
Identification of Protein–Excipient Interaction Hotspots Using Computational Approaches


Abstract
:
1. Introduction
2. Results and Discussion
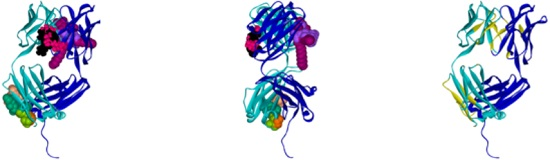
2.1. Hotspots for Protein Interactions with Excipients—Flexible Docking
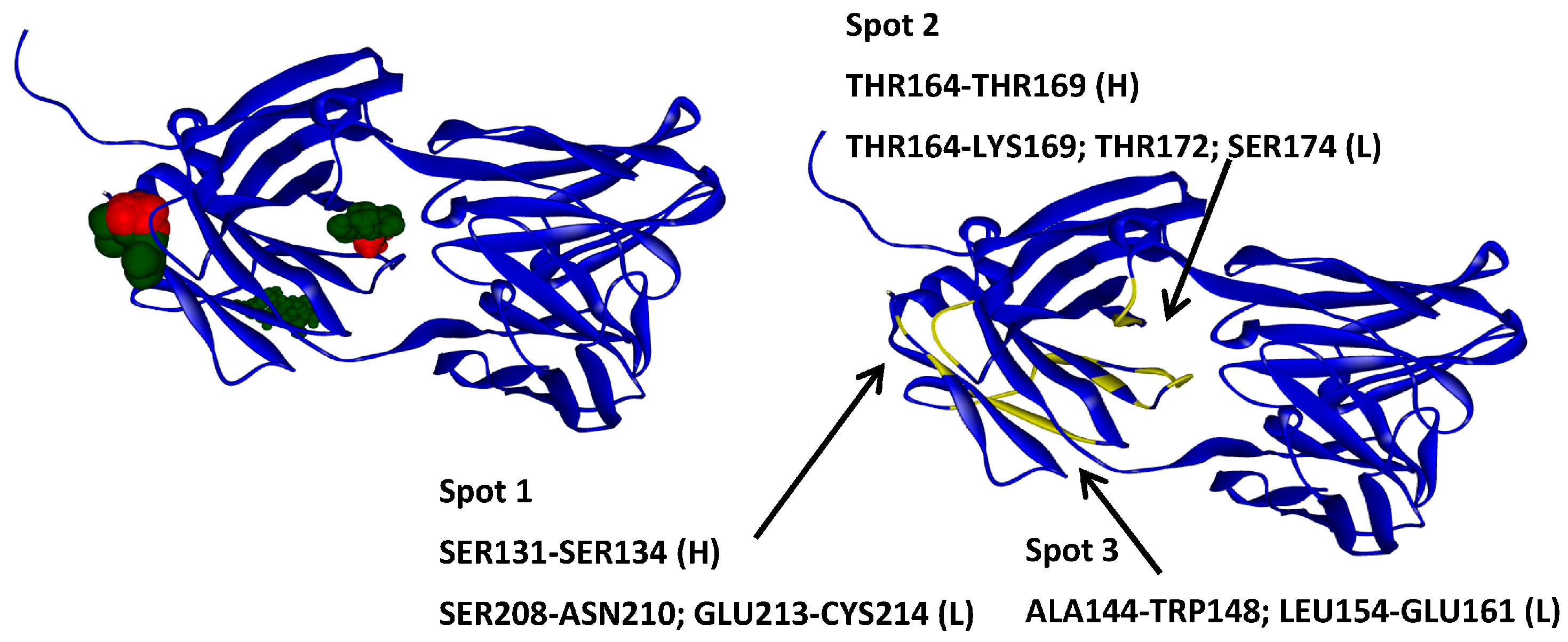
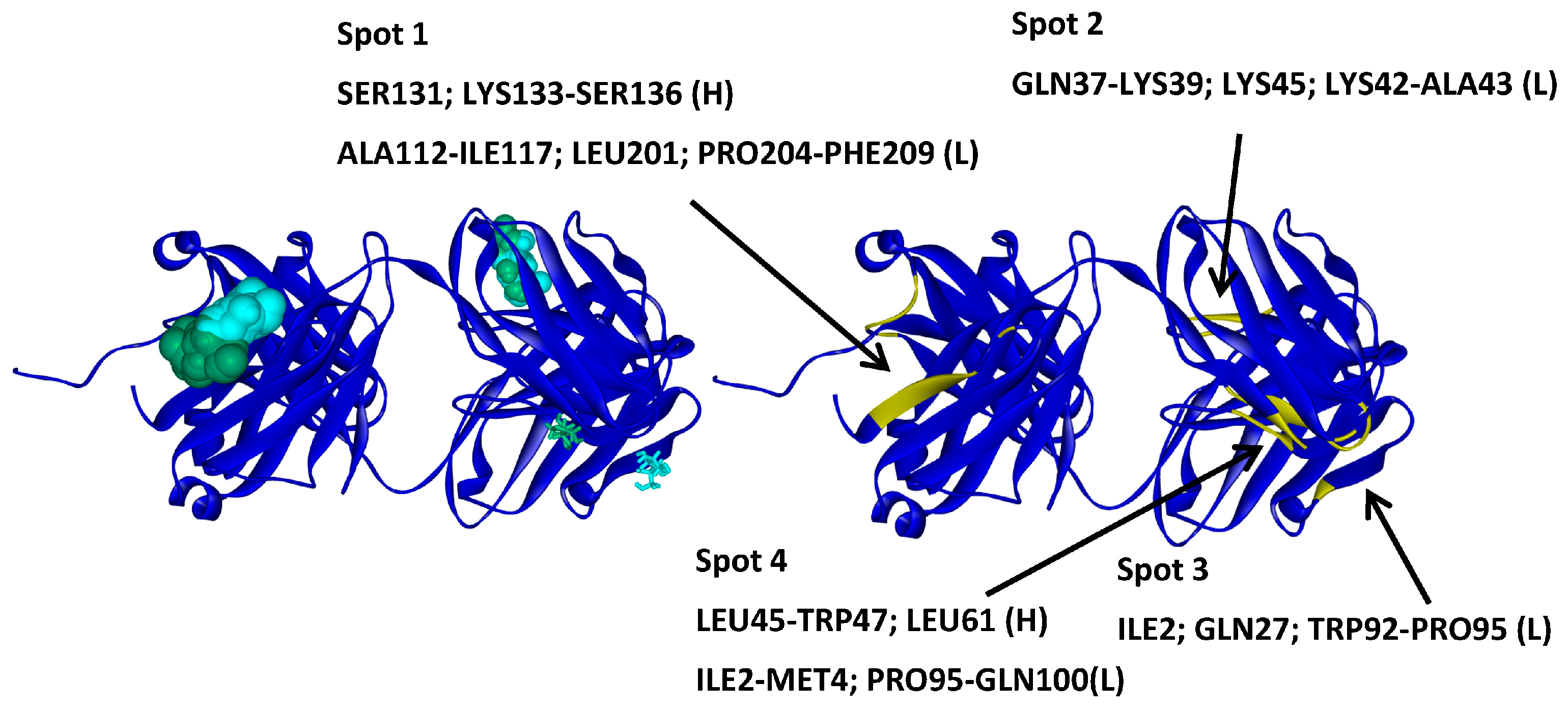
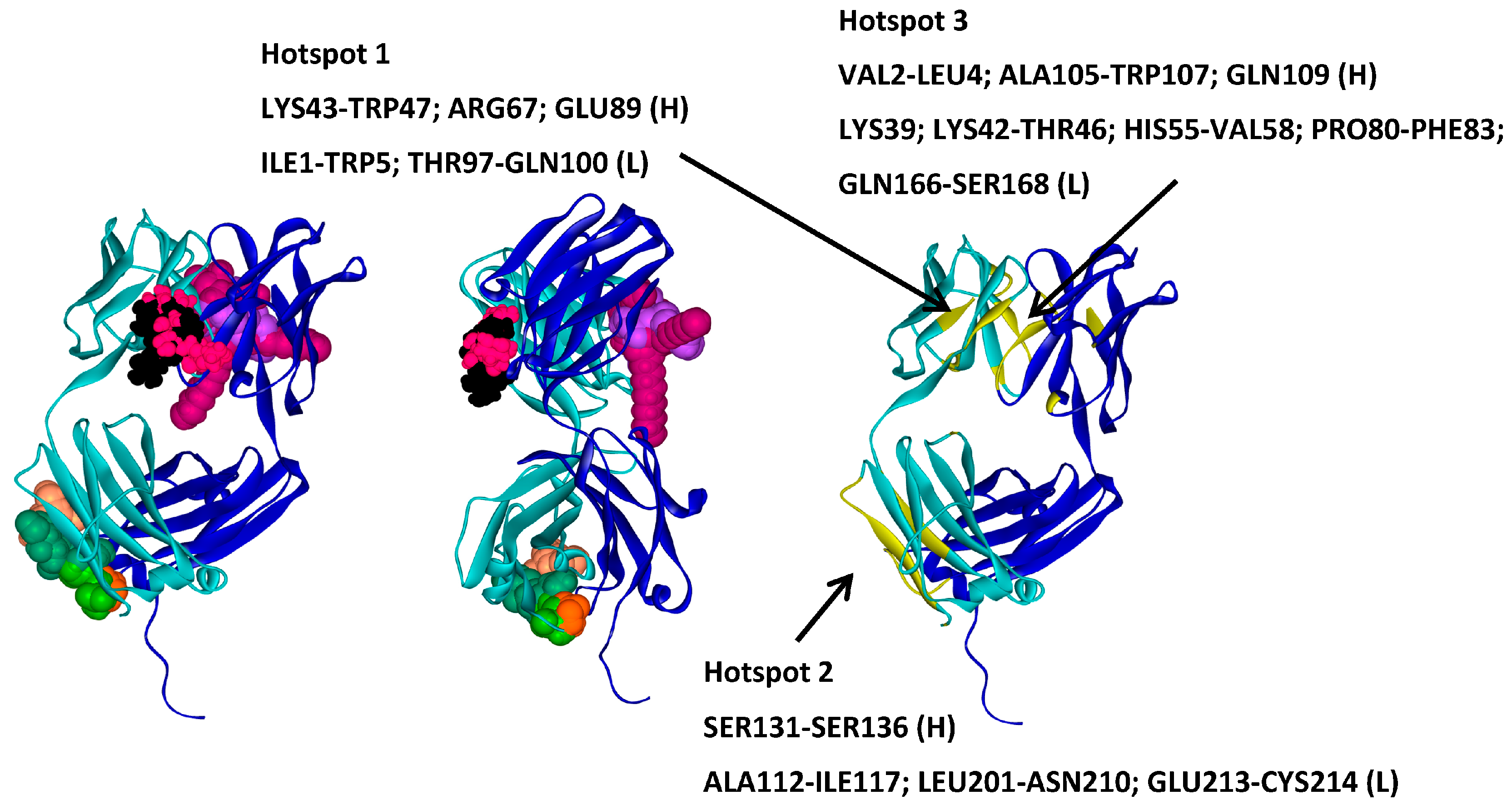
2.2. Hotspots for Protein–Protein Interactions—Rigid Docking
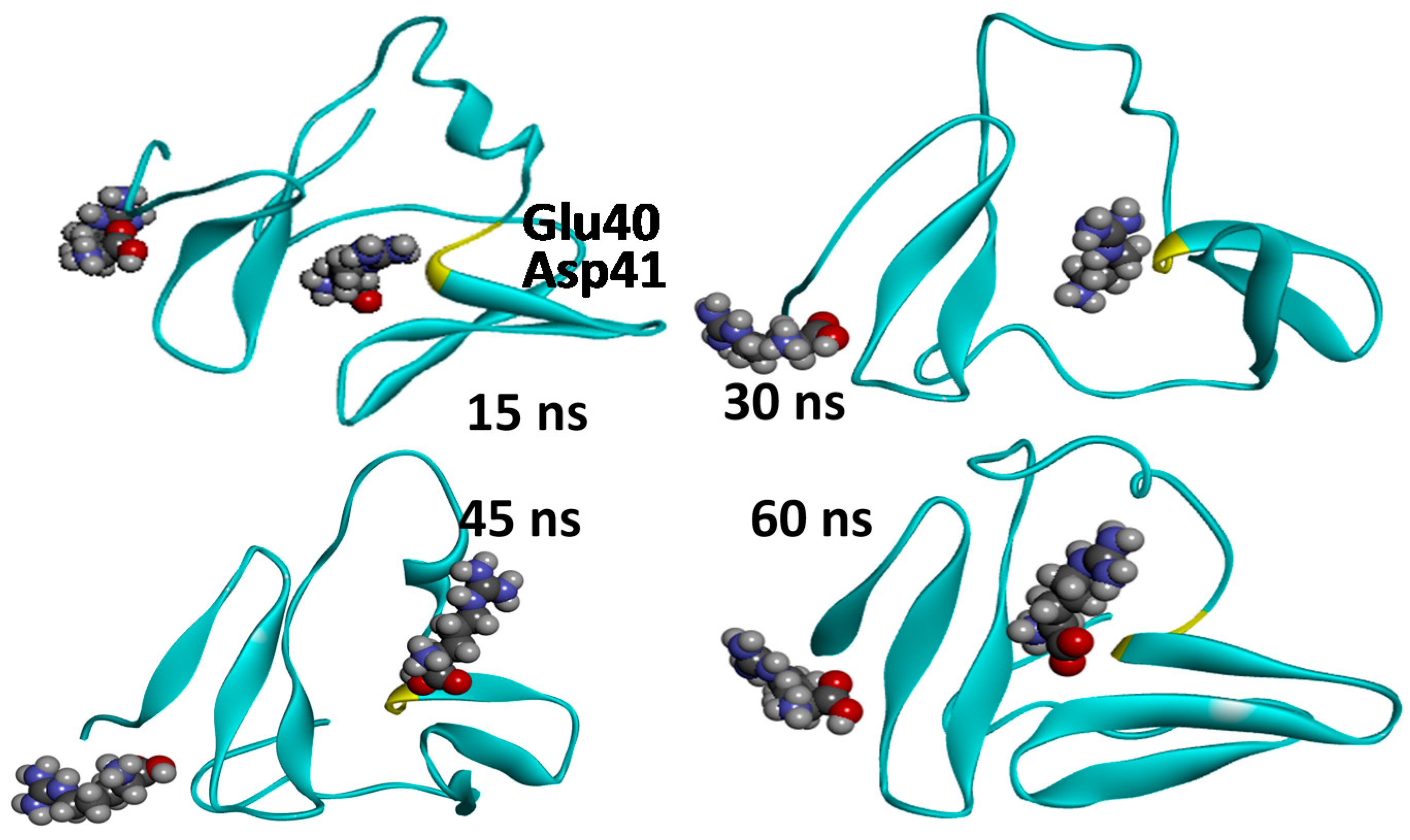
2.3. Molecular Dynamics Simulations—Validation of Identified Hotspots
