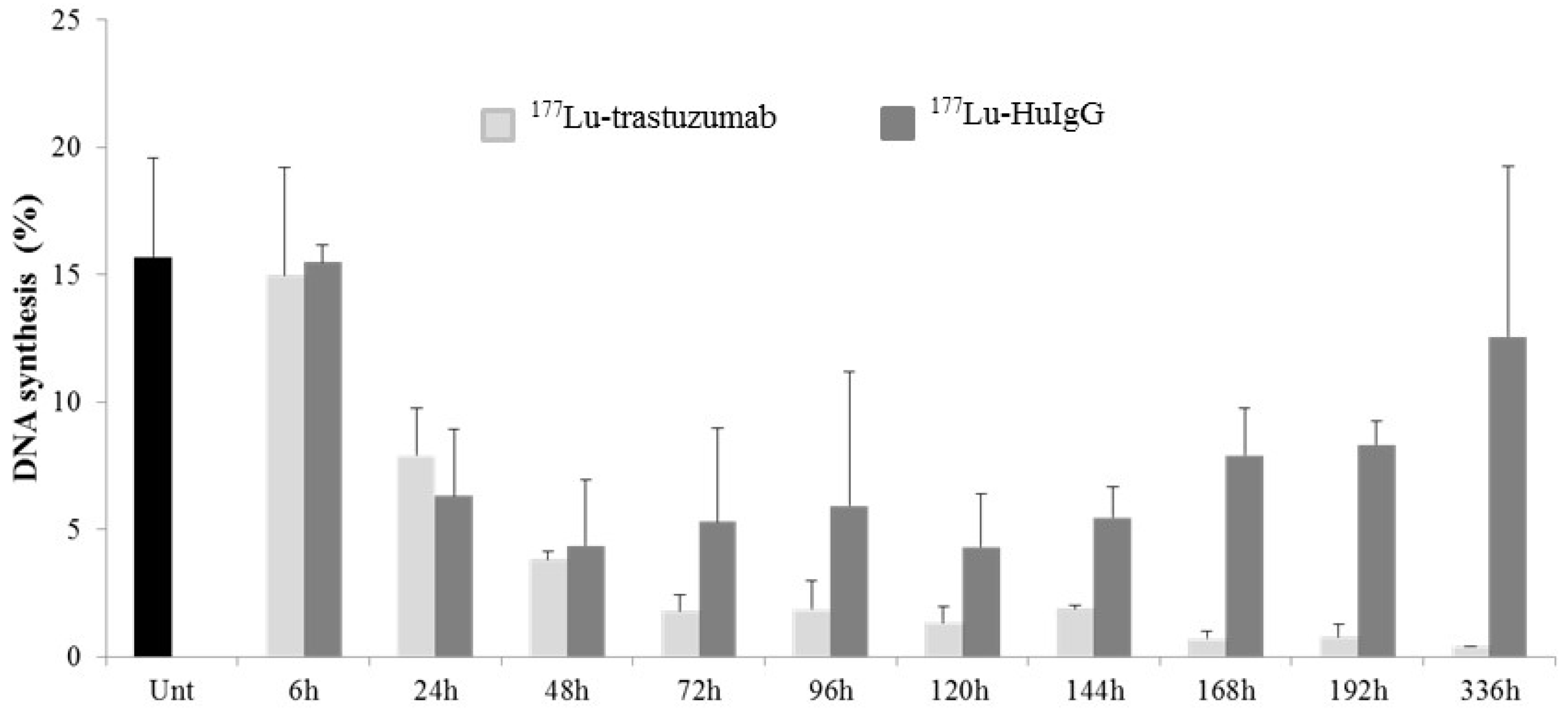
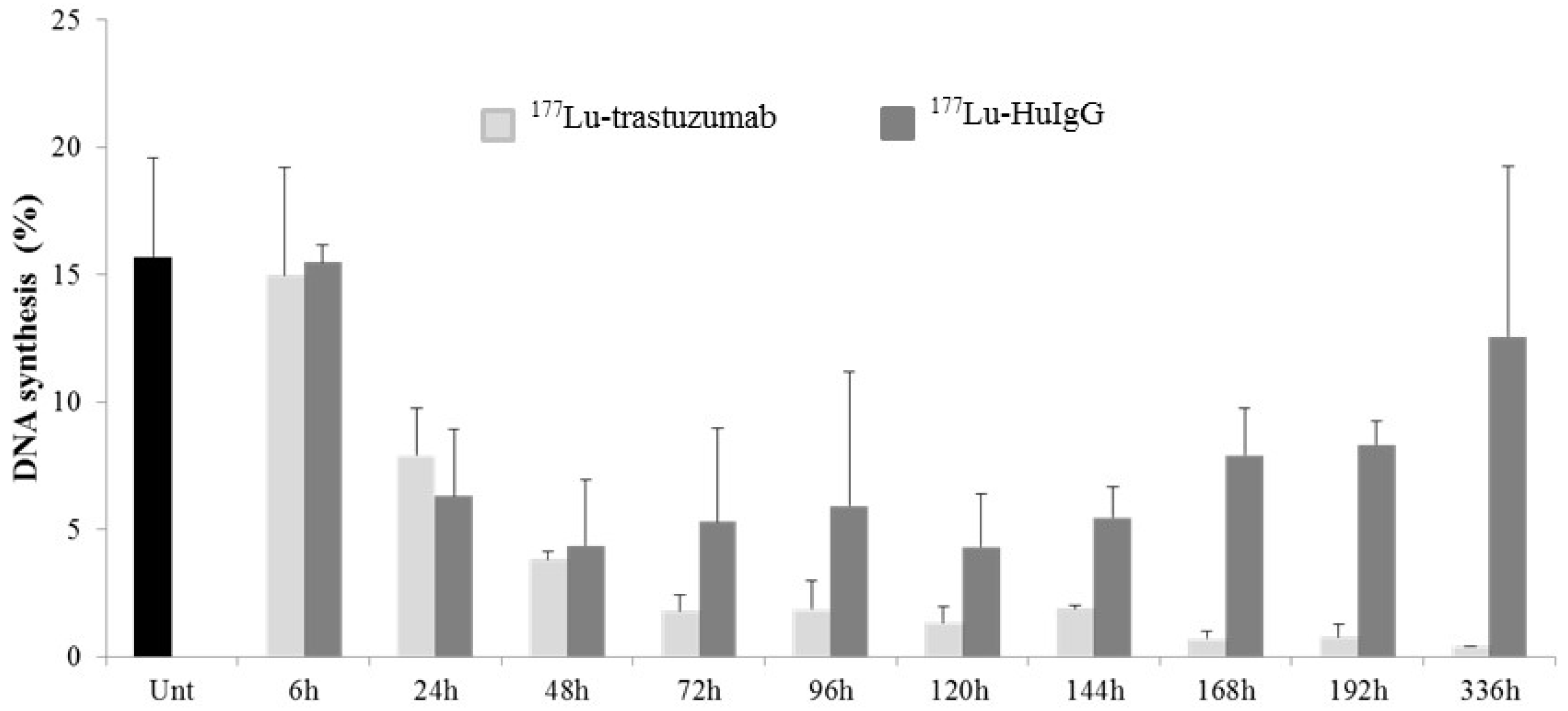
2.4. 177Lu-Trastuzumab Attenuates Proliferation in S Phase and Induces G2 Cell Cycle Arrest
2.5. Up-Regulation of Genes Involved in Apoptosis Is Delayed by 177Lu-Trastuzumab Therapy
2.6. 177Lu-Trastuzumab-Induced Tumor Cytotoxicity May Be Associated with Differential Expression of Genes Involved in Cell Cycle Arrest and Cell Cycle Check Point Regulation
2.7. Expression of Genes Involved in Repair of Damaged DNA Induced by 177Lu-Trastuzumab
3. Discussion
4. Materials and Methods
4.1. Cell Line
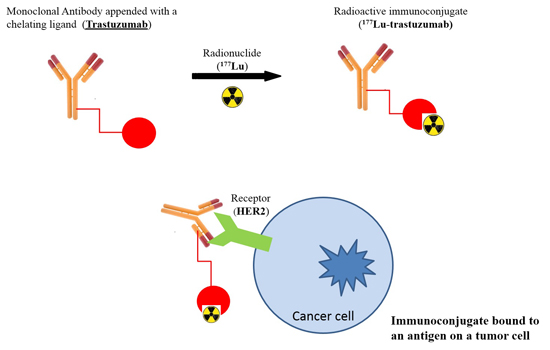
4.2. Preparation of 177Lu-Trastuzumab and 177Lu-HuIgG
4.3. Tumor Model, Treatment, and Tumor Harvesting
4.4. Flow Cytometry Measurements
4.5. Immunohistochemistry
4.6. Immunoblotting
4.7. Metaphase Spread
4.8. RNA Purification
4.9. Human DNA Damage PCR Array
4.10. Statistics
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Milenic, D.E. Monoclonal antibody-based therapy strategies: Providing options for the cancer patient. Curr. Pharm. Des. 2002, 8, 1749–1764. [Google Scholar] [CrossRef] [PubMed]
- Brady, E.D.; Milenic, D.E.; Brechbiel, M.W. Antibody guided precision radiation therapy. Discov. Med. 2004, 4, 213–219. [Google Scholar] [PubMed]
- Kotzerke, J.; Bunjes, D.; Scheinberg, D.A. Radioimmunoconjugates in acute leukemia treatment: The future is radiant. Bone Marrow Transplant. 2005, 36, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Milenic, D.E.; Garmestani, K.; Brady, E.D.; Albert, P.S.; Ma, D.; Abdulla, A.; Brechbiel, M.W. Targeting of HER2 antigen for the treatment of disseminated peritoneal disease. Clin. Cancer Res. 2004, 10, 7834–7841. [Google Scholar] [CrossRef] [PubMed]
- Milenic, D.E.; Garmestani, K.; Brady, E.D.; Albert, P.S.; Ma, D.; Abdulla, A.; Brechbiel, M.W. α-Particle radioimmunotherapy of disseminated peritoneal disease using a 212Pb-labeled radioimmunoconjugate targeting HER2. Cancer Biother. Radiopharm. 2005, 20, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Meredith, R.F.; Torgue, J.; Azure, M.T.; Shen, S.; Saddekni, S.; Banaga, E.; Carlise, R.; Bunch, P.; Yoder, D.; Alvarez, R. Pharmacokinetics and imaging of 212Pb-TCMC-trastuzumab after intraperitoneal administration in ovarian cancer patients. Cancer Biother. Radiopharm. 