
A Novel Technique to Detect EGFR Mutations in Lung Cancer

Abstract
:
1. Introduction
2. Results
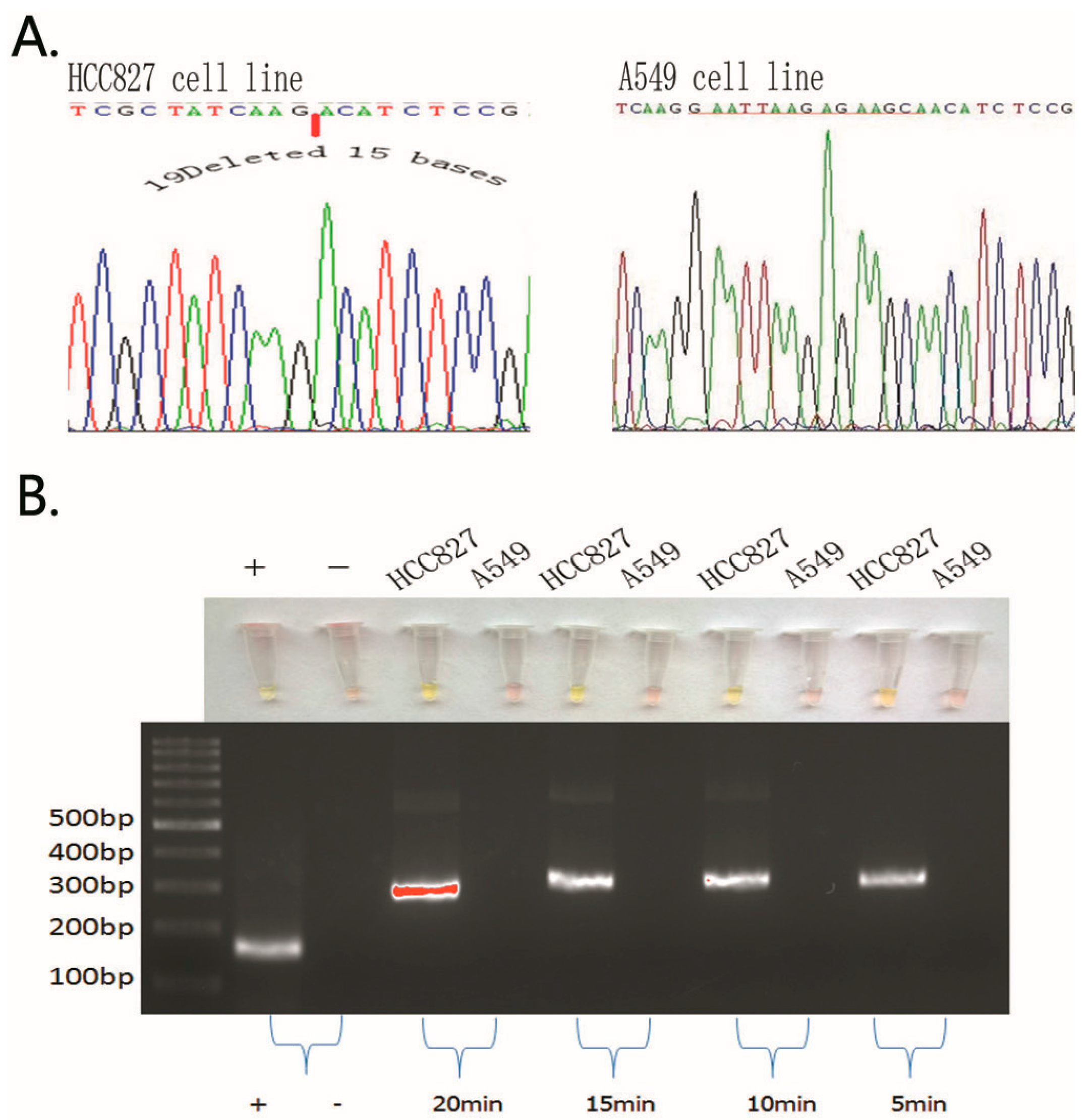
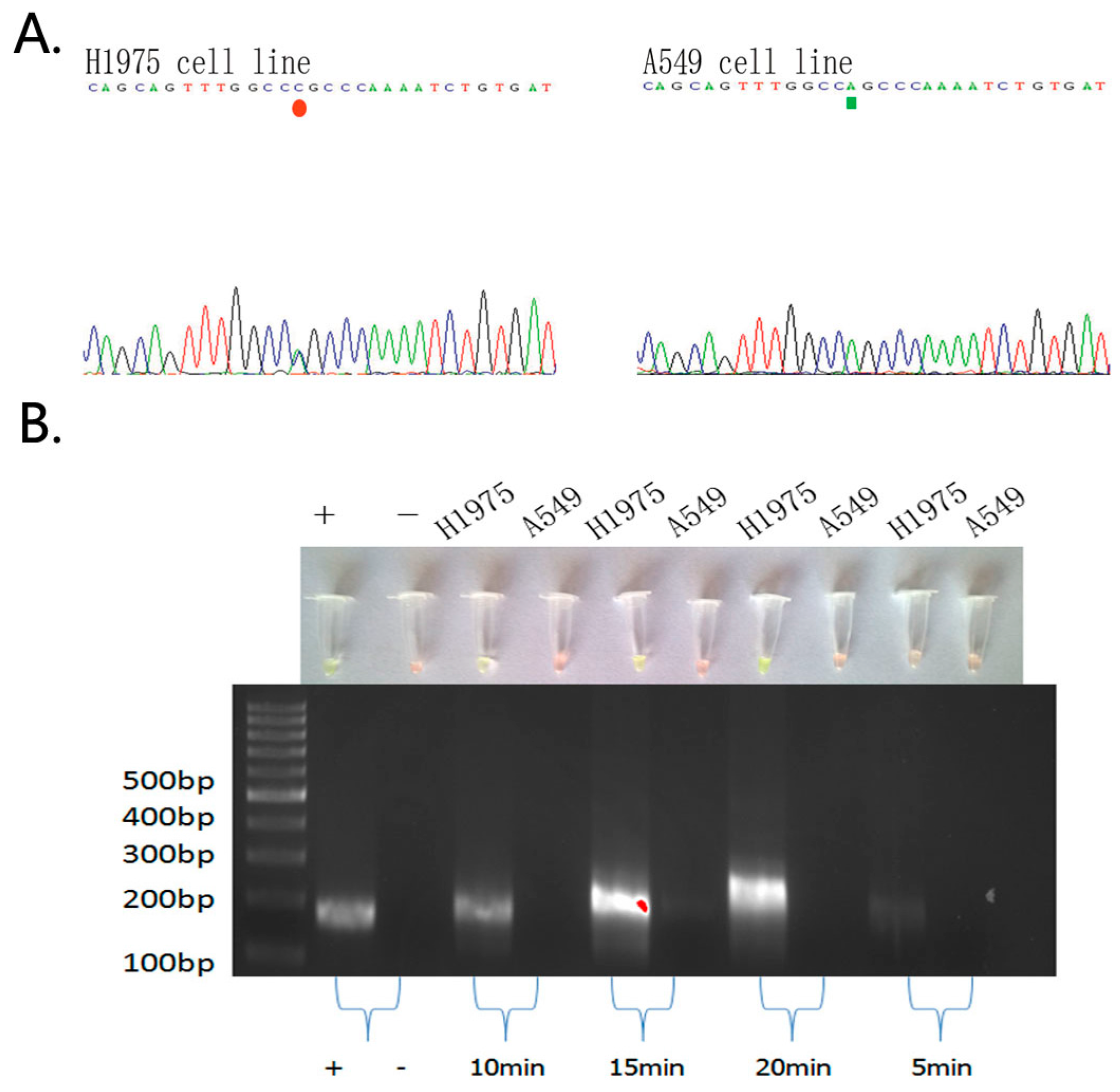
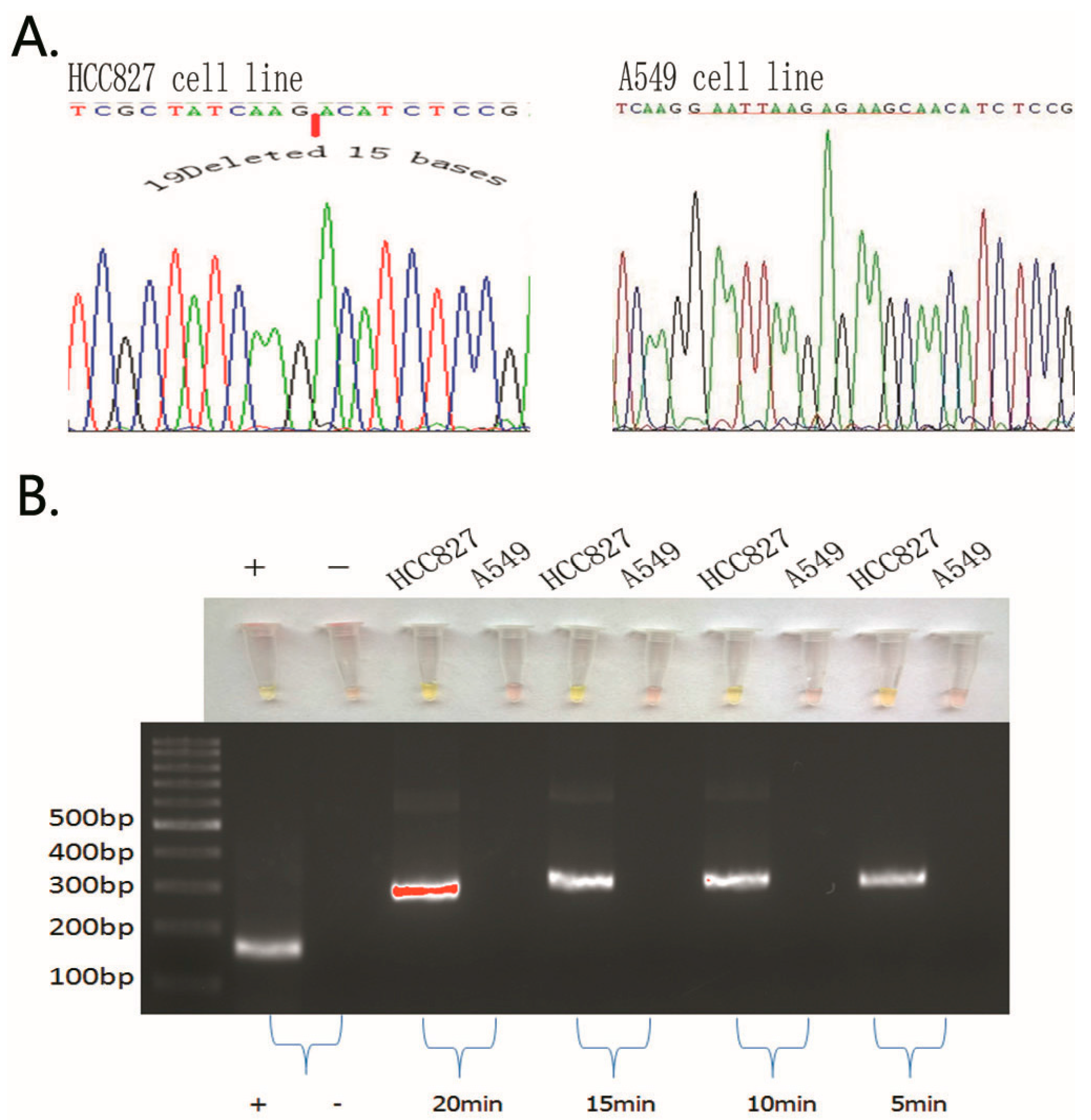
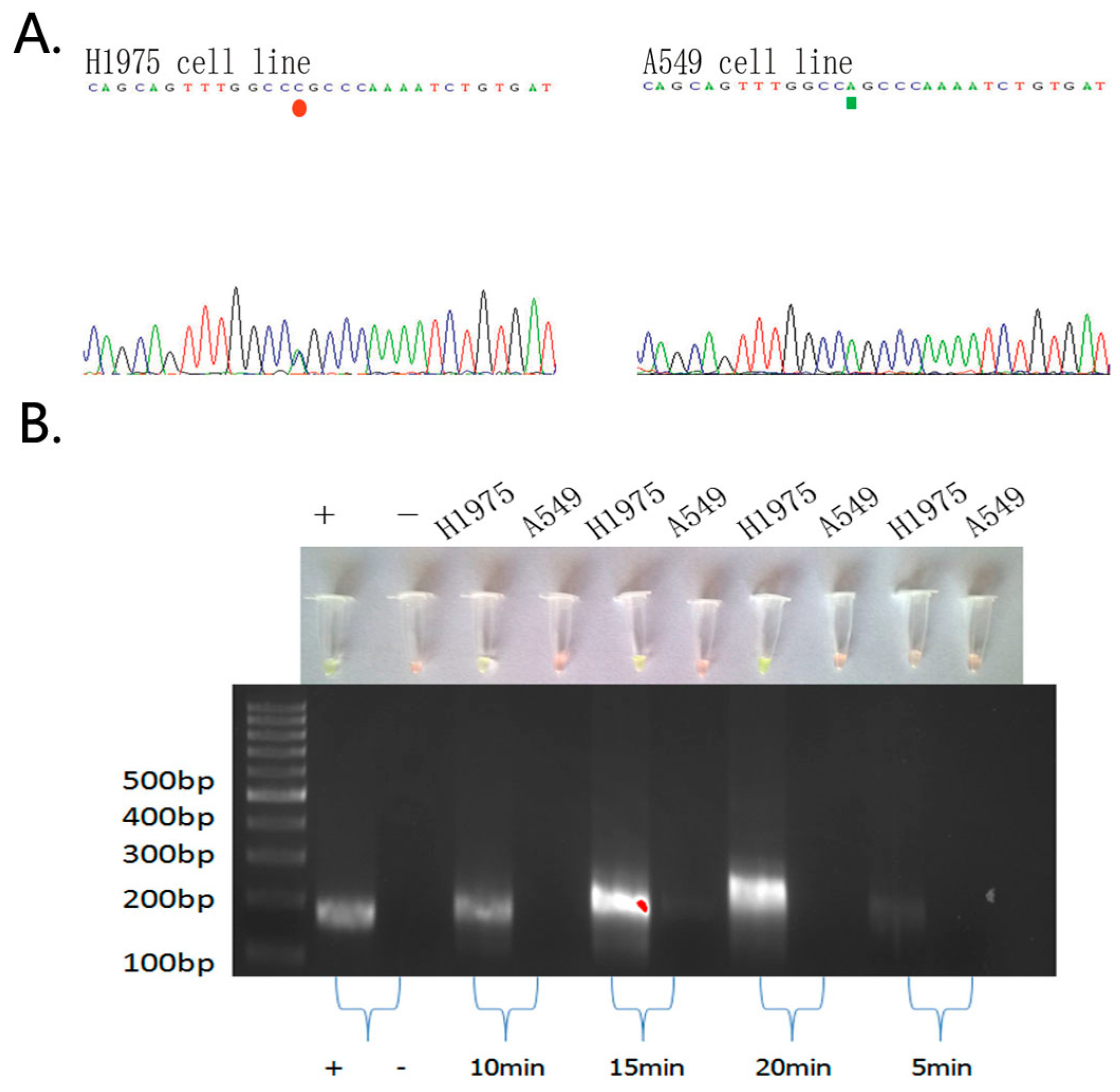
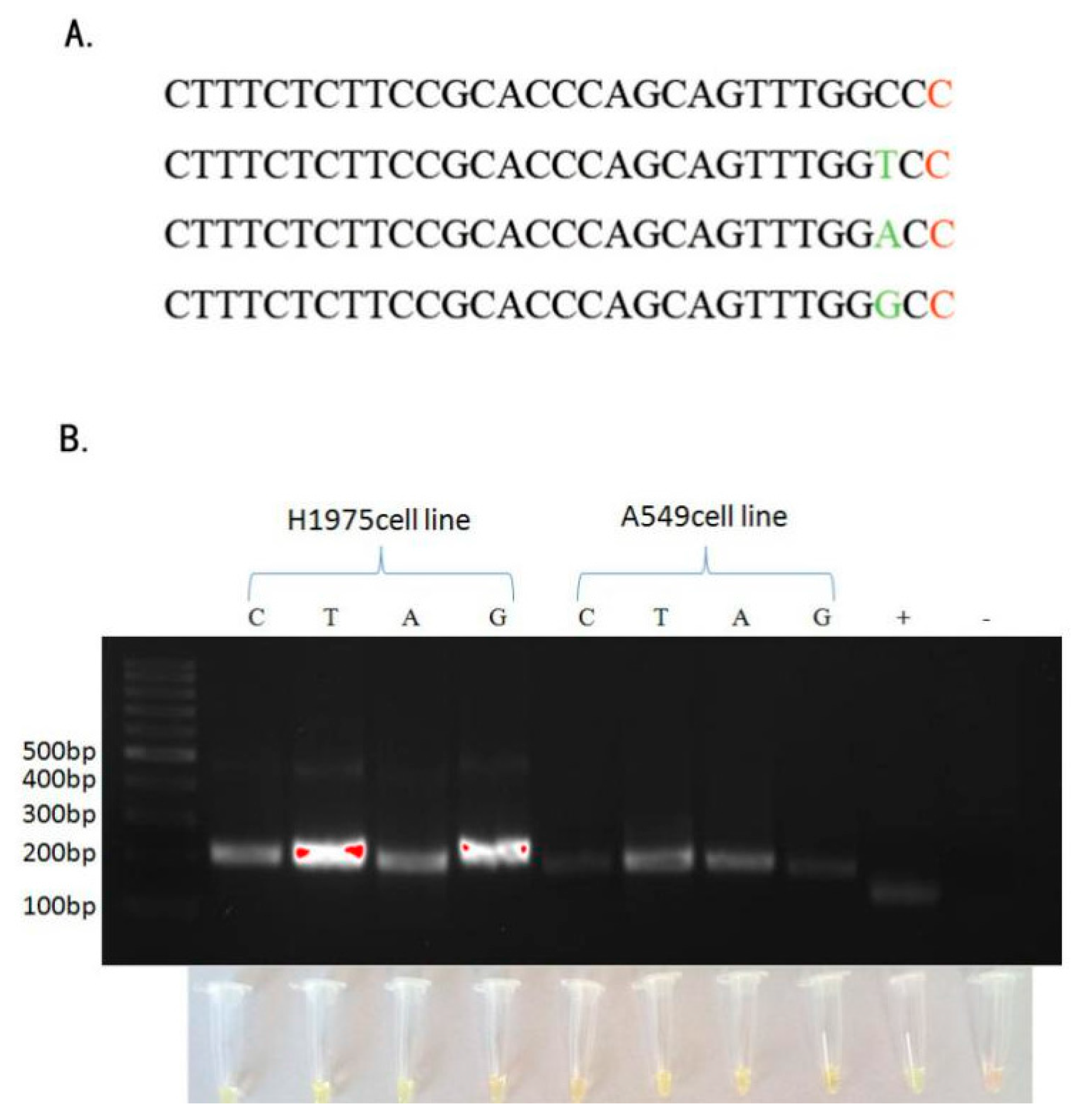
2.1. Detection of EGFR Mutations in NSCLC Cell Lines and Comparison of Gold Standard PCR and Direct DNA Sequencing with Our Novel Detection Technique
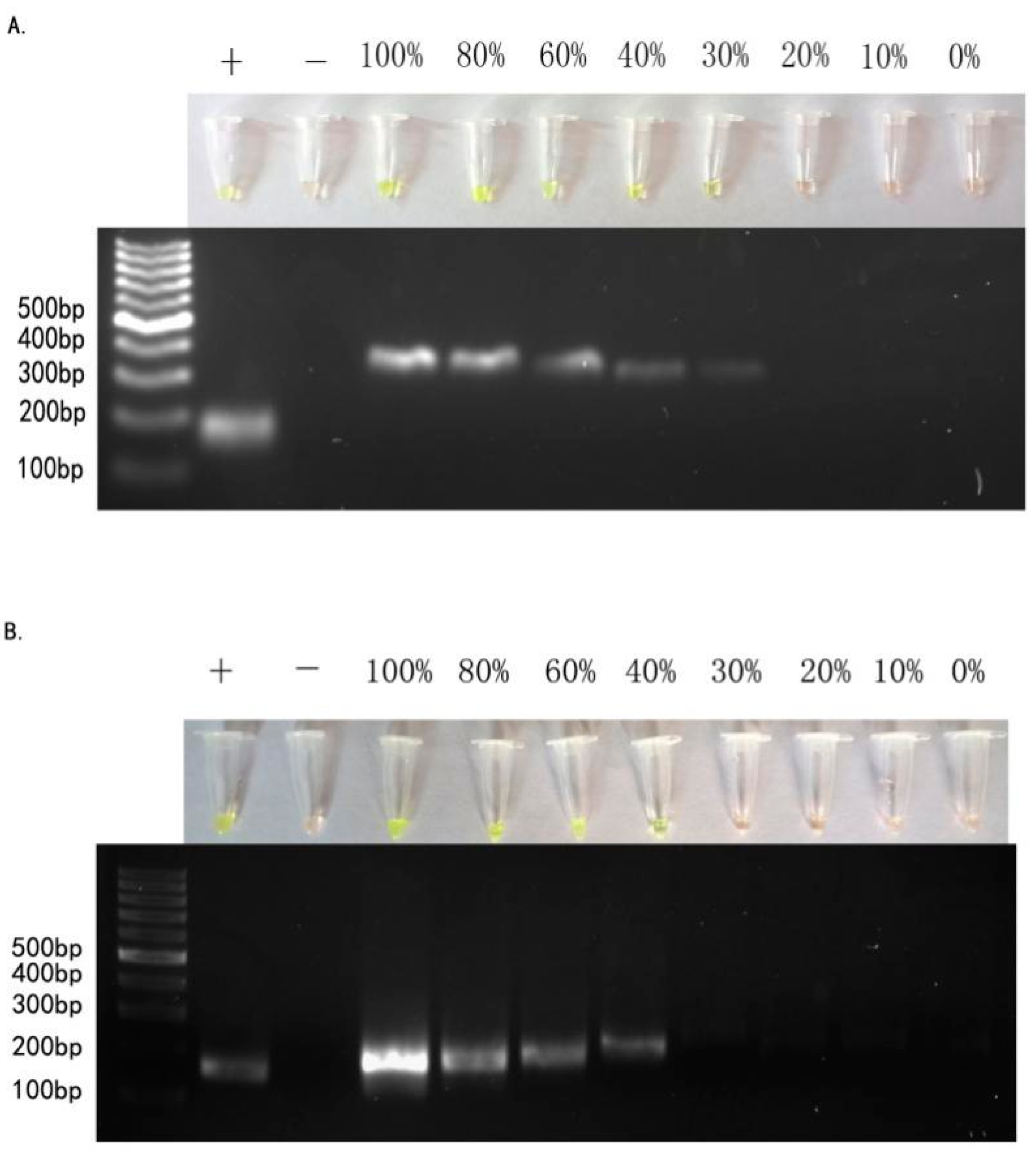
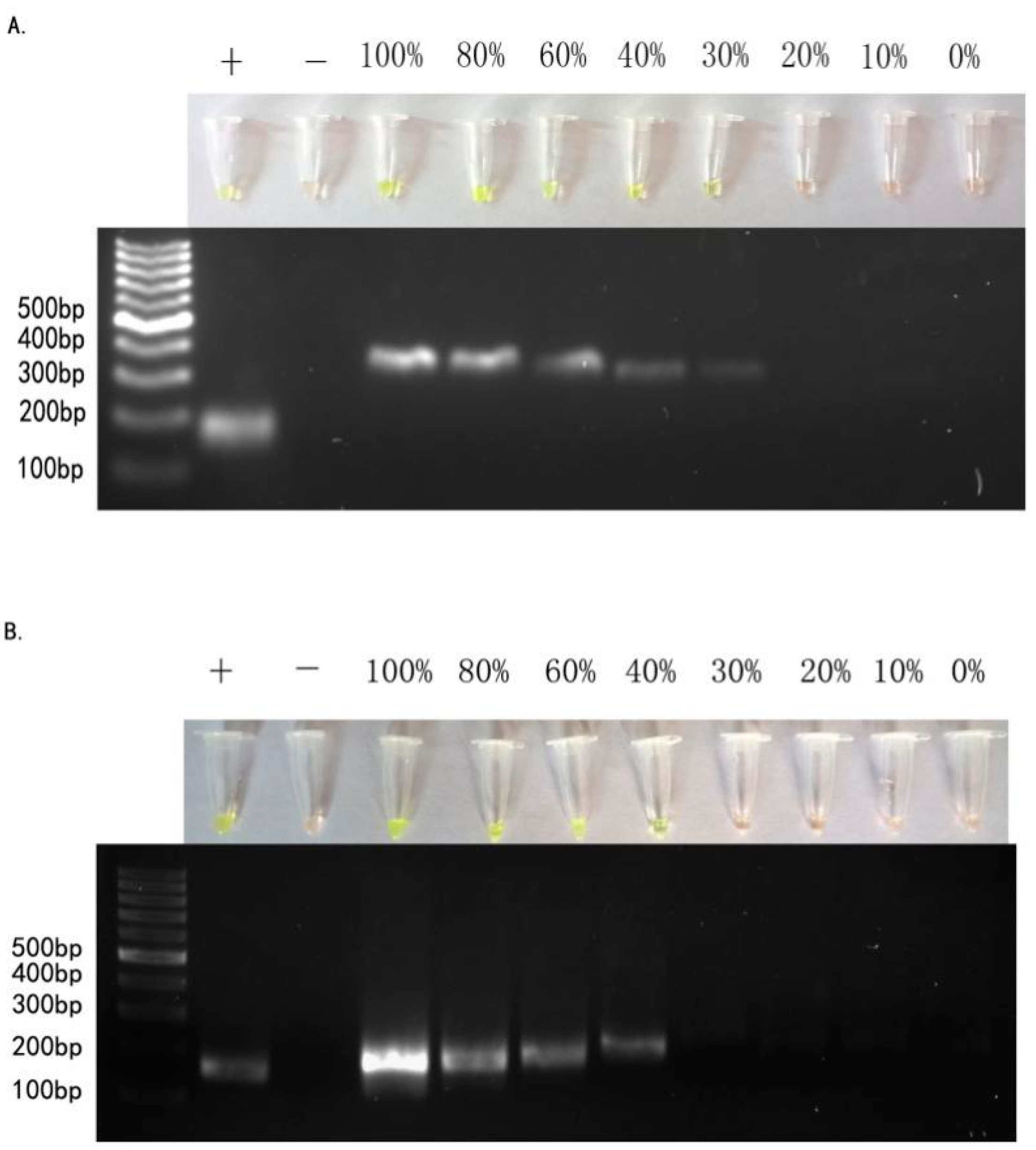
2.2. Sensitivity of ARPS in Mixed-DNAs to Detect EGFR Mutations
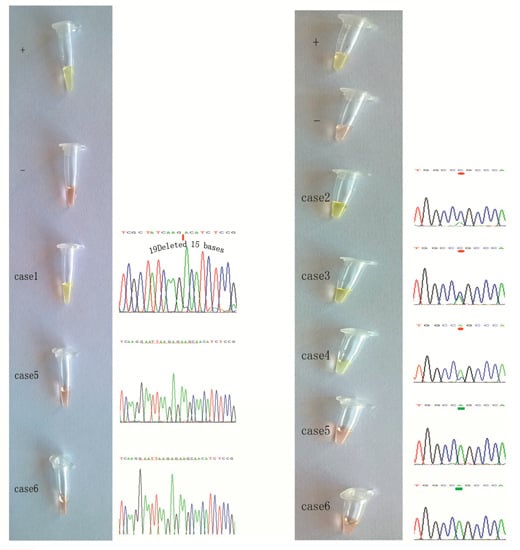
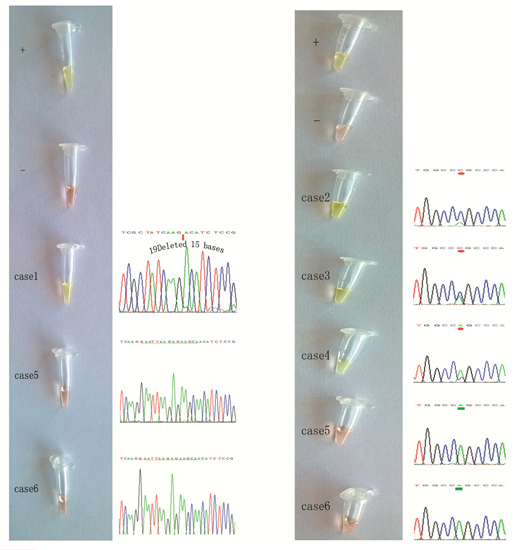
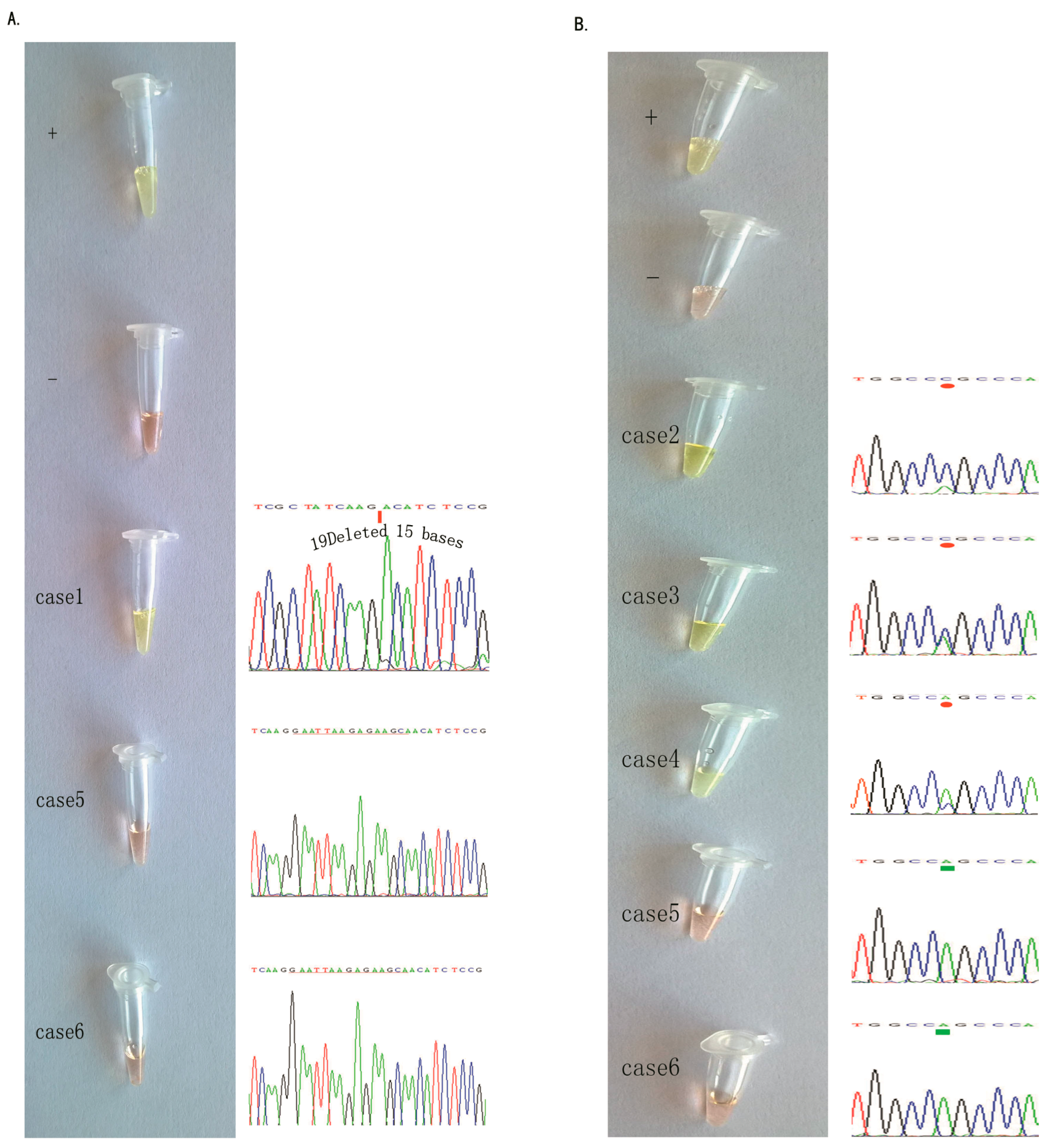
2.3. Detection of EGFR Mutations in NSCLC Tissue Samples Using our Novel ARPS Method vs. the PCR and DNA Sequencing Method
2.4. Specific Considerations While Performing the ARPS Assay to Detect EGFR Mutations
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. NSCLC Tissue Samples
4.3. Genomic DNA Extraction and Detection of EGFR Mutations Using PCR and DNA Sequencing
4.4. The Novel Technique to Detect EGFR Mutations
Principle of RPA primer design
4.5. Probe Design with PNA to Prevent Non-Target Sequence Amplification