2.4. Application to the Fab A33 and a Set of Commercial Excipients
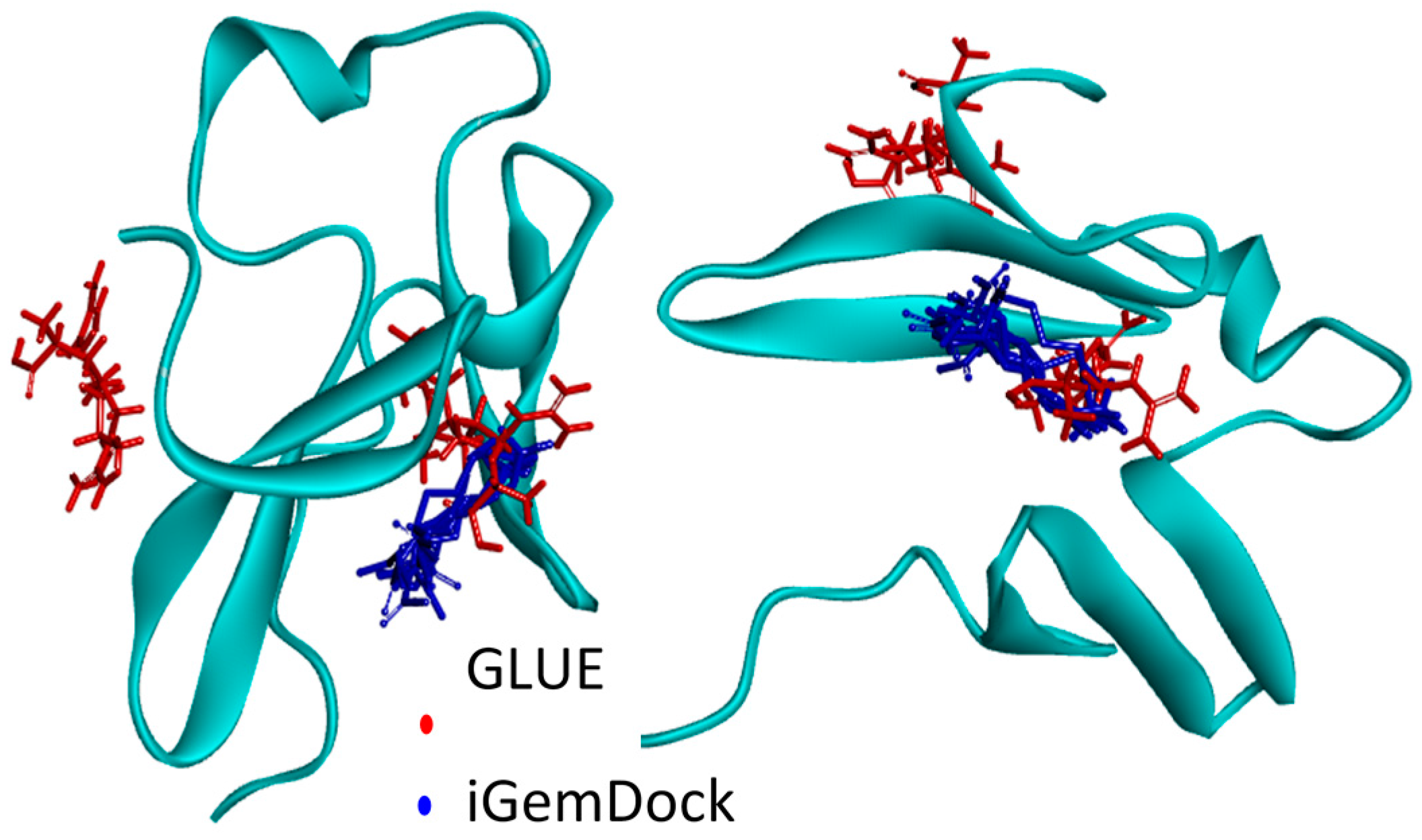
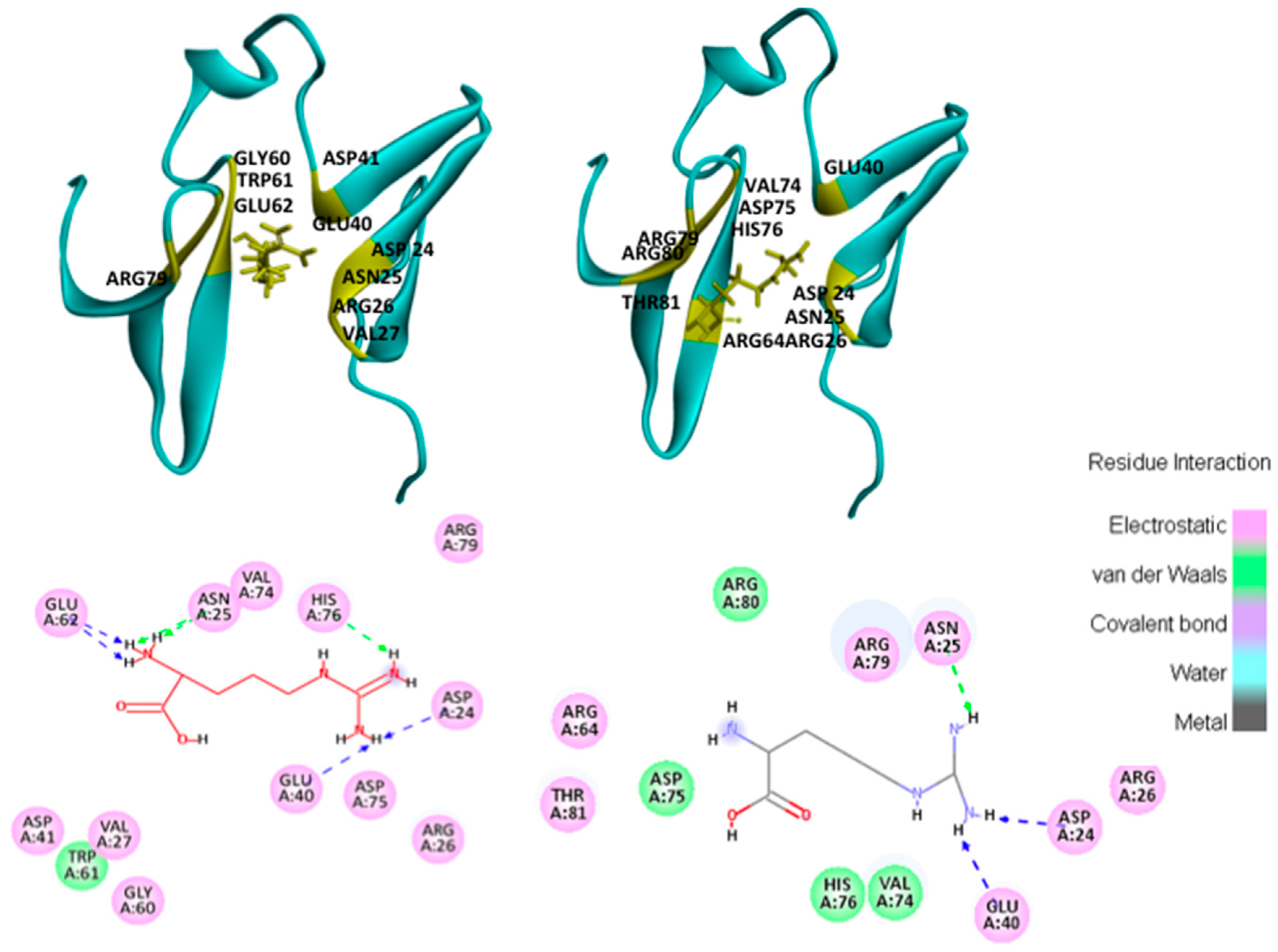
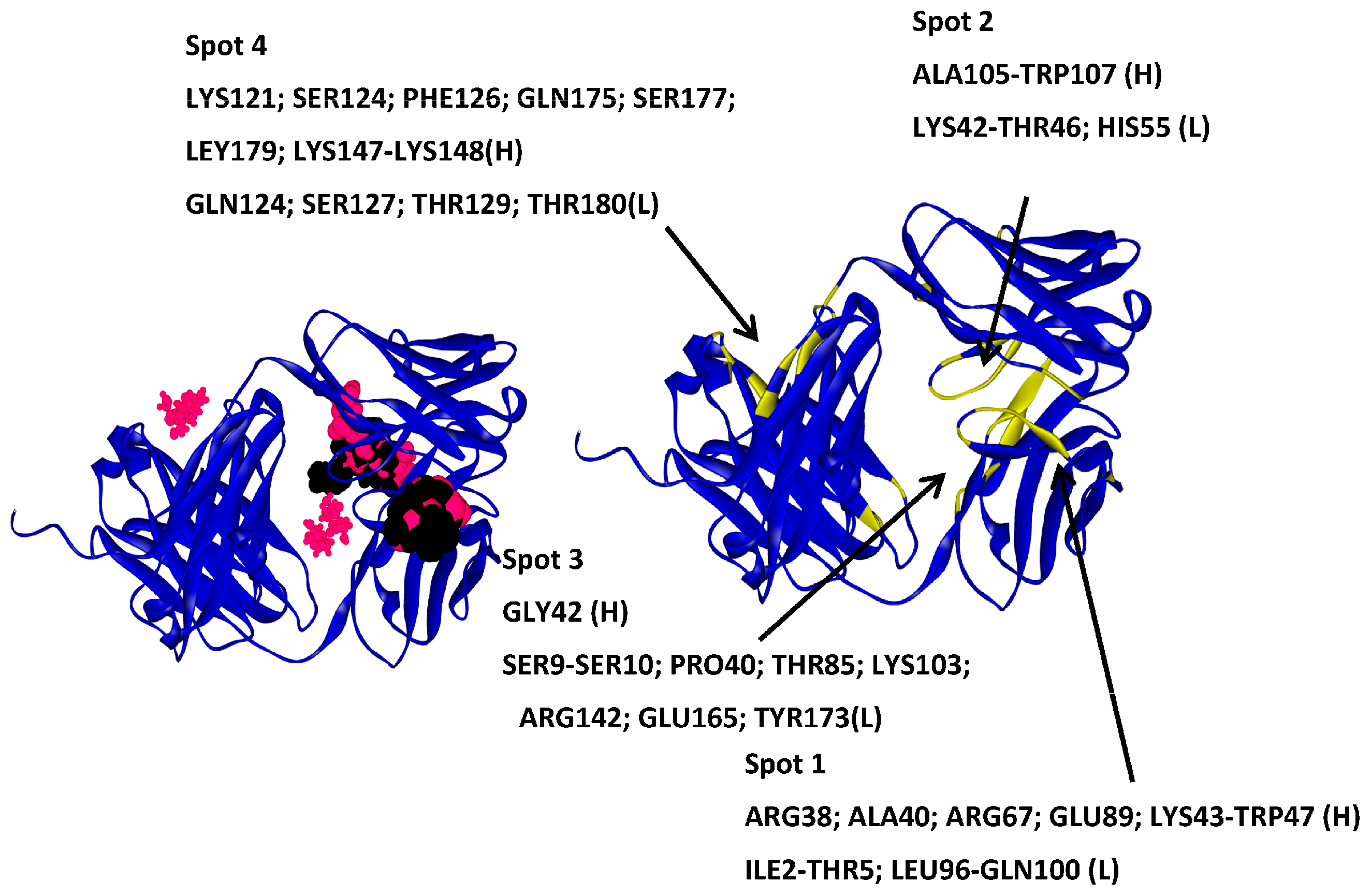
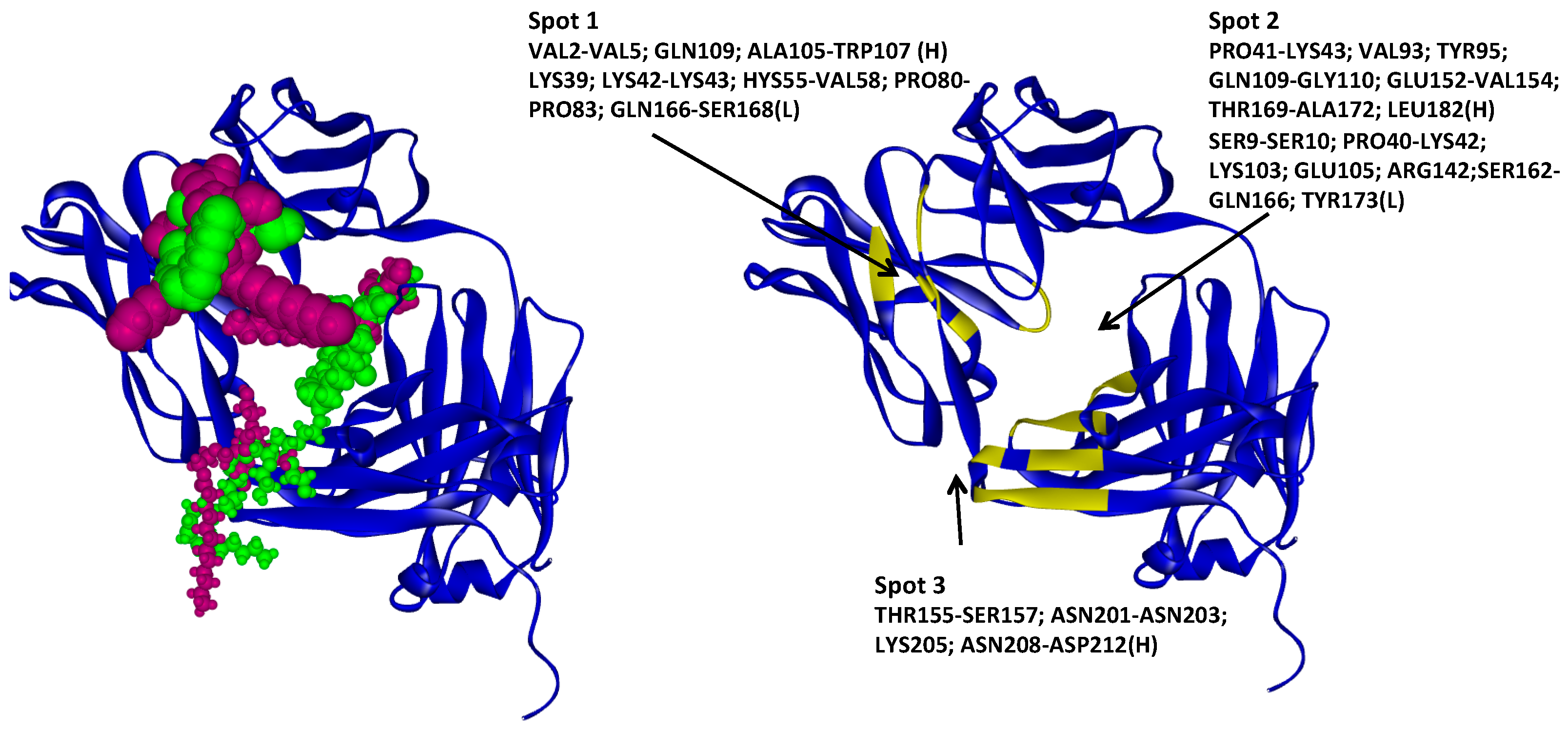
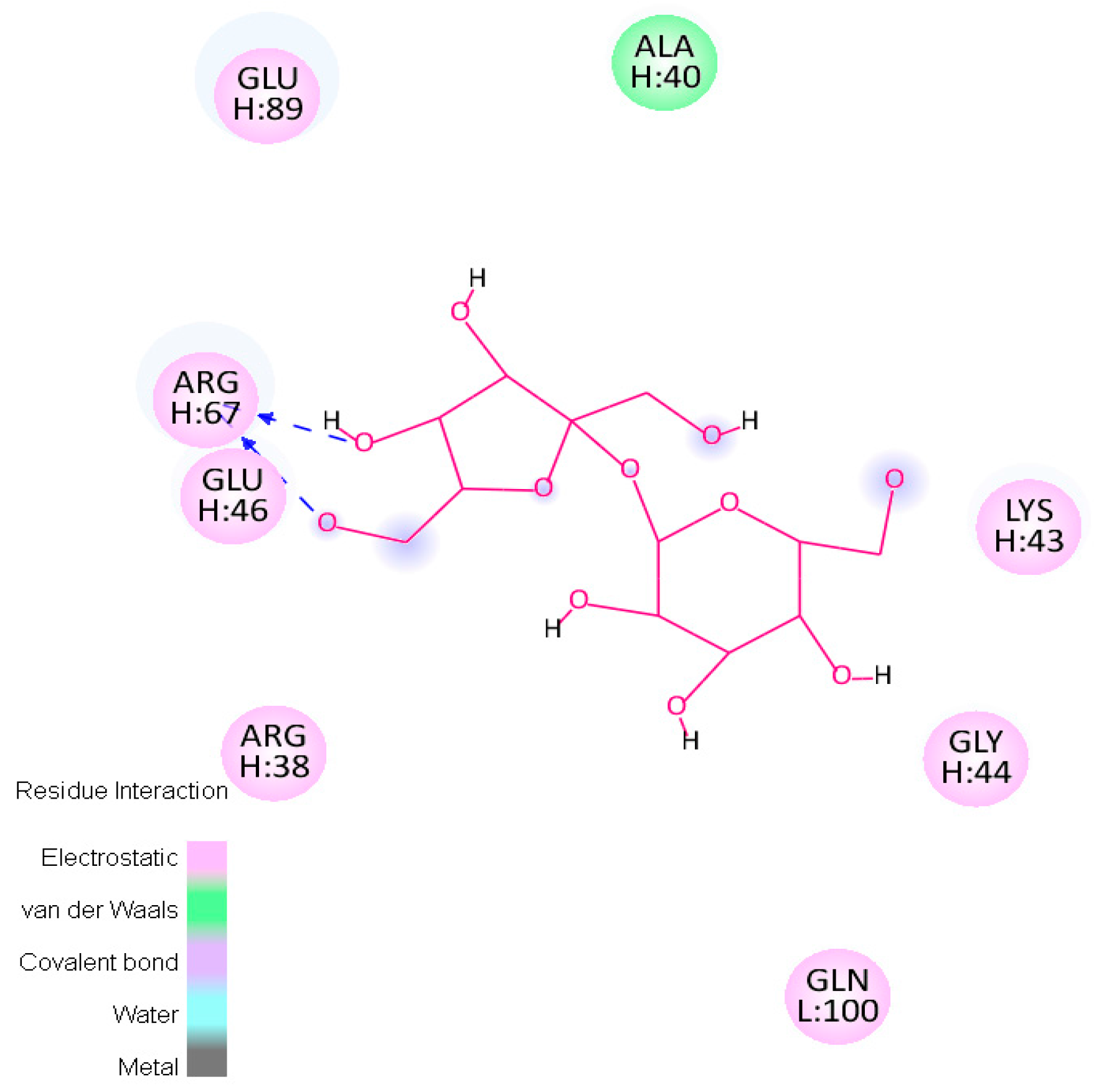
2.4.1. Hotspots for Fab A33 Interactions with Two Amino Acids
2.4.2. Hotspots for Fab A33 Interactions with Two Saccharides
2.4.3. Hotspots for Fab A33 Interactions with Two Sugar Alcohol Isomers
2.4.4. Hotspots for Fab A33 Interactions with Two Surfactants
2.5. Hotspots for Fab A33 Interactions with Excipients
