
Molecular Mechanisms of Pulmonary Vascular Remodeling in Pulmonary Arterial Hypertension

Abstract
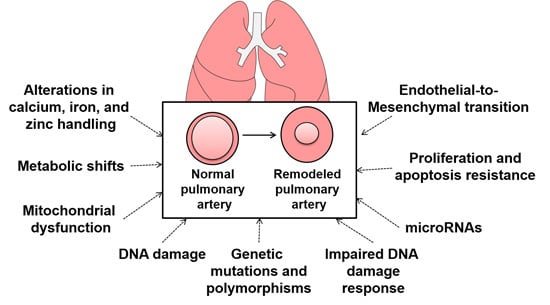
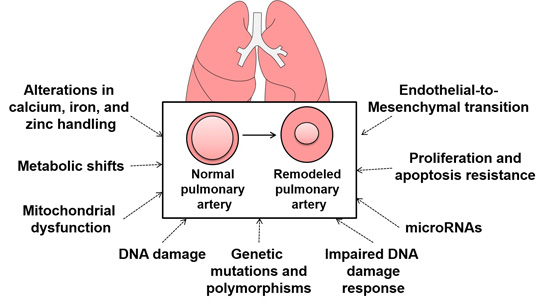
:
1. Introduction
2. Genetic and Epigenetic Regulation of Pulmonary Arterial Hypertension (PAH)
3. DNA Damage in PAH and the DNA Damage Response
4. MicroRNAs Regulate Gene Expression in PAH
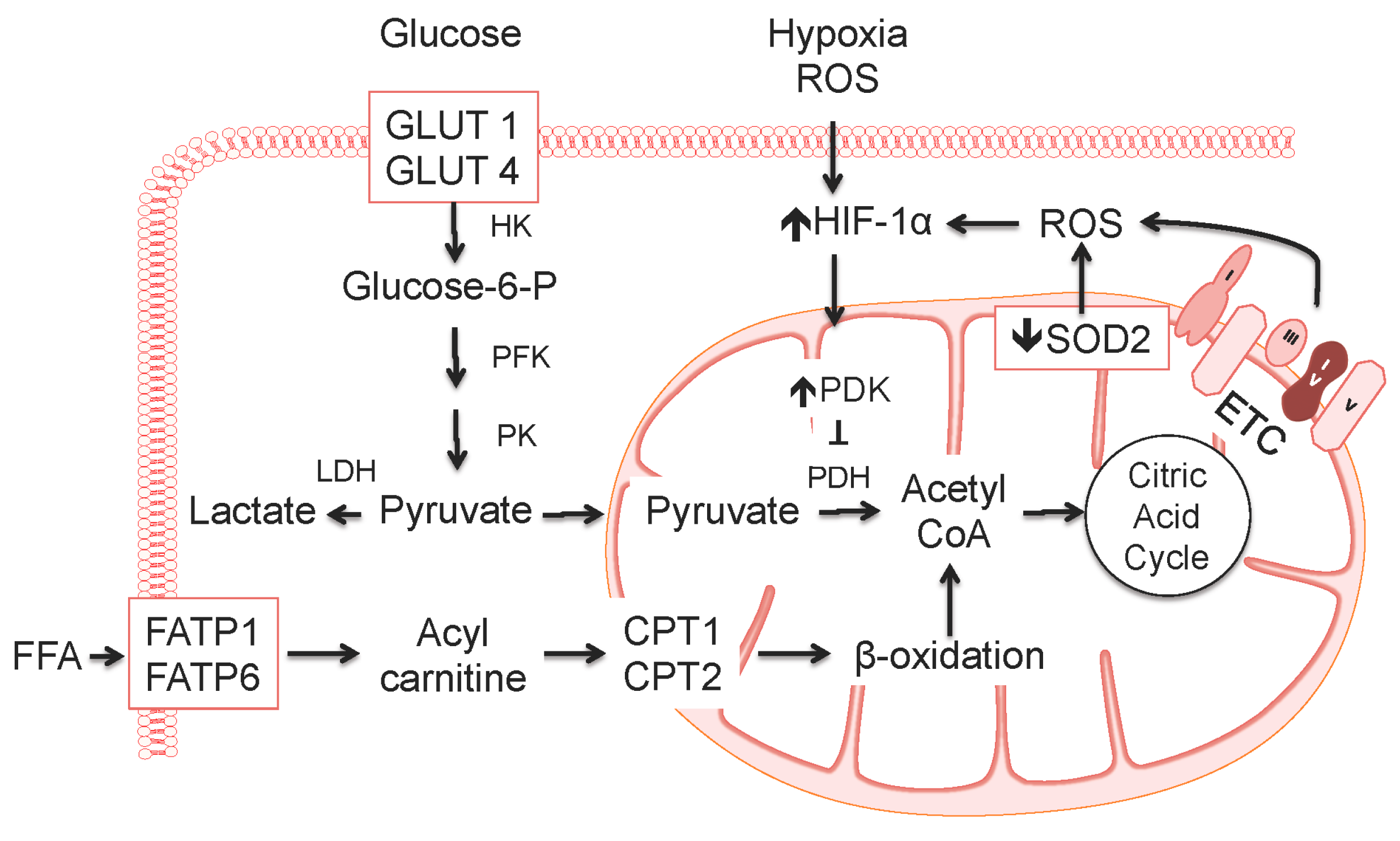
5. Changes in Cellular Metabolism, Metabolic Flux, and Mitochondrial Function
6. Zinc, Iron, and Calcium Handling in Pulmonary Hypertension
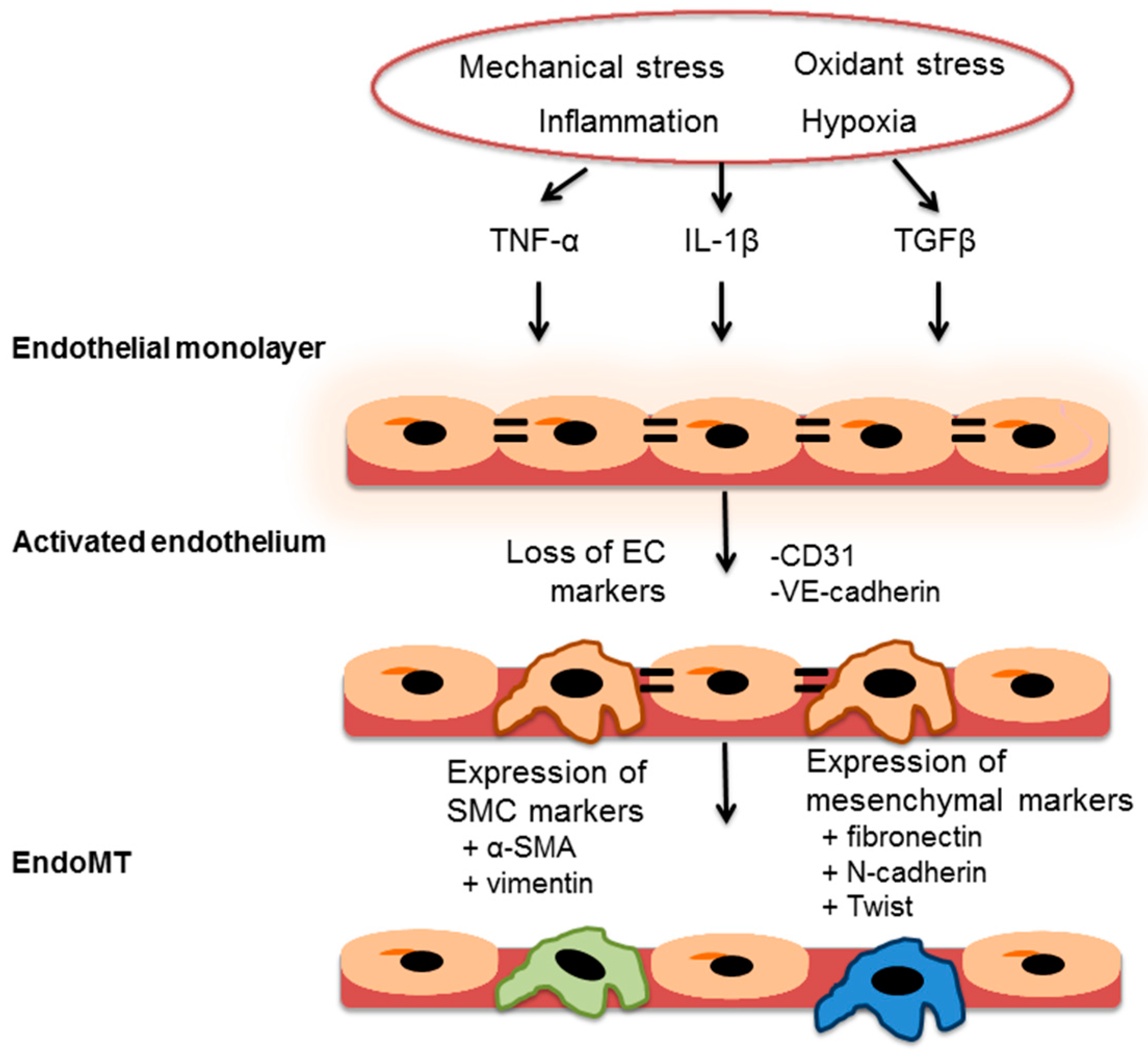
7. Endothelial-to-Mesenchymal Transition
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hoeper, M.M.; Bogaard, H.J.; Condliffe, R.; Frantz, R.; Khanna, D.; Kurzyna, M.; Langleben, D.; Manes, A.; Satoh, T.; Torres, F.; et al. Definitions and diagnosis of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D42–D50. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Johnson, M.K.; Kiely, D.G.; Condliffe, R.; Elliot, C.A.; Gibbs, J.S.; Howard, L.S.; Pepke-Zaba, J.; Sheares, K.K.; Corris, P.A.; et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: Results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am. J. Respir. Crit. Care Med. 2012, 186, 790–796. [Google Scholar] [CrossRef] [PubMed]
- McGoon, M.D.; Benza, R.L.; Escribano-Subias, P.; Jiang, X.; Miller, D.P.; Peacock, A.J.; Pepke-Zaba, J.; Pulido, T.; Rich, S.; Rosenkranz, S.; et al. Pulmonary arterial hypertension: Epidemiology and registries. J. Am. Coll. Cardiol. 2013, 62, D51–D59. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Archer, S.L.; Dorfmuller, P.; Erzurum, S.C.; Guignabert, C.; Michelakis, E.; Rabinovitch, M.; Schermuly, R.; Stenmark, K.R.; Morrell, N.W. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D4–D12. [Google Scholar] [CrossRef] [PubMed]
- International, P.P.H.C.; Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A., 3rd; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar]
- Deng, Z.; Haghighi, F.; Helleby, L.; Vanterpool, K.; Horn, E.M.; Barst, R.J.; Hodge, S.E.; Morse, J.H.; Knowles, J.A. Fine mapping of PPH1, a gene for familial primary pulmonary hypertension, to a 3-cM region on chromosome 2q33. Am. J. Respir. Crit. Care Med. 2000, 161, 1055–109. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.; Machado, R.; Pauciulo, M.; Morgan, N.; Yacoub, M.; Corris, P.; McNeil, K.; Loyd, J.; Nichols, W.; Trembath, R. Familial and sporadic primary pulmonary hypertension is caused by BMPR2 gene mutations resulting in haploinsufficiency of the bone morphogenetic protein tuype II receptor. J. Heart Lung Transplant. 2001, 20, 149. [Google Scholar] [CrossRef]