2014, 29, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Meredith, R.F.; Torgue, J.; Shen, S.; Fisher, D.R.; Banaga, E.; Carlise, R.; Bunch, P.; Yoder, D.; Alvarez, R. Dose escalation and dosimetry of first-in-human α-radioimmunotherapy with 212Pb-TCMC-trastuzumab. J. Nucl. Med. 2014, 55, 1636–1642. [Google Scholar] [CrossRef] [PubMed]
- Rasaneh, S.; Rajabi, H.; Babaei, M.H.; Daha, F.J. 177Lu labeling of Herceptin and preclinical validation as a new radiopharmaceutical for radioimmunotherapy of breast cancer. Nucl. Med. Biol. 2010, 37, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Schlom, J.; Siler, K.; Milenic, D.E.; Eggensperger, D.; Colcher, D.; Miller, L.S.; Houchens, D.; Cheng, R.; Kaplan, D.; Goeckeler, W. Monoclonal antibody-based therapy of a human tumor xenograft with a 177Lu-labeled immunoconjugate. Cancer Res. 1991, 51, 2889–2896. [Google Scholar] [PubMed]
- Ray, G.L.; Baidoo, K.E.; Keller, L.M.; Albert, P.S.; Brechbiel, M.W.; Milenic, D.E. Pre-clinical assessment of Lu-labeled trastuzumab targeting HER2 for treatment and management of cancer patients with disseminated intraperitoneal disease. Phamaceuticals 2011, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Soyland, C.; Hassfjell, S.P. Survival lung epithelial cells following in vitro α-particle irradiation with absolute determination of the number of α-particle traversals of individual cells. Int. J. Radiat. Biol. 2000, 76, 1315–1322. [Google Scholar] [PubMed]
- Yong, K.; Brechbiel, M.W. Towards translation of 212Pb as a clinical therapeutic; getting the lead in! Dalton Trans. 2011, 40, 6068–6076. [Google Scholar] [CrossRef] [PubMed]
- Yong, K.J.; Milenic, D.E.; Baidoo, K.E.; Brechbiel, M.W. 212Pb-radioimmunotherapy induces G2 cell cycle arrest and delays DNA damage repair in tumor xenografts in a model for disseminated intraperitoneal disease. Mol. Cancer Ther. 2012, 11, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Yong, K.J.; Milenic, D.E.; Baidoo, K.E.; Kim, Y.S.; Brechbiel, M.W. Gene expression profiling upon 212Pb-TCMC-trastuzumab treatment in the LS-174T i.p. xenograft. Cancer Med. 2013, 2, 646–653. [Google Scholar] [CrossRef] [PubMed]