4.6. Biosensor SYBR Green I
4.7. ARPS Reactions
4.8. Agarose Gel Electrophoresis of RPA Products
4.9. Assessment of Specificity and Sensitivity of the ARPS Assay for EGFR Mutations
4.10. Quality Control
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Castillo, L.; Etienne-Grimaldi, M.C.; Fischel, J.L.; Formento, P.; Magne, N.; Milano, G. Pharmacological background of EGFR targeting. Ann. Oncol. 2004, 15, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Woodburn, J.R. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol. Ther. 1999, 82, 241–250. [Google Scholar] [CrossRef]
- Salomon, D.S.; Brandt, R.; Ciardiello, F.; Normanno, N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit. Rev. Oncol. Hematol. 1995, 19, 183–232. [Google Scholar] [CrossRef]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rush, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004, 305, 1163167. [Google Scholar] [CrossRef] [PubMed]
- Califano, R.; Abidin, A.Z.; Peck, R.; Faivre-Finn, C.; Lorigan, P. Management of small cell lung cancer: recent developments for optimal care. Drugs 2012, 72, 471–490. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, A.; Martella, C.; Felicioni, L.; Barassi, F.; Salvatore, S.; Chella, A.; Camplese, P.P.; Iarussi, T.; Mucilli, F.; Mezzetti, A.; et al. EGFR mutations in non-small-cell lung cancer: Analysis of a large series of cases and development of a rapid and sensitive method for diagnostic screening with potential implications on pharmacologic treatment. J. Clin. Oncol. 2005, 23, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Asano, H.; Toyooka, S.; Tokumo, M.; Ichimura, K.; Aoe, K.; Ito, S.; Tsukuda, K.; Ouchida, M.; Aoe, M.; Katayama, H.; et al. Detection of EGFR gene mutation in lung cancer by mutant-enriched polymerase chain reaction assay. Clin. Cancer Res. 2006, 12, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Oshita, F.; Matsukuma, S.; Yoshihara, M.; Sakuma, Y.; Ohgane, N.; Kameda, Y.; Saito, H.; Yamada, K.; Tsuchiya, E.; Miyagi, Y. Novel heteroduplex method using small cytology specimens with a remarkably high success rate for analysing EGFR gene mutations with a significant correlation to gefitinib efficacy in non-small-cell lung cancer. Br. J. Cancer 2006, 95, 1070–1075. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Borras, A.M.; Kuang, Y.; Rogers, A.M.; Joshi, V.A.; Liyanage, H.; Lindeman, N.; Lee, J.C.; Halmos, B.; Maher, E.A.; et al. A rapid