
A Study of Single Nucleotide Polymorphisms of the SLC19A1/RFC1 Gene in Subjects with Autism Spectrum Disorder

Abstract
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Genotyping
4.3. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Mannual of Mental Disorders (DSM-V), 5th ed.; American Psychiatric Pub.: Washington, DC, USA, 2013. [Google Scholar]
- Chakrabarti, B.; Dudbridge, F.; Kent, L.; Wheelwright, S.; Hill-Cawthorne, G.; Allison, C.; Banerjee-Basu, S.; Baron-Cohen, S. Genes related to sex steroids, neural growth, and social–emotional behavior are associated with autistic traits, empathy, and Asperger syndrome. Autism Res. 2009, 2, 157–177. [Google Scholar] [CrossRef] [PubMed]
- Moscow, J.A.; Gong, M.; He, R.; Sgagias, M.K.; Dixon, K.H.; Anzick, S.L.; Meltzer, P.S.; Cowan, K.H. Isolation of a gene encoding a human reduced folate carrier (RFC1) and analysis of its expression in transport-deficient, methotrexate-resistant human breast cancer cells. Cancer Res. 1995, 55, 3790–3794. [Google Scholar] [PubMed]
- Tolner, B.; Roy, K.; Sirotnak, F.M. Structural analysis of the human RFC-1 gene encoding a folate transporter reveals multiple promoters and alternatively spliced transcripts with 5′ end heterogeneity. Gene 1998, 211, 331–341. [Google Scholar] [CrossRef]
- Williams, F.M.; Flintoff, W.F. Structural organization of the human reduced folate carrier gene: Evidence for 5′ heterogeneity in lymphoblast mRNA. Somat. Cell Mol. Genet. 1998, 24, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wong, S.C.; Matherly, L.H. Structure and organization of the human reduced folate carrier gene1. Biochim. Biophys. Acta 1998, 1442, 389–393. [Google Scholar] [CrossRef]
- Prasad, P.D.; Ramamoorthy, S.; Leibach, F.H.; Ganapathy, V. Molecular Cloning of the Human Placental Folate Transporter. Biochem. Biophys. Res. Commun. 1995, 206, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Williams, F.M.R.; Flintoff, W.F. Isolation of a Human cDNA that Complements a Mutant Hamster Cell Defective in Methotrexate Uptake. J. Biol. Chem. 1995, 270, 2987–2992. [Google Scholar] [PubMed]
- Wong, S.C.; Proefke, S.A.; Bhushan, A.; Matherly, L.H. Isolation of human cDNAs that restore methotrexate sensitivity and reduced folate carrier activity in methotrexate transport-defective Chinese Hamster ovary cells. J. Biol. Chem. 1995, 270, 17468–17475. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, V.; Smith, S.; Prasad, P. SLC19: The folate/thiamine transporter family. Pflugers Arch. 2004, 447, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Main, P.A.; Angley, M.T.; Thomas, P.; O’Doherty, C.E.; Fenech, M. Folate and methionine metabolism in autism: A systematic review. Am. J. Clin. Nutr. 2010, 91, 1598–1620. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, W.; Wang, J.; Tan, Y.; Zhou, Y.; Ding, W.; Hua, Z.; Shen, J.; Xu, Y.; Shen, H. Reduced folate carrier gene G80A polymorphism is associated with an increased risk of gastroesophageal cancers in a Chinese population. Eur. J. Cancer 2006, 42, 3206–3211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xu, Y.; Zhou, J.; Wang, X.; Wang, L.; Hu, X.; Guo, J.; Wei, Q.; Shen, H. Polymorphisms of thymidylate synthase in the 5′- and 3′-untranslated regions associated with risk of gastric cancer in South China: A case-control analysis. Carcinogenesis 2005, 26, 1764–1769. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.; Lucock, M.; Stuart, J.; Fardell, S.; Baker, K.; Ng, X. Preliminary evidence for involvement of the folate gene polymorphism 19 bp deletion-DHFR in occurrence of autism. Neurosci. Lett. 2007, 422, 24–29. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Melnyk, S.; Jernigan, S.; Cleves, M.A.; Halsted, C.H.; Wong, D.H.; Cutler, P.; Bock, K.; Boris, M.; Bradstreet, J.J.; et al. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2006, 141B, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Paşca, S.P.; Nemeş, B.; Vlase, L.; Gagyi, C.E.; Dronca, E.; Miu, A.C.; Dronca, M. High levels of homocysteine and low serum paraoxonase 1 arylesterase activity in children with autism. Life Sci. 2006, 78, 2244–2248. [Google Scholar] [CrossRef] [PubMed]