- de Jong, M.; Breeman, W.A.; Valkema, R.; Bernard, B.F.; Krenning, E.P. Combination radionuclide therapy using 177Lu- and 90Y-labeled somatostain analogs. J. Nucl. Med. 2005, 46, 13S–17S. [Google Scholar] [PubMed]
- Yong, K.J.; Milenic, D.E.; Baidoo, K.E.; Brechbiel, M.W. Sensitization of tumor to 212Pb radioimmunotherapy by gemcitabine involves initial abrogation of G2 arrest and blocked DNA damage repair by interference with Rad51. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Lo, J.; van Gent, D.C.; Engelward, B.P. DNA double-strand break repair: From mechanistic understanding to cancer treatment. DNA Repair 2007, 6, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Friesen, C.; Fulda, S.; Debatin, K.M. Cytotoxic drugs and the CD95 pathway. Leukemia 1999, 13, 1854–1858. [Google Scholar] [CrossRef] [PubMed]
- Friesen, C.; Lubatschofski, A.; Kotzerke, J.; Buchmann, I.; Reske, S.N.; Debatin, K.M. Beta irradiation used for systemic radioimmunotherapy induces apoptosis and activates apoptosis pathways in leukemia cells. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.; Earnshaw, W.C. Induction of apoptosis by cancer therapy. Exp. Cell Res. 2000, 256, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Sgouros, G.; Knox, S.J.; Jonier, M.C.; Morgan, W.F.; Kassis, A.L. MIRD continuing education: Bystander and low dose late effects: Are these relevant to radionuclide therapy? J. Nucl. Med. 2007, 48, 1683–1691. [Google Scholar] [CrossRef] [PubMed]
- Asaithamby, A.; Hu, B.; Chen, D.J. Unrepaired clustered DNA lesions induce chromosome breakage in human cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8293–8298. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, B.M.; Bennett, P.V.; Sidorkira, O.; Laval, J. Clustered damages and total lesions induced in DNA by ionizing radiation: Oxidized bases and strand breaks. Biochemistry 2000, 39, 8026–8031. [Google Scholar] [CrossRef] [PubMed]