- Machado, R.D.; Southgate, L.; Eichstaedt, C.A.; Aldred, M.A.; Austin, E.D.; Best, D.H.; Chung, W.K.; Benjamin, N.; Elliott, C.G.; Eyries, M.; et al. Pulmonary Arterial Hypertension: A Current Perspective on Established and Emerging Molecular Genetic Defects. Hum. Mutat. 2015, 36, 1113–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, E.D.; Ma, L.; LeDuc, C.; Rosenzweig, E.B.; Borczuk, A.; Phillips, J.A., III; Palomero, T.; Sumazin, P.; Kim, H.R.; Talati, M.H.; et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ. Cardiovasc. Genet. 2012, 5, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Roman-Campos, D.; Austin, E.D.; Eyries, M.; Sampson, K.S.; Soubrier, F.; Germain, M.; Tregouet, D.A.; Borczuk, A.; Rosenzweig, E.B.; et al. A novel channelopathy in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Eyries, M.; Montani, D.; Girerd, B.; Perret, C.; Leroy, A.; Lonjou, C.; Chelghoum, N.; Coulet, F.; Bonnet, D.; Dorfmuller, P.; et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat. Genet. 2014, 46, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Germain, M.; Eyries, M.; Montani, D.; Poirier, O.; Girerd, B.; Dorfmuller, P.; Coulet, F.; Nadaud, S.; Maugenre, S.; Guignabert, C.; et al. Genome-wide association analysis identifies a susceptibility locus for pulmonary arterial hypertension. Nat. Genet. 2013, 45, 518–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Knight, L.; Ji, R.; Lawrence, P.; Kanaan, U.; Li, L.; Das, A.; Cui, B.; Zou, W.; Penny, D.J.; et al. Early onset severe pulmonary arterial hypertension with “two-hit” digenic mutations in both BMPR2 and KCNA5 genes. Int. J. Cardiol. 2014, 177, e167–e169. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.D.; Shroyer, K.R.; Markham, N.E.; Cool, C.D.; Voelkel, N.F.; Tuder, R.M. Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J. Clin. Investig. 1998, 101, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Lee, S.D.; Cool, C.C. Histopathology of pulmonary hypertension. Chest 1998, 114, 1S–6S. [Google Scholar] [CrossRef] [PubMed]
- Yeager, M.E.; Halley, G.R.; Golpon, H.A.; Voelkel, N.F.; Tuder, R.M. Microsatellite instability of endothelial cell growth and apoptosis genes within plexiform lesions in primary pulmonary hypertension. Circ. Res. 2001, 88, E2–E11. [Google Scholar] [CrossRef] [PubMed]
- Aldred, M.A.; Comhair, S.A.; Varella-Garcia, M.; Asosingh, K.; Xu, W.; Noon, G.P.; Thistlethwaite, P.A.; Tuder, R.M.; Erzurum, S.C.; Geraci, M.W.; et al. Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2010, 182, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Meloche, J.; Pflieger, A.; Vaillancourt, M.; Paulin, R.; Potus, F.; Zervopoulos, S.; Graydon, C.; Courboulin, A.; Breuils-Bonnet, S.; Tremblay, E.; et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 2014, 129, 786–797. [Google Scholar] [CrossRef] [PubMed]