- Gordon, N. Cerebral folate deficiency. Dev. Med. Child Neurol. 2009, 51, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Moretti, P.; Peters, S.U.; del Gaudio, D.; Sahoo, T.; Hyland, K.; Bottiglieri, T.; Hopkin, R.J.; Peach, E.; Min, S.H.; Goldman, D.; et al. Brief Report: Autistic Symptoms, Developmental Regression, Mental Retardation, Epilepsy, and Dyskinesias in CNS Folate Deficiency. J. Autism Dev. Disord. 2008, 38, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Ramaekers, V.T.; Blau, N.; Sequeira, J.M.; Nassogne, M.C.; Quadros, E.V. Folate receptor autoimmunity and cerebral folate deficiency in low-functioning autism with neurological deficits. Neuropediatrics 2007, 38, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Munesue, T.; Yokoyama, S.; Nakamura, K.; Anitha, A.; Yamada, K.; Hayashi, K.; Asaka, T.; Liu, H.-X.; Jin, D.; Koizumi, K.; et al. Two genetic variants of CD38 in subjects with autism spectrum disorder and controls. Neurosci. Res. 2010, 67, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, S.; Al Mahmuda, N.; Munesue, T.; Hayashi, K.; Yagi, K.; Yamagishi, M.; Higashida, H. Association Study between the CD157/BST1 Gene and Autism Spectrum Disorders in a Japanese Population. Brain Sci. 2015, 5, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Akey, J.M.; Zhang, K.; Chakraborty, R.; Jin, L. Distribution of recombination crossovers and the origin of haplotype blocks: The interplay of population history, recombination, and mutation. Am. J. Hum. Genet. 2002, 71, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, V.B.; Pangilinan, F.; Cox, C.; Parle-McDermott, A.; Conley, M.; Molloy, A.M.; Kirke, P.N.; Mills, J.L.; Brody, L.C.; Scott, J.M. Reduced folate carrier polymorphisms and neural tube defect risk. Mol. Genet. Metab. 2006, 87, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Pei, L.J.; Li, Z.W.; Zhang, W.; Ren, A.G.; Zhu, H.P.; Hao, L.; Zhu, J.H.; Li, Z. Epidemiological study on reduced folate carrier gene (RFC1 A80G) polymorphism and other risk factors of neural tube defects. J. Peking Univ. Health Sci. 2005, 37, 341–345. [Google Scholar]
- Pei, L.J.; Zhu, H.P.; Li, Z.W.; Zhang, W.; Ren, A.G.; Zhu, J.H.; Li, Z. Interaction between maternal periconceptional supplementation of folic acid and reduced folate carrier gene polymorphism of neural tube defects. Chin. J. Med. Genet. 2005, 22, 284–287. [Google Scholar]
- Zhang, T.; Lou, J.; Zhong, R.; Wu, J.; Zou, L.; Sun, Y.; Lu, X.; Liu, L.; Miao, X.; Xiong, G. Genetic variants in the folate pathway and the risk of neural tube defects: A meta-analysis of the published literature. PLoS ONE 2013, 8, e59570. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Zhao, H.; Niu, B.; Li, W.I.; Zhou, R.; Zhang, T.; Xie, J. Correlation of polymorphism of MTHFRs and RFC-1 genes with neural tube defects in China. Birth Defects Res. A Clin. Mol. Teratol. 2008, 82, 3–7. [Google Scholar] [CrossRef] [PubMed]
- De Marco, P.; Calevo, M.G.; Moroni, A.; Merello, E.; Raso, A.; Finnell, R.H.; Zhu, H.; Andreussi, L.; Cama, A.; Capra, V. Reduced folate carrier polymorphism (80A→G) and neural tube defects. Eur. J. Hum. Genet. 2003, 11, 245–252. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Melnyk, S.; Jernigan, S.; Lehman, S.; Seidel, L.; Gaylor, D.W.; Cleves, M.A. A functional polymorphism in the reduced folate carrier gene and DNA hypomethylation in mothers of children with autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153B, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Said, H.M.; Chatterjee, N.; Haq, R.U.; Subramanian, V.S.; Ortiz, A.; Matherly, L.H.; Sirotnak, F.M.; Halsted, C.; Rubin, S.A. Adaptive regulation of intestinal folate uptake: Effect of dietary folate deficiency. Am. J. Physiol. Cell Physiol. 2000, 279, C1889–C1895. [Google Scholar] [PubMed]