and sensitive enzymatic method for epidermal growth factor receptor mutation screening. Clin. Cancer Res. 2006, 12, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Compton, J. Nucleic acid sequence-based amplification. Nature 1991, 350, 91–92. [Google Scholar] [CrossRef] [PubMed]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, E63. [Google Scholar] [CrossRef] [PubMed]
- Gusev, Y.; Sparkowski, J.; Raghunathan, A.; Ferguson, H., Jr.; Montano, J.; Bogdan, N.; Schweitzer, B.; Wiltshire, S.; Kingsmore, S.F.; Maltzman, W.; et al. Rolling circle amplification: A new approach to increase sensitivity for immunohistochemistry and flow cytometry. Am. J. Pathol. 2001, 159, 63–69. [Google Scholar] [CrossRef]
- Vincent, M.; Xu, Y.; Kong, H. Helicase-dependent isothermal DNA amplification. EMBO Rep. 2004, 5, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, K.; Takakura, H.; Mitani, Y.; Tatsumi, K.; Momiyama, N.; Ichikawa, Y.; Togo, S.; Miyagi, T.; Kawai, Y.; Kogo, Y.; et al. Rapid detection of epidermal growth factor receptor mutations in lung cancer by the Smart-Amplification Process. Clin. Cancer Res. 2007, 13, 4974–4983. [Google Scholar] [CrossRef] [PubMed]