- De Jesus Perez, V.A.; Yuan, K.; Lyuksyutova, M.A.; Dewey, F.; Orcholski, M.E.; Shuffle, E.M.; Mathur, M.; Yancy, L., Jr.; Rojas, V.; Li, C.G.; et al. Whole-exome sequencing reveals TopBP1 as a novel gene in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 1260–1272. [Google Scholar] [CrossRef] [PubMed]
- Federici, C.; Drake, K.M.; Rigelsky, C.M.; McNelly, L.N.; Meade, S.L.; Comhair, S.A.; Erzurum, S.C.; Aldred, M.A. Increased Mutagen Sensitivity and DNA Damage in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Vattulainen, S.; Aho, J.; Orcholski, M.; Rojas, V.; Yuan, K.; Helenius, M.; Taimen, P.; Myllykangas, S.; de Jesus Perez, V.; et al. Loss of bone morphogenetic protein receptor 2 is associated with abnormal DNA repair in pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. microRNAs: Tiny regulators with great potential. Cell 2001, 107, 823–826. [Google Scholar] [CrossRef]
- Courboulin, A.; Paulin, R.; Giguere, N.J.; Saksouk, N.; Perreault, T.; Meloche, J.; Paquet, E.R.; Biardel, S.; Provencher, S.; Cote, J.; et al. Role for miR-204 in human pulmonary arterial hypertension. J. Exp. Med. 2011, 208, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J.; Wharton, J.; Boon, R.A.; Roexe, T.; Tsang, H.; Wojciak-Stothard, B.; Chakrabarti, A.; Howard, L.S.; Gibbs, J.S.; Lawrie, A.; et al. Reduced microRNA-150 is associated with poor survival in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2013, 187, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Bockmeyer, C.L.; Maegel, L.; Janciauskiene, S.; Rische, J.; Lehmann, U.; Maus, U.A.; Nickel, N.; Haverich, A.; Hoeper, M.M.; Golpon, H.A.; et al. Plexiform vasculopathy of severe pulmonary arterial hypertension and microRNA expression. J. Heart Lung Transplant. 2012, 31, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Chen, T.; Raj, J.U. MicroRNAs in pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2015, 52, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Caruso, P.; MacLean, M.R.; Khanin, R.; McClure, J.; Soon, E.; Southgate, M.; MacDonald, R.A.; Greig, J.A.; Robertson, K.E.; Masson, R.; et al. Dynamic Changes in Lung MicroRNA Profiles During the Development of Pulmonary Hypertension due to Chronic Hypoxia and Monocrotaline. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 716–723. [Google Scholar] [CrossRef] [PubMed]
- White, K.; Lu, Y.; Annis, S.; Hale, A.E.; Chau, B.N.; Dahlman, J.E.; Hemann, C.; Opotowsky, A.R.; Vargas, S.O.; Rosas, I.; et al. Genetic and hypoxic alterations of the microRNA-210-ISCU1/2 axis promote iron-sulfur deficiency and pulmonary hypertension. EMBO Mol. Med. 2015, 7, 695–713. [Google Scholar] [CrossRef] [PubMed]