- Frye, R.E.; Rossignol, D.A. Cerebral Folate Deficiency in Autism Spectrum Disorders. Autism Sci. Dig. J. Autsmone 2011. Available online: http://www.autismone.org/content/cerebral-folate-deficiency-autism-spectrum-disorders-richard-frye-md-phd-and-daniel-rossigno (accessed on 19 May 2016). [Google Scholar]
- Ramaekers, V.; Sequeira, J.M.; Quadros, E.V. Clinical recognition and aspects of the cerebral folate deficiency syndromes. Clin. Chem. Lab. Med. 2013, 51, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Matherly, L.; Hou, Z.; Deng, Y. Human reduced folate carrier: Translation of basic biology to cancer etiology and therapy. Cancer Metastasis Rev. 2007, 26, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Stanislawska-Sachadyn, A.; Mitchell, L.E.; Woodside, J.V.; Buckley, P.T.; Kealey, C.; Young, I.S.; Scott, J.M.; Murray, L.; Boreham, C.A.; McNulty, H.; et al. The reduced folate carrier (SLC19A1) c.80G>A polymorphism is associated with red cell folate concentrations among women. Ann. Hum. Genet. 2009, 73 Pt 5, 484–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, P.; Sahoo, T.; Hyland, K.; Bottiglieri, T.; Peters, S.; del Gaudio, D.; Roa, B.; Curry, S.; Zhu, H.; Finnell, R.H.; et al. Cerebral folate deficiency with developmental delay, autism, and response to folinic acid. Neurology 2005, 64, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.L.; Cohen, D.J.; Miller, S.; Young, J.G. Folic acid and B12 in autism and neuropsychiatric disturbances of childhood. J. Am. Acad. Child Psychiatry 1981, 20, 104–111. [Google Scholar] [CrossRef]
- Ramaekers, V.T.; Rothenberg, S.P.; Sequeira, J.M.; Opladen, T.; Blau, N.; Quadros, E.V.; Selhub, J. Autoantibodies to folate receptors in the cerebral folate deficiency syndrome. N. Eng. J. Med. 2005, 352, 1985–1991. [Google Scholar] [CrossRef] [PubMed]
- Ramaekers, V.T.; Sequeira, J.M.; Blau, N.; Quadros, E.V. A milk-free diet downregulates folate receptor autoimmunity in cerebral folate deficiency syndrome. Dev. Med. Child Neurol. 2008, 50, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.-J.; Hashii, M.; Munesue, T.; Hayashi, K.; Yagi, K.; Yamagishi, M.; Higashida, H.; Yokoyama, S. Non-synonymous single-nucleotide variations of the human oxytocin receptor gene and autism spectrum disorders: A case-control study in a Japanese population and functional analysis. Mol. Autism 2013, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Gillberg, C.; Gillberg, C.; Råstam, M.; Wentz, E. The Asperger Syndrome (and High-Functioning Autism) Diagnostic Interview (ASDI): A Preliminary Study of a New Structured Clinical Interview. Autism 2001, 5, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Rutter, M.; le Couteur, A. Autism Diagnostic Interview-Revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J. Autism Dev. Disord. 1994, 24, 659–685. [Google Scholar] [CrossRef] [PubMed]
- Autism Society Japan. Pervasive Developmental Disorders Autism Society Japan Rating Scale (PARS); Spectrum Publishing Company: Tokyo, Japan, 2006. [Google Scholar]
- Wing, L.; Leekam, S.R.; Libby, S.J.; Gould, J.; Larcombe, M. The diagnostic interview for social and communication disorders: Background, inter-rater reliability and clinical use. J. Child Psychol. Psychiatry 2002, 43, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Kurita, H.; Miyake, Y. The Reliability and Validity of the Tokyo Autistic Behaviour Scale. Psychiatry Clin. Neurosci. 1990, 44, 25–32. [Google Scholar] [CrossRef]
- Nishida, N.; Tanabe, T.; Takasu, M.; Suyama, A.; Tokunaga, K. Further development of multiplex single nucleotide polymorphism typing method, the DigiTag2 assay. Anal. Biochem. 2007, 364, 78–85. [Google Scholar] [CrossRef] [PubMed]
- dbSNP: Short Genetic Variations. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/SNP/ (accessed on 24 June 2015).