- Podhorecka, M.; Skiadanowski, A.; Bozko, P. H2AX phosphorylation: Its role in DNA damage response and cancer therapy. J. Nucleic Acids 2010. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, M.A. Biomarkers for human radiation exposure. J. Biomed. Sci. 2008, 15, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, B.; Cooper, B.; Cooper, P.K.; Holley, W.R.; Chatterjee, A. Dose-dependent misrejoining of radiation induced DNA double strand breaks in human fibroblasts: Experimental and theoretical study for high- and low-LET radiation. Radiat. Res. 2005, 163, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Zlotorynski, E.; Goldberg, M.; Ozeri, E.; Rahat, A.; le Sage, C.; Chen, D.J.; Agami, R.; Kerem, B. Homologous recombination and nonhomologous end-joining repair pathways regulate fragile site stability. Genes Dev. 2005, 19, 2715–2726. [Google Scholar] [CrossRef] [PubMed]
- Schler, E.; Rudqvist, N.; Paris, T.Z.; Langen, B.; Spetz, J.; Helou, K.; Forssell-Aronsson, E. Time and dose rate-related effects of internal 177Lu exposure on gene expression in mouse kidney tissue. Nucl. Med. Biol. 2014, 41, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Prieto, R.; Sanchez-Arevalo, V.J.; Servitja, J.M.; Gutkind, J.S. Regulation of p73 by cAbl through the p38 MAP kinase pathway. Oncogene 2002, 21, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Shao, Y.; Xu, L.; Yu, L.; Sun, L. Gadd45-α and Gadd45-γ utilize p38 and INK signaling pathways to induce cell cycle G2/M arrest in Hep-G2 heptoma cells. Mol. Biol. Rep. 2009, 36, 2075–2085. [Google Scholar] [CrossRef] [PubMed]
- Rufini, A.; Agostini, M.; Grespi, F.; Tomasini, R.; Sayan, B.S.; Niklison-Chirou, M.V.; Conforti, F.; Velletri, T.; Mastino, A.; Mak, T.W.; et al. p73 in cancer. Genes Cancer 2011, 2, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, K.; Inoue, T.; Tokino, T.; Nakamura, Y.; Akiyama, T. Overexpression of GML promotes radiation-induced cell cycle arrest and apoptosis. Biochem. Biophys. Res. Commun. 1997, 241, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Chehab, N.H.; Malikzay, A.; Appel, M.; Halazonetis, T.D. Chk2/hCds1 functions as a DNA damage checkpoint in G1 by stabilizing p53. Genes Dev. 2000, 14, 278–288. [Google Scholar] [PubMed]