- Mitani, Y.; Lezhava, A.; Kawai, Y.; Kikuchi, T.; Oguchi-Katayama, A.; Kogo, Y.; Itoh, M.; Miyagi, T.; Takakura, H.; Hoshi, K.; et al. Rapid SNP diagnostics using asymmetric isothermal amplification and a new mismatch-suppression technology. Nat. Methods 2007, 4, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA detection using recombination proteins. PLoS Biol. 2006, 4, e204. [Google Scholar] [CrossRef] [PubMed]
- Del Río, J.S.; Yehia Adly, N.; Acero-Sánchez, J.L.; Henry, O.Y.; O'Sullivan, C.K. Electrochemical detection of Francisella tularensis genomic DNA using solid-phase recombinase polymerase amplification. Biosens Bioelectron. 2014, 54, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Boyle, D.S.; Lehman, D.A.; Lillis, L.; Peterson, D.; Singhal, M.; Armes, N.; Parker, M.; Piepenburg, O.; Overbaugh, J. Rapid detection of HIV-1 proviral DNA for early infant diagnosis using recombinase polymerase amplification. MBio 2013, 4, e00135-13. [Google Scholar] [CrossRef] [PubMed]
- Euler, M.; Wang, Y.; Heidenreich, D.; Patel, P.; Strohmeier, O.; Hakenberg, S.; Niedrig, M.; Hufert, F.T.; Weidmann, M. Development of a panel of recombinase polymerase amplification assays for detection of biothreat agents. J. Clin. Microbiol. 