- Nyholt, D.R. A simple correction for multiple testing for SNPs in linkage disequilibrium with each other. Am. J. Hum. Genet. 2004, 74, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Cherny, S.S.; Sham, P.C. Genetic power calculator: Design of linkage and association genetic mapping studies of complex traits. Bioinformatics (Oxf. Engl.) 2003, 19, 149–150. [Google Scholar] [CrossRef]
- Genetic Power Calculator. Available online: http://pngu.mgh.harvard.edu/purcell/gpc/cc2.html (accessed on 27 June 2015).
- Developmental Disabilities Monitoring Network Surveillance Year 2010 Principal Investigators; Centers for Disease Control and Prevention (CDC). Prevalence of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill. Summ. 2014, 63, 1–21. [Google Scholar]
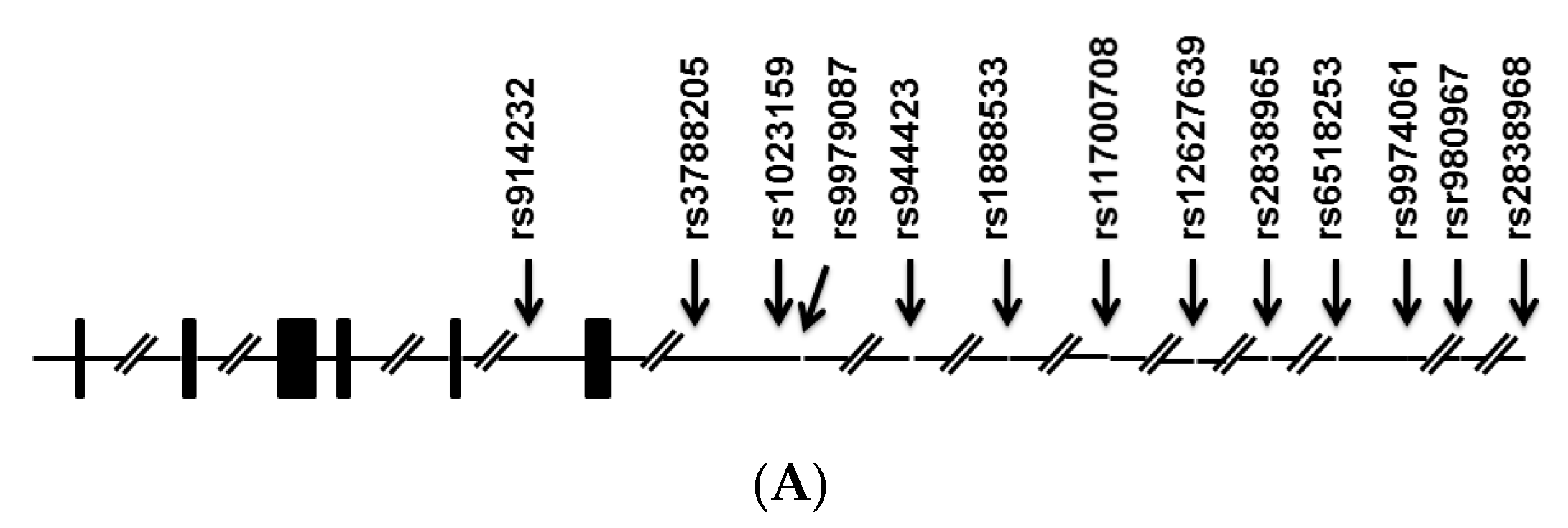
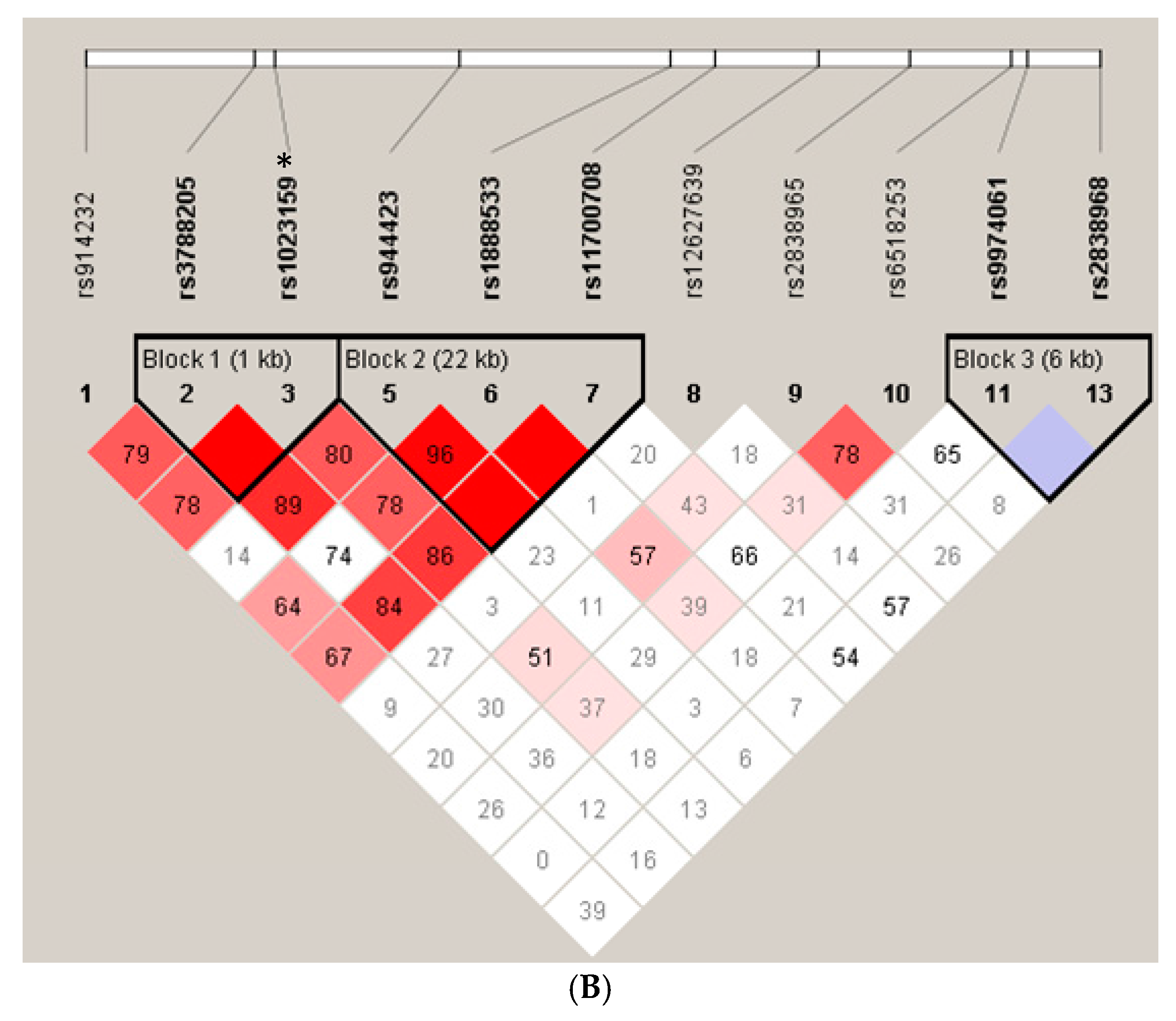

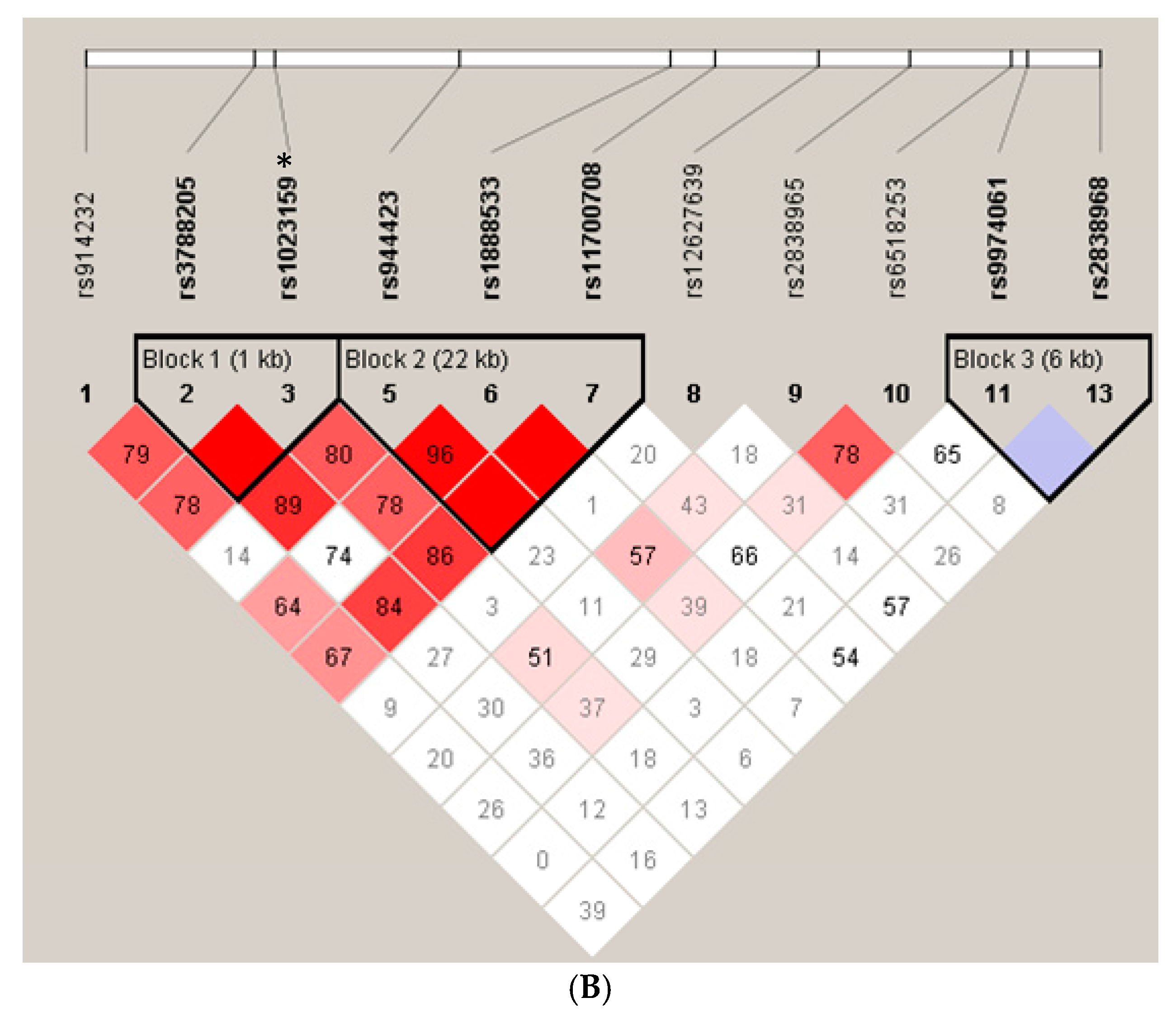
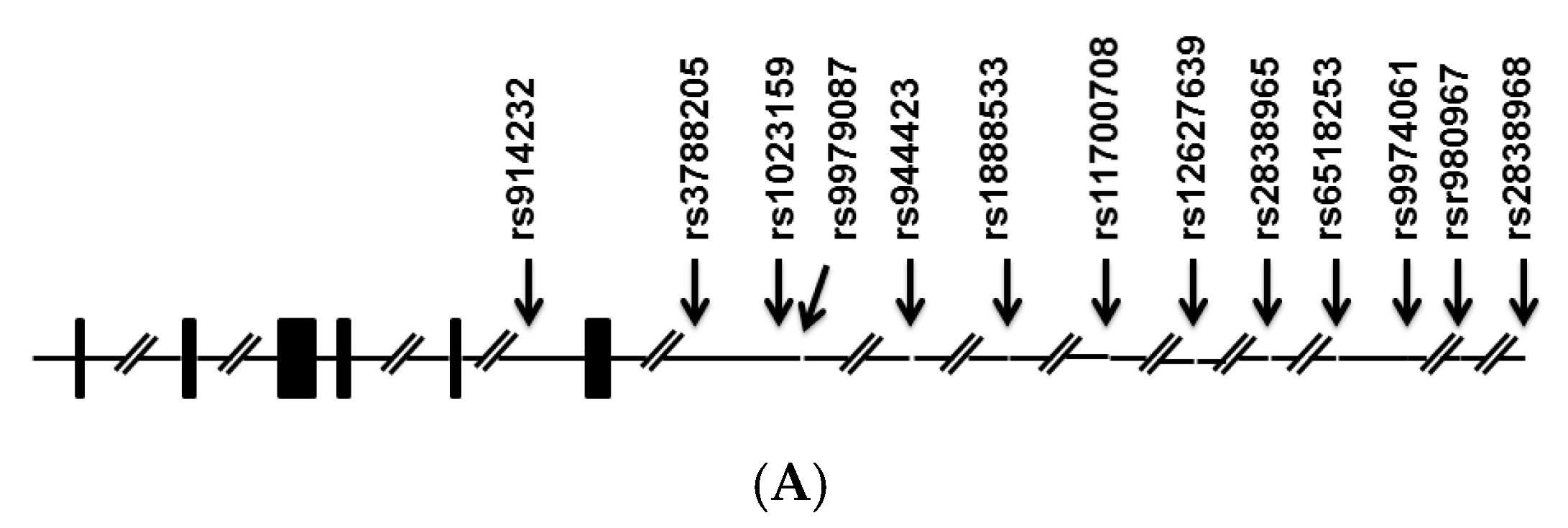{kind=link}
{kind=link}
| rs1023159 | Cases | Control | Odds Ratio (95% CI) | p |
|---|---|---|---|---|
| Genotype | (n = 144) | (n = 146) | ||
| G/G | 72 (50.0%) | 62 (42.5%) | Reference | |
| A/G | 63 (43.8%) | 64 (43.8%) | 0.85 (0.52–1.4) | 0.5368 |
| A/A | 9 (6.3%) | 20 (13.7%) | 0.39 (0.16–0.91) | 0.0394 |
| Allele | (n = 288) | (n = 292) | ||
| G | 207 (71.9%) | 188 (64.4%) | Reference | |
| A | 81 (28.1%) | 104 (35.6%) | 0.71 (0.50–1.0) | 0.0613 |
| rs1023159 | KU | AGRE |