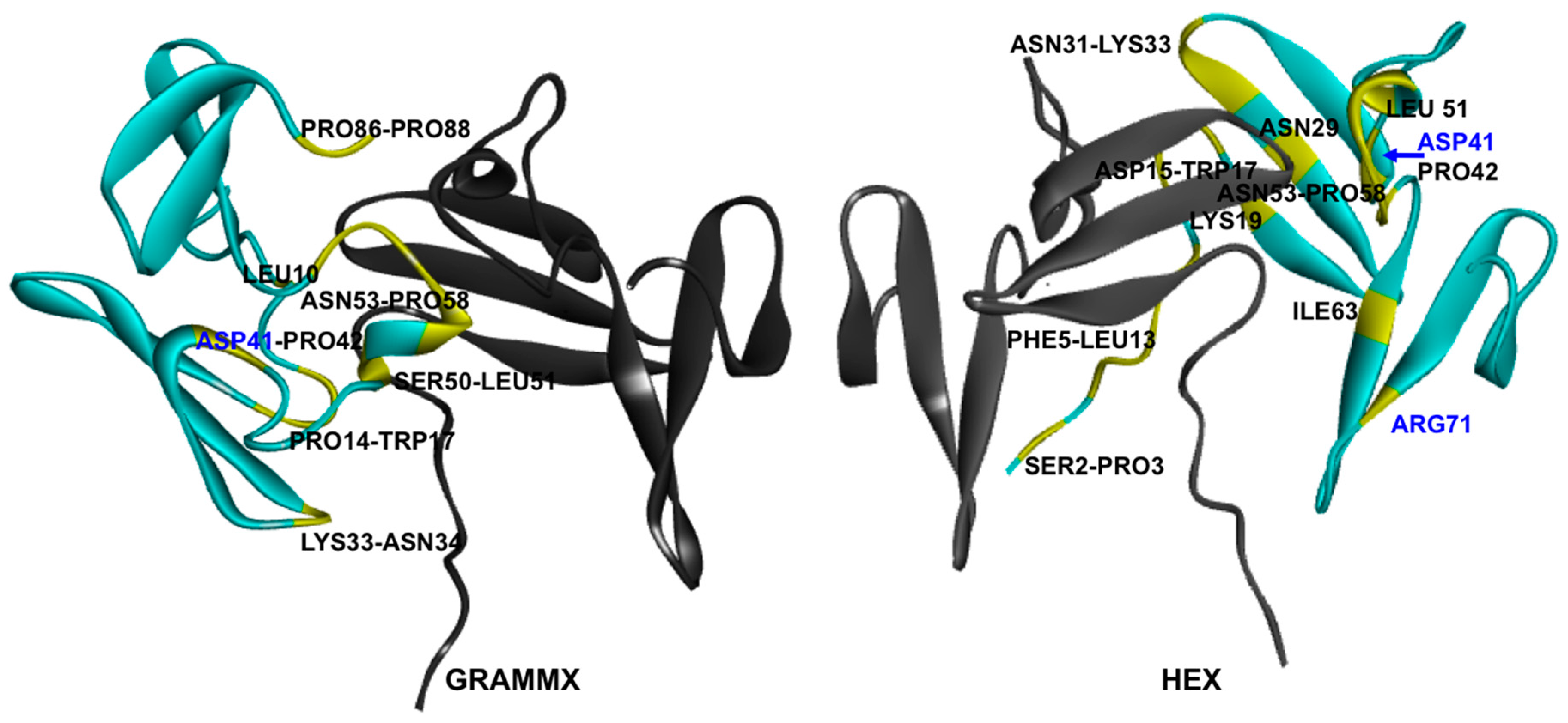
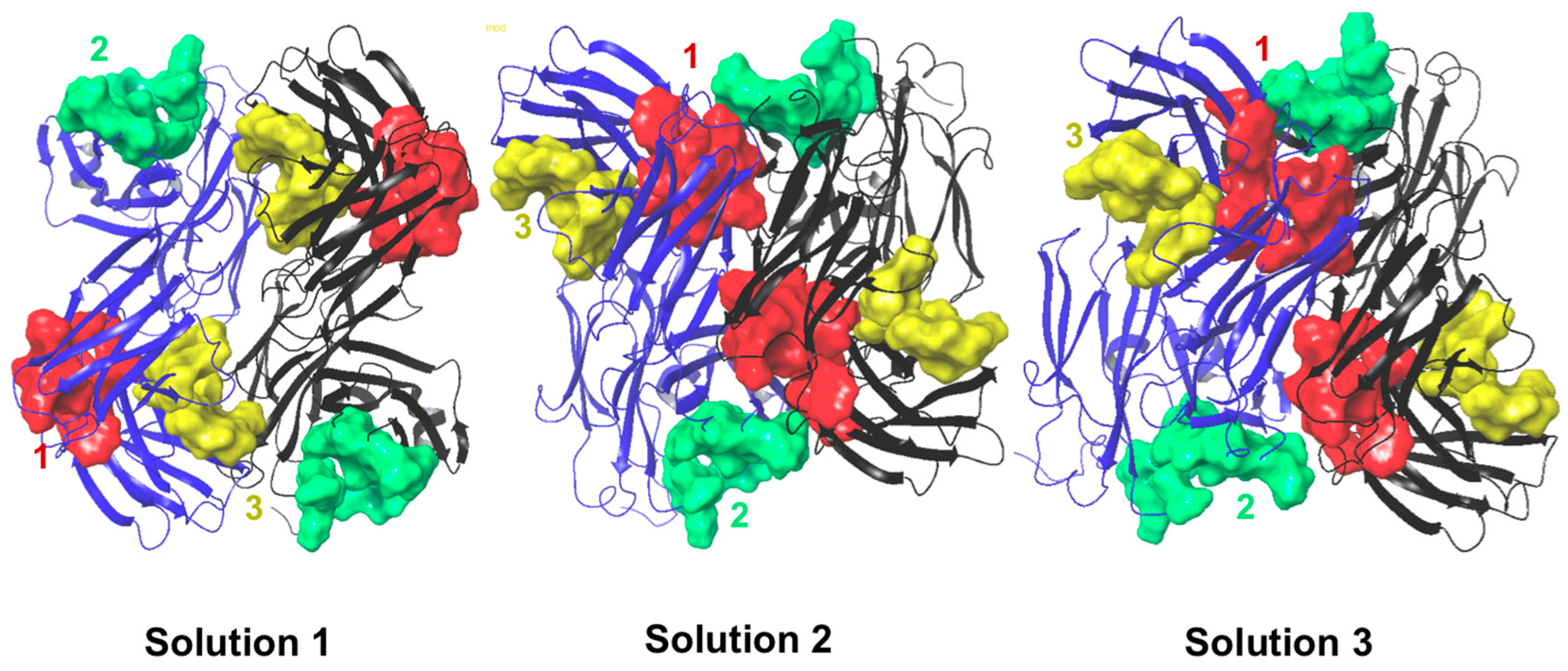
2.6. Protein–Protein Interaction Surfaces between Two Fab A33 Molecules
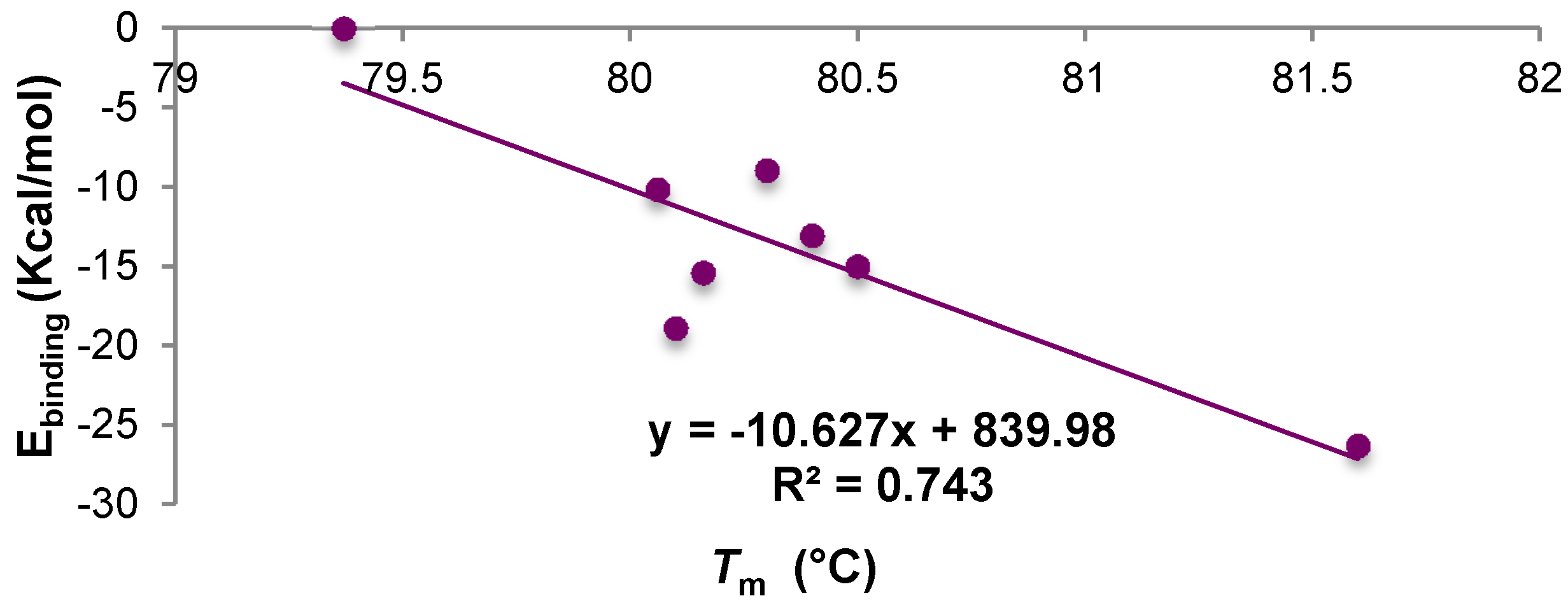
2.7. Experimental Confirmation of the Presence of Interaction between A33 and the Commercial Excipients
3. Materials and Methods
3.1. Molecular Structures Setup
3.2. Protein-Protein Molecular Docking
3.3. Protein–Excipient Molecular Docking
3.4. Molecular Dynamic Simulations
3.5. Stepped Thermal Experiments to Determine Tm Values