- Potus, F.; Ruffenach, G.; Dahou, A.; Thebault, C.; Breuils-Bonnet, S.; Tremblay, È.; Nadeau, V.; Paradis, R.; Graydon, C.; Wong, R.; et al. Downregulation of MicroRNA-126 Contributes to the Failing Right Ventricle in Pulmonary Arterial Hypertension. Circulation 2015, 132, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Stevens, H.C.; Deng, L.; Grant, J.S.; Pinel, K.; Thomas, M.; Morrell, N.W.; MacLean, M.R.; Baker, A.H.; Denby, L. Regulation and function of miR-214 in pulmonary arterial hypertension. Pulm. Circ. 2016, 6, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Brock, M.; Trenkmann, M.; Gay, R.E.; Michel, B.A.; Gay, S.; Fischler, M.; Ulrich, S.; Speich, R.; Huber, L.C. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ. Res. 2009, 104, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Pullamsetti, S.S.; Doebele, C.; Fischer, A.; Savai, R.; Kojonazarov, B.; Dahal, B.K.; Ghofrani, H.A.; Weissmann, N.; Grimminger, F.; Bonauer, A.; et al. Inhibition of microRNA-17 improves lung and heart function in experimental pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Cordes, K.R.; Sheehy, N.T.; White, M.P.; Berry, E.C.; Morton, S.U.; Muth, A.N.; Lee, T.H.; Miano, J.M.; Ivey, K.N.; Srivastava, D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009, 460, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Boettger, T.; Beetz, N.; Kostin, S.; Schneider, J.; Kruger, M.; Hein, L.; Braun, T. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the miR143/145 gene cluster. J. Clin. Investig. 2009, 119, 2634–2647. [Google Scholar] [CrossRef] [PubMed]
- Courboulin, A.; Tremblay, V.L.; Barrier, M.; Meloche, J.; Jacob, M.H.; Chapolard, M.; Bisserier, M.; Paulin, R.; Lambert, C.; Provencher, S.; et al. Kruppel-like factor 5 contributes to pulmonary artery smooth muscle proliferation and resistance to apoptosis in human pulmonary arterial hypertension. Respir. Res. 2011, 12, 128. [Google Scholar] [CrossRef] [PubMed]
- Caruso, P.; Dempsie, Y.; Stevens, H.C.; McDonald, R.A.; Long, L.; Lu, R.; White, K.; Mair, K.M.; McClure, J.D.; Southwood, M.; et al. A role for miR-145 in pulmonary arterial hypertension: evidence from mouse models and patient samples. Circ. Res. 2012, 111, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Mitsialis, S.A.; Aslam, M.; Vitali, S.H.; Vergadi, E.; Konstantinou, G.; Sdrimas, K.; Fernandez-Gonzalez, A.; Kourembanas, S. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation 2012, 126, 2601–2611. [Google Scholar] [CrossRef] [PubMed]
- Parikh, V.N.; Jin, R.C.; Rabello, S.; Gulbahce, N.; White, K.; Hale, A.; Cottrill, K.A.; Shaik, R.S.; Waxman, A.B.; Zhang, Y.Y.; et al. MicroRNA-21 integrates pathogenic signaling to control pulmonary hypertension: results of a network bioinformatics approach. Circulation 2012, 125, 1520–1532. [Google Scholar] [CrossRef] [PubMed]
- Bertero, T.; Cottrill, K.; Krauszman, A.; Lu, Y.; Annis, S.; Hale, A.; Bhat, B.; Waxman, A.B.; Chau, B.N.; Kuebler, W.M.; et al. The microRNA-130/301 family controls vasoconstriction in pulmonary hypertension. J. Biol. Chem. 2015, 290, 2069–2085. [Google Scholar] [CrossRef] [PubMed]