|---|---|---|
| Genotype | (n = 144) | (n = 191) |
| G/G | 72 (50.0%) | 63 (31.4%) |
| A/G | 63 (43.8%) | 104 (53.1%) |
| A/A | 9 (6.3%) | 30 (15.5%) |
| Allele | (n = 288) | (n = 394) |
| G | 207 (71.9%) | 230 (58.4%) |
| A | 81 (28.1%) | 164 (41.6%) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmuda, N.A.; Yokoyama, S.; Huang, J.-J.; Liu, L.; Munesue, T.; Nakatani, H.; Hayashi, K.; Yagi, K.; Yamagishi, M.; Higashida, H. A Study of Single Nucleotide Polymorphisms of the SLC19A1/RFC1 Gene in Subjects with Autism Spectrum Disorder. Int. J. Mol. Sci. 2016, 17, 772. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17050772
Mahmuda NA, Yokoyama S, Huang J-J, Liu L, Munesue T, Nakatani H, Hayashi K, Yagi K, Yamagishi M, Higashida H. A Study of Single Nucleotide Polymorphisms of the SLC19A1/RFC1 Gene in Subjects with Autism Spectrum Disorder. International Journal of Molecular Sciences. 2016; 17(5):772. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17050772
Chicago/Turabian StyleMahmuda, Naila Al, Shigeru Yokoyama, Jian-Jun Huang, Li Liu, Toshio Munesue, Hideo Nakatani, Kenshi Hayashi, Kunimasa Yagi, Masakazu Yamagishi, and Haruhiro Higashida. 2016. "A Study of Single Nucleotide Polymorphisms of the SLC19A1/RFC1 Gene in Subjects with Autism Spectrum Disorder" International Journal of Molecular Sciences 17, no. 5: 772. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17050772