- Lee, J.S.; Collins, K.M.; Brown, A.L.; Lee, C.H.; Chung, J.H. hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature 2000, 404, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Monte, M.; Benetti, R.; Buscemi, G.; Sandy, P.; Del Sal, G.; Schneider, C. The cell cycle-regulated protein human GTSE-1 controls DNA damage-induced apoptosis by affecting p53 function. J. Biol. Chem. 2003, 278, 30356–30364. [Google Scholar] [CrossRef] [PubMed]
- De Wind, N.; Dekker, M.; Berns, A.; Radman, M.; te Riele, H. Inactivation of the mouse MSH2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell 1995, 82, 321–330. [Google Scholar] [CrossRef]
- Meek, K.; Dang, V.; Lees-Miller, S.P. DNA-PK: The means to justify the ends? Adv. Immunol. 2008, 99, 33–58. [Google Scholar] [PubMed]
- Tom, B.H.; Rutzky, L.P.; Jakstys, M.M.; Oyasu, R.; Kaye, C.L.; Kahan, B.D. Human colonic adenocarcinoma cells. I. Establishment and description of a new cell line. Vitro 1976, 12, 180–191. [Google Scholar] [CrossRef]
- Brechbiel, M.W.; Gansow, O.A. Synthesis of C-functionalized trans-cyclohexyldiethylene-triamine-pentaacetic acids for labeling of monoclonal antibodies with the bismuth-212 α-particle emitter. J. Chem. Soc. Perkin Trans. 1992, 1173–1178. [Google Scholar] [CrossRef]
- Pippin, C.G.; Parker, T.A.; McMurry, T.J.; Brechbiel, M.W. Spectrometric method for the determination of a bifunctional DTPA ligand in DTPA-monoclonal antibody conjugates. Bioconjug. Chem. 1992, 3, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Chappell, L.L.; Dadachova, E.; Milenic, D.E.; Garmestani, K.; Wu, C.; Brechbiel, M.W. Synthesis, characterization, and evaluation of a novel bifunctional chelating agent for the lead isotopes 203Pb and 212Pb. Nucl. Med. Biol. 2000, 27, 93–100. [Google Scholar] [CrossRef]
- Gregoire, V.; Van, N.T.; Stephens, L.C.; Brock, W.A.; Milas, L.; Plunkett, W.; Hittelman, W.N. The role of fludarabine-induced apoptosis and cell cycle synchronization in enhanced murine tumor radiation responses in vivo. Cancer Res. 1994, 54, 6201–6209. [Google Scholar] [PubMed]