- Bertero, T.; Lu, Y.; Annis, S.; Hale, A.; Bhat, B.; Saggar, R.; Saggar, R.; Wallace, W.D.; Ross, D.J.; Vargas, S.O.; et al. Systems-level regulation of microRNA networks by miR-130/301 promotes pulmonary hypertension. J. Clin. Investig. 2014, 124, 3514–3528. [Google Scholar] [CrossRef] [PubMed]
- Paulin, R.; Michelakis, E.D. The metabolic theory of pulmonary arterial hypertension. Circ. Res. 2014, 115, 148–164. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.P.; Lehninger, A.L. Oxidation of fatty acids and tricarboxylic acid cycle intermediates by isolated rat liver mitochondria. J. Biol. Chem. 1949, 179, 957–972. [Google Scholar] [PubMed]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L. Mitochondrial dynamics—Mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [PubMed]
- Yu, A.Y.; Shimoda, L.A.; Iyer, N.V.; Huso, D.L.; Sun, X.; McWilliams, R.; Beaty, T.; Sham, J.S.; Wiener, C.M.; Sylvester, J.T.; et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J. Clin. Investig. 1999, 103, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Sidhu, V.K.; Fang, Y.H.; Ryan, J.J.; Parikh, K.S.; Hong, Z.; Toth, P.T.; Morrow, E.; Kutty, S.; Lopaschuk, G.D.; et al. FOXO1-mediated upregulation of pyruvate dehydrogenase kinase-4 (PDK4) decreases glucose oxidation and impairs right ventricular function in pulmonary hypertension: therapeutic benefits of dichloroacetate. J. Mol. Med. 2013, 3, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Koeck, T.; Lara, A.R.; Neumann, D.; DiFilippo, F.P.; Koo, M.; Janocha, A.J.; Masri, F.A.; Arroliga, A.C.; Jennings, C.; et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc. Natl. Acad. Sci. USA 2007, 104, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Marsboom, G.; Toth, P.T.; Ryan, J.J.; Hong, Z.; Wu, X.; Fang, Y.H.; Thenappan, T.; Piao, L.; Zhang, H.J.; Pogoriler, J.; et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ. Res. 2012, 110, 1484–1497. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Marsboom, G.; Kim, G.H.; Zhang, H.J.; Toth, P.T.; Svensson, E.C.; Dyck, J.R.; Gomberg-Maitland, M.; Thébaud, B.; Husain, A.N.; et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation 2010, 121, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Guignabert, C.; Tu, L.; Izikki, M.; Dewachter, L.; Zadigue, P.; Humbert, M.; Adnot, S.; Fadel, E.; Eddahibi, S. Dichloroacetate treatment partially regresses established pulmonary hypertension in mice with SM22α-targeted overexpression of the serotonin transporter. FASEB J. 2009, 23, 4135–4147. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Michelakis, E.D.; Porter, C.J.; Andrade-Navarro, M.A.; Thebaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; et al. An abnormal mitochondrial-hypoxia inducible factor-1α-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation 2006, 113, 2630–2641. [Google Scholar] [CrossRef] [PubMed]