2013, 51, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Newton, C.R.; Graham, A.; Heptinstall, L.E.; Powell, S.J.; Summers, C.; Kalsheker, N.; Smith, J.C.; Markham, A.F. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 1989, 17, 2503–2516. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Guo, J.; Xu, L.; Gao, S.; Lin, Q.; Wu, Q.; Wu, L.; Que, Y. Establishment and application of a loop-mediated isothermal amplification(LAMP) system for detection of cry1Actransgenic sugarcane. Sci. Rep. 2014, 4, 4912. [Google Scholar] [PubMed]
- Wagner, R.; Debbie, P.; Radman, M. Mutation detection using immobilized mismatch binding protein (MutS). Nucleic Acids Res. 1995, 23, 3944–3948. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, K.; Mitani, Y.; Watanabe, J.; Takakura, H.; Hoshi, K.; Kawai, Y.; Kikuchi, T.; Kogo, Y.; Oguchi-Katayama, A.; Tomaru, Y.; et al. Rapid screening assay for KRAS mutations by the modified smart amplification process. J. Mol. Diagn. 2008, 10, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Daher, R.K.; Stewart, G.; Boissinot, M.; Bergeron, M.G. Isothermal recombinase polymerase amplification assay applied to the detection of group B streptococci invaginal/anal samples. Clin Chem. 2014, 60, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Crannell, Z.A.; Cabada, M.M.; Castellanos-Gonzalez, A.; Irani, A.; White, A.C.; Richards-Kortum, R. Recombinase polymerase amplification-based assay to diagnose Giardia in stool samples. Am. J. Trop. Med. Hyg. 2015, 92, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Lutz, S.; Weber, P.; Focke, M.; Faltin, B.; Hoffmann, J.; Müller, C.; Mark, D.; Roth, G.; Munday, P.; Armes, N.; et al. Microfluidic lab-on-a-foil for nucleic acid analysis based on isothermal recombinase polymerase amplification(RPA). Lab. Chip. 2010, 10, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Goldstraw, P.; Crowley, J.; Chansky, K.; Giroux, D.J.; Groome, P.A.; Rami-Porta, R.; Postmus, P.E.; Rusch, V.; Sobin, L.; et al. The IASLC lung cancer staging project: Proposals for the revision of the TNM stage groupings in the forth coming (seventh) edition of the TNM classification of malignant tumours. J. Thorac Oncol 2007, 2, 706–714. [Google Scholar] [CrossRef] [PubMed]
- PNA Design Web. Available online: http://www6.appliedbiosystems.com/support/pnadesigner.cfm (accessed on 1 March 2016).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EGFR Mutation Type Primers Amplicon (bp) |
|---|