- Eriksson, S.E.; Elgstrom, E.; Back, T.; Ohlsson, T.; Jenssen, H.; Nilsson, R.; Lindegren, S.; Tennvall, J. Sequential radioimmunotherapy with 177Lu- and 211At-labeled monoclonal antibody BR96 in a syngeneric rat colon carcinoma model. Cancer Biother. Radiopharm. 2014, 29, 238–246. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Phase | Time Point (h) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 6 | 24 | 48 | 72 | 96 | 120 | 144 | 168 | 192 | 336 | ||
| None | G1 | 76.2 ± 1.3 | ||||||||||
| S | 15.4 ± 1.2 | |||||||||||
| G2/M | 8.5 ± 0.1 | |||||||||||
| 177Lu-trastuzumab | G1 | 75.7 ± 6.6 | 76.0 ± 3.9 | 77.8 ± 3.9 | 74.9 ± 2.9 | 78.6 ± 2.1 | 77.9 ± 5.0 | 82.5 ± 3.7 | 77.7 ± 2.6 | 80.2 ± 3.3 | 84.4 ± 3.2 | |
| S | 15.0 ± 5.7 | 10.0 ± 3.5 | 6.10 ± 1.6 | 4.60 ± 1.1 | 4.7 ± 0.7 | 3.6 ± 0.5 | 3.4 ± 0.5 | 3.3 ± 0.5 | 3.5 ± 0.5 | 2.5 ± 0.1 | ||
| G2/M | 9.4 ± 3.1 | 14.0 ± 2.4 | 16.1 ± 2.6 | 20.5 ± 2.0 | 16.6 ± 1.9 | 18.5 ± 4.8 | 14.1 ± 3.9 | 19.1 ± 2.5 | 16.3 ± 2.9 | 13.1 ± 3.1 | ||
| 177Lu-HuIgG | G1 | 73.1 ± 5.5 | 74.7 ± 2.0 | 77.0 ± 5.0 | 71.8 ± 4.4 | 75.5 ± 1.7 | 78.0 ± 3.5 | 80.2 ± 4.9 | 76.2 ± 4.6 | 77.6 ± 3.1 | 75.0 ± 2.7 | |
| S | 18.8 ± 3.2 | 10.6 ± 3.1 | 6.60 ± 2.0 | 8.30 ± 4.4 | 7.9 ± 3.6 | 6.4 ± 1.2 | 7.3 ± 2.5 | 11.1 ± 2.8 | 9.5 ± 2.9 | 12.9 ± 3.0 | ||
| G2/M | 8.1 ± 3.3 | 14.7 ± 2.0 | 16.3 ± 6.0 | 19.9 ± 8.5 | 16.6 ± 4.2 | 15.7 ± 3.0 | 12.5 ± 2.7 | 12.7 ± 2.5 | 12.9 ± 1.2 | 12.2 ± 3.4 | ||
| Symbol | GeneBank ID | Fold Change | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 24 h | 168 h | ||||||||
| 177Lu-Trastuzumab | p | 177Lu-HuIgG | p | 177Lu-Trastuzumab | p | 177Lu-HuIgG | p | ||
| ABL | NM_005157 | −1.6 | 0.001 | −2.2 | 0.036 | 4.9 | 0.012 | −2.6 | 0.001 |
| BRCA1 | NM_007294 | −2.9 | 0.013 | −3.2 | 0.011 | −6.6 | 0.010 | −4.8 | 0.006 |
| CIDEA | NM_001279 | −7.0 | 0.001 | −3.3 | 0.002 | 1.8 | 0.219 | −4.3 | 0.001 |
| GADD45α | NM_001924 | 1.8 | 0.007 | 1.9 | 0.040 | 2.2 | 0.013 | 3.7 | 0.000 |
| GADD45γ | NM_006705 | 1.2 | 0.669 | 1.3 | 0.690 | 2.5 | 0.050 | 2.1 | 0.040 |
| GML | NM_002066 | −1.6 | 0.002 | −2.2 | 0.046 | 5.0 | 0.010 | −2.7 | 0.008 |
| IP6K3 | NM_054111 | −1.1 | 0.931 | −1.2 | 0.899 | 5.0 | 0.010 | −1.6 | 0.210 |