- McMurtry, M.S.; Bonnet, S.; Wu, X.; Dyck, J.R.; Haromy, A.; Hashimoto, K.; Michelakis, E.D. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ. Res. 2004, 95, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Gomberg-Maitland, M.; Maitland, M.L.; Rich, S.; Garcia, J.G.; Weir, E.K. Mitochondrial metabolism, redox signaling, and fusion: A mitochondria-ROS-HIF-1α-Kv1.5O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H570–H578. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Oliver, E.; Maratou, K.; Atanur, S.S.; Dubois, O.D.; Cotroneo, E.; Chen, C.N.; Wang, L.; Arce, C.; Chabosseau, P.L.; et al. The zinc transporter ZIP12 regulates the pulmonary vascular response to chronic hypoxia. Nature 2015, 524, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Cotroneo, E.; Ashek, A.; Wang, L.; Wharton, J.; Dubois, O.; Bozorgi, S.; Busbridge, M.; Alavian, K.N.; Wilkins, M.R.; Zhao, L. Iron homeostasis and pulmonary hypertension: Iron deficiency leads to pulmonary vascular remodeling in the rat. Circ. Res. 2015, 116, 1680–1690. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, G.; Manders, E.; Happe, C.M.; Schalij, I.; Groepenhoff, H.; Howard, L.S.; Wilkins, M.R.; Bogaard, H.J.; Westerhof, N.; van der Laarse, W.J.; et al. Intravenous iron therapy in patients with idiopathic pulmonary arterial hypertension and iron deficiency. Pulm. Circ. 2015, 5, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Rochefort, G.; Sutendra, G.; Archer, S.L.; Haromy, A.; Webster, L.; Hashimoto, K.; Bonnet, S.N.; Michelakis, E.D. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc. Natl. Acad. Sci. USA 2007, 104, 11418–11423. [Google Scholar] [CrossRef] [PubMed]
- Kuga, T.; Kobayashi, S.; Hirakawa, Y.; Kanaide, H.; Takeshita, A. Cell cycle—Dependent expression of L- and T-type Ca2+ currents in rat aortic smooth muscle cells in primary culture. Circ. Res. 1996, 79, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Li, S.; Hong, W.; Hu, J.; Jiang, Y.; Hu, G.; Zou, Y.; Zhou, Y.; Xu, J.; Ran, P. Chronic Hypoxia Increases Intracellular Ca2+ Concentration via Enhanced Ca2+ Entry Through Receptor-Operated Ca2+ Channels in Pulmonary Venous Smooth Muscle Cells. Circ. J. 2015, 79, 2058–2068. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, R.A.; Wan, J.; Song, S.; Smith, K.A.; Gu, Y.; Tauseef, M.; Tang, H.; Makino, A.; Mehta, D.; Yuan, J.X. Upregulated expression of STIM2, TRPC6, and Orai2 contributes to the transition of pulmonary arterial smooth muscle cells from a contractile to proliferative phenotype. Am. J. Physiol. Cell Physiol. 2015, 308, C581–C593. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, G.; Ducret, T.; Marthan, R.; Savineau, J.P.; Quignard, J.F. Stretch-induced Ca2+ signalling in vascular smooth muscle cells depends on Ca2+ store segregation. Cardiovasc. Res. 2014, 103, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Hadri, L.; Kratlian, R.G.; Benard, L.; Maron, B.A.; Dorfmuller, P.; Ladage, D.; Guignabert, C.; Ishikawa, K.; Aguero, J.; Ibanez, B.; et al. Therapeutic efficacy of AAV1.SERCA2a in monocrotaline-induced pulmonary arterial hypertension. Circulation 2013, 128, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Aguero, J.; Ishikawa, K.; Hadri, L.; Santos-Gallego, C.; Fish, K.; Kohlbrenner, E.; Hammoudi, N.; Kho, C.; Lee, A.; Ibanez, B.; et al. Intratracheal gene delivery of SERCA2a amerliorates chronic post-capillary pulmonary hypertension: A large animal model. J. Am. Coll. Cardiol. 2016, 67, 2032–2046. [Google Scholar] [CrossRef] [PubMed]
- Aguero, J.; Ishikawa, K.; Hadri, L.; Santos-Gallego, C.; Fish, K.; Hammoudi, N.; Chaanine, A.; Torquato, S.; Naim, C.; Ibanez, B.; et al. Characterization of right ventricular remodeling and failure in a chronic pulmonary hypertension model. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1204–H1215. [Google Scholar] [CrossRef] [PubMed]
- Frid, M.G.; Kale, V.A.; Stenmark, K.R. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: in vitro analysis. Circ. Res. 2002, 90, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Krenning, G.; Barauna, V.G.; Krieger, J.E.; Harmsen, M.C.; Moonen, J.R. Endothelial Plasticity: Shifting Phenotypes through Force Feedback. Stem Cells Int. 2016, 2016, 9762959. [Google Scholar] [CrossRef] [PubMed]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Pechoux, C.; Bogaard, H.J.; Dorfmuller, P.; Remy, S.; Lecerf, F.; Plante, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Good, R.B.; Gilbane, A.J.; Trinder, S.L.; Denton, C.P.; Coghlan, G.; Abraham, D.J.; Holmes, A.M. Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am. J. Pathol. 2015, 185, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Nishimura, T.; Shi, L.; Sessions, D.; Thrasher, A.; Trudell, J.R.; Berry, G.J.; Pearl, R.G.; Kao, P.N. Endothelial fate mapping in mice with pulmonary hypertension. Circulation 2014, 129, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.Q.; Lighthouse, J.K.; Greif, D.M. Recapitulation of developing artery muscularization in pulmonary hypertension. Cell Rep. 2014, 6, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.Q.; Misra, A.; Rosas, I.O.; Adams, R.H.; Greif, D.M. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci. Transl. Med. 2015, 7, 308ra159. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| WHO Group | Clinical Group | Clinical Definition | Hemodynamic Definition |
|---|---|---|---|
| 1 | Pulmonary arterial hypertension | Precapillary PH | mPA ≥ 25 mmHg |