| 19Del(2)FP 5’-GTGAGAAAGTTAAAATTCCCGTCGCTATCAAGACATCTC-3’ 266 |
| 19Del(2)RP 5’-GATACCAGCATGGGAGAGGCCAGTGCTGTCTCTAAG-3’ |
| L858R FP 5’-CTGAATTCGGATGCAGAGCTTCTTCCCATGA-3’ 201 |
| L858R RP 5’-CTTTCTCTTCCGCACCCAGCAGTTTGGCCC-3’ |
| PNA 5’-GCCAGCCCAA-3’ |
| EGFR Mutation Type Primers Amplicon (bp) |
|---|
| 19Del FP 5’-AACGTCTTCCTTCTCTCTCTGTCA-3’ 135(Mut)/150(Wild) |
| 19Del RP 5’-CCACACAGCAAAGCAGAAACT-3’ |
| L858R FP 5’-CTGAATTCGGATGCAGAGCTT-3’ 291 |
| L858R RP 5’-CTAGTGGGAAGGCAGCCTGGT-3’ |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Lei, T.; Liu, Z.; Kuang, Y.; Lyu, J.; Wang, Q. A Novel Technique to Detect EGFR Mutations in Lung Cancer. Int. J. Mol. Sci. 2016, 17, 792. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17050792
Liu Y, Lei T, Liu Z, Kuang Y, Lyu J, Wang Q. A Novel Technique to Detect EGFR Mutations in Lung Cancer. International Journal of Molecular Sciences. 2016; 17(5):792. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17050792
Chicago/Turabian StyleLiu, Yuanbin, Ting Lei, Zhiyu Liu, Yanbin Kuang, Jianxin Lyu, and Qi Wang. 2016. "A Novel Technique to Detect EGFR Mutations in Lung Cancer" International Journal of Molecular Sciences 17, no. 5: 792. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17050792