| PCBP4 | NM_020418 | 1.1 | 0.439 | 1.6 | 0.061 | 2.1 | 0.110 | 3.3 | 0.000 |
| RAD21 | NM_006265 | −1.2 | 0.212 | −1.3 | 0.198 | −2.8 | 0.011 | −2.4 | 0.010 |
| p73 | NM_005427 | −1.0 | 0.962 | −1.7 | 0.936 | 2.8 | 0.023 | 2.6 | 0.026 |
| Symbol | GeneBank ID | Fold Change | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 24 h | 168 h | ||||||||
| 177Lu-Trastuzumab | p | 177Lu-HuIgG | p | 177Lu-Trastuzumab | p | 177Lu-HuIgG | p | ||
| BRCA1 | NM_007294 | −2.9 | 0.013 | −3.2 | 0.011 | −6.6 | 0.010 | −4.8 | 0.006 |
| CHK1 | NM_001274 | −1.7 | 0.004 | −1.9 | 0.001 | −4.6 | 0.001 | −3.7 | 0.001 |
| CHK2 | NM_007194 | −1.7 | 0.023 | −1.9 | 0.010 | −7.1 | 0.001 | −2.5 | 0.003 |
| DDIT3 | NM_004083 | −2.5 | 0.008 | −2.9 | 0.006 | −2.5 | 0.053 | −1.6 | 0.037 |
| FANCG | NM_004629 | −2.6 | 0.003 | −2.4 | 0.004 | −1.7 | 0.022 | −4.3 | 0.001 |
| GADD45α | NM_001924 | 1.8 | 0.007 | 1.9 | 0.040 | 2.2 | 0.013 | 3.7 | 0.000 |
| GML | NM_002066 | −1.6 | 0.001 | −2.2 | 0.046 | 5.0 | 0.010 | −2.7 | 0.008 |
| GTSE1 | NM_016426 | −1.7 | 0.032 | −2.4 | 0.009 | −9.0 | 0.001 | −3.5 | 0.004 |
| MAPK12 | NM_002969 | −4.7 | 0.005 | −5.0 | 0.005 | 1.5 | 0.053 | −3.6 | 0.007 |
| NBN | NM_002485 | −1.1 | 0.037 | −1.2 | 0.035 | −2.8 | 0.015 | −1.3 | 0.731 |
| PCBP4 | NM_020418 | 1.1 | 0.043 | 1.5 | 0.061 | 2.1 | 0.116 | 3.2 | 0.000 |
| SESN1 | NM_014454 | 4.5 | 0.001 | 5.4 | 0.000 | 2.3 | 0.008 | 4.0 | 0.002 |
| Symbol | GeneBank ID | Fold Change | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 24 h | 168 h | ||||||||
| 177Lu-Trastuzumab | p | 177Lu-HuIgG | p | 177Lu-Trastuzumab | p | 177Lu-HuIgG | p | ||
| BRCA1 | NM_007294 | −2.9 | 0.013 | −3.2 | 0.010 | −6.6 | 0.010 | −4.8 | 0.006 |
| BTG2 | NM_006763 | 3.4 | 0.000 | 4.8 | 0.000 | 3.9 | 0.004 | 4.8 | 0.000 |
| ERCC1 | NM_001983 | 1.1 | 0.486 | 1.2 | 0.491 | 1.8 | 0.160 | 2.8 | 0.000 |
| EXO1 | NM_130398 | −1.8 | 0.061 | −2.4 | 0.014 | −3.4 | 0.008 | −3.8 | 0.006 |
| FANCG | NM_004629 | −2.6 | 0.002 | −2.4 | 0.004 | −1.7 | 0.022 | −4.3 | 0.001 |
| FEN1 | NM_004111 | −1.4 | 0.219 | −2.1 | 0.085 | −4.7 | 0.023 | −2.2 | 0.059 |