| mPAWP < 15 mmHg | |||
| 2 | PH due to left heart disease | Postcapillary PH | mPA ≥ 25 mmHg |
| mPAWP > 15 mmHg | |||
| Isolated postcapillary PH | DPG < 7 mmHg and/or | ||
| PVR ≤ 3 Wood units | |||
| Combined postcapillary and precapillary PH | DPG < 7 mmHg and/or | ||
| PVR ≥ 3 Wood units | |||
| 3 | PH due to lung disease or hypoxia | Precapillary PH | mPA ≥ 25 mmHg |
| mPAWP < 15 mmHg | |||
| 4 | Chronic thromboembolic pulmonary hypertension | Precapillary PH | mPA ≥ 25 mmHg |
| mPCWP < 15 mmHg | |||
| 5 | PH associated with miscellaneous diseases | Precapillary PH | mPA ≥ 25 mmHg |
| mPAWP < 15 mmHg | |||
| Postcapillary PH | mPA ≥ 25 mmHg | ||
| mPAWP > 15 mmHg | |||
| Isolated postcapillary PH | DPG < 7 mmHg and/or | ||
| PVR ≤ 3 Wood units | |||
| Combined postcapillary and precapillary PH | DPG < 7 mmHg and/or | ||
| PVR ≥ 3 Wood units |
| MicroRNA | Expression in PAH | Species and Model | Reference |
|---|---|---|---|
| miR-17-92 | ↑ | Mouse—hypoxia | [27,32] |
| Rat—monocrotaline, hypoxia | |||
| miR-21 | ↑ | Mouse—hypoxia, Sugen5416/hypoxia, VHL null | [25,38] |
| Interleukin-6 transgenic | |||
| Rat—monocrotaline | |||
| Human PAH—pulmonary arteries, plexiform lesions | |||
| miR-126 | ↓ | Rat—monocrotaline | [29] |
| Human PAH—right ventricle | |||
| miR-145 | ↑ | Mouse—hypoxia, BMPR2 mutation | [25,36] |
| Human PAH—lung tissue, plexiform lesions | |||
| miR-150 | ↓ | Human PAH—plasma | [24] |
| miR-204 | ↓ | Mouse—hypoxia | [23,25,37] |
| Rat—monocrotaline, Sugen5416/hypoxia | |||
| Human PAH—lung, pulmonary arteries | |||
| miR-210 | ↑ | Mouse—Sugen5416/hypoxia | [28] |
| Human PAH—pulmonary arteries | |||
| miR-214 | ↑ | Mouse—hypoxia, Sugen5416/hypoxia | [27,30] |
| Rat—monocrotaline, Sugen5416/hypoxia | |||
| miR-130/310 | ↑ | Mouse—hypoxia, Sugen5416/hypoxia, VHL null, Interleukin-6 transgenic, BMPR2X transgenic, Schistosoma mansoni-infected | [39,40] |
| Rat—monocrotaline | |||
| Juvenile lamb—pulmonary artery-aorta shunt | |||
| Human PH—pulmonary artery plasma |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leopold, J.A.; Maron, B.A. Molecular Mechanisms of Pulmonary Vascular Remodeling in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2016, 17, 761. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17050761
Leopold JA, Maron BA. Molecular Mechanisms of Pulmonary Vascular Remodeling in Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2016; 17(5):761. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17050761
Chicago/Turabian StyleLeopold, Jane A., and Bradley A. Maron. 2016. "Molecular Mechanisms of Pulmonary Vascular Remodeling in Pulmonary Arterial Hypertension" International Journal of Molecular Sciences 17, no. 5: 761. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17050761