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | directory of open access journals |
| TLA | three-letter acronym |
| LD | linear dichroism |
References
- Bhambhani, A.; Kissmann, J.M.; Joshi, S.B.; Volkin, D.B.; Kashi, R.S.; Middaugh, C.R. Formulation design and high-throughput excipient selection based on structural integrity and conformational stability of dilute and highly concentrated IGG1 monoclonal antibody solutions. J. Pharm. Sci. 2012, 101, 1120–1135. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, S.V.; Joshi, S.B.; Jones, M.E.; Sathish, H.A.; Bishop, S.M.; Volkin, D.B.; Middaugh, C.R. Excipients differentially influence the conformational stability and pretransition dynamics of two IGG1 monoclonal antibodies. J. Pharm. Sci. 2012, 101, 3062–3077. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, J.X.; Le, Y.; Chen, J.F. Studies of bicalutamide-excipients interaction by combination of molecular docking and molecular dynamics simulation. Mol. Pharm. 2013, 10, 2362–2369. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, V.; Murnane, D.; Hammond, R.B.; Pickering, J.; Roberts, K.J.; Soufian, M.; Forbes, B.; Jaffari, S.; Martin, G.P.; Collins, E.; et al. Formulation pre-screening of inhalation powders using computational atom-atom systematic search method. Mol. Pharm. 2015, 12, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Rawat, S.; Gupta, P.; Kumar, A.; Garg, P.; Suri, C.R.; Sahoo, D.K. Molecular mechanism of poly(vinyl alcohol) mediated prevention of aggregation and stabilization of insulin in nanoparticles. Mol. Pharm. 2015, 12, 1018–1030. [Google Scholar] [PubMed]
- Westermaier, Y.; Veurink, M.; Riis-Johannessen, T.; Guinchard, S.; Gurny, R.; Scapozza, L. Identification of aggregation breakers for bevacizumab (avastin®) self-association through similarity searching and interaction studies. Eur. J. Pharm. Biopharm. 2013, 85, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Golovanov, A.P.; Hautbergue, G.M.; Wilson, S.A.; Lian, L.Y. A simple method for improving protein solubility and long-term stability. J. Am. Chem. Soc. 2004, 126, 8933–8939. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Trout, B.L. Understanding the synergistic effect of arginine and glutamic acid mixtures on protein solubility. J. Phys. Chem. B 2011, 115, 11831–11839. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.K.; Shameem, M. 1–Commercial therapeutic protein drug products. In Therapeutic Protein Drug Products; Meyer, B.K., Ed.; Woodhead Publishing: Cambridge, UK, 2012; pp. 1–11. [Google Scholar]
- Kamal, M.Z.; Kumar, V.; Satyamurthi, K.; Das, K.K.; Rao, N.M. Mutational probing of protein aggregates to design aggregation-resistant proteins. FEBS Open Bio 2016, 6, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Kastelic, M.; Kalyuzhnyi, Y.V.; Hribar-