| LIG1 | NM_00234 | −1.9 | 0.009 | −1.6 | 0.015 | −1.5 | 0.048 | −3.3 | 0.012 |
| MRE11A | NM_005590 | 2.4 | 0.001 | 1.2 | 0.434 | −3.7 | 0.006 | −3.6 | 0.035 |
| MSH2 | NM_00251 | −1.2 | 0.229 | −1.6 | 0.081 | −5.4 | 0.014 | −2.1 | 0.030 |
| NBN | NM_002485 | −1.1 | 0.392 | −1.2 | 0.357 | −2.8 | 0.015 | −1.3 | 0.731 |
| PNKP | NM_007254 | −3.2 | 0.006 | −2.7 | 0.011 | 1.9 | 0.003 | −1.8 | 0.027 |
| PRKDC | NM_006904 | −1.1 | 0.392 | −1.0 | 0.933 | −5.4 | 0.002 | −1.7 | 0.016 |
| RAD18 | NM_020165 | −2.1 | 0.014 | −2.5 | 0.006 | −1.5 | 0.006 | −3.3 | 0.002 |
| RAD21 | NM_006265 | −1.1 | 0.007 | −1.3 | 0.198 | −2.8 | 0.011 | −2.4 | 0.010 |
| RAD51B | NM_133509 | −1.6 | 0.372 | −1.8 | 0.897 | −5.0 | 0.018 | −1.8 | 0.040 |
| p73 | NM_005427 | −1.0 | 0.962 | −1.7 | 0.936 | 2.8 | 0.023 | 2.6 | 0.026 |
| UNG | NM_003362 | −2.3 | 0.002 | −1.9 | 0.014 | −1.7 | 0.024 | −2.4 | 0.001 |
| XPC | NM_004628 | 2.7 | 0.001 | 3.5 | 0.003 | 2.3 | 0.003 | 3.0 | 0.000 |
| XRCC2 | NM_005431 | 1.0 | 0.988 | −1.3 | 0.270 | −1.3 | 0.271 | −2.6 | 0.031 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yong, K.J.; Milenic, D.E.; Baidoo, K.E.; Brechbiel, M.W. Mechanisms of Cell Killing Response from Low Linear Energy Transfer (LET) Radiation Originating from 177Lu Radioimmunotherapy Targeting Disseminated Intraperitoneal Tumor Xenografts. Int. J. Mol. Sci. 2016, 17, 736. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17050736
Yong KJ, Milenic DE, Baidoo KE, Brechbiel MW. Mechanisms of Cell Killing Response from Low Linear Energy Transfer (LET) Radiation Originating from 177Lu Radioimmunotherapy Targeting Disseminated Intraperitoneal Tumor Xenografts. International Journal of Molecular Sciences. 2016; 17(5):736. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17050736
Chicago/Turabian StyleYong, Kwon Joong, Diane E. Milenic, Kwamena E. Baidoo, and Martin W. Brechbiel. 2016. "Mechanisms of Cell Killing Response from Low Linear Energy Transfer (LET) Radiation Originating from 177Lu Radioimmunotherapy Targeting Disseminated Intraperitoneal Tumor Xenografts" International Journal of Molecular Sciences 17, no. 5: 736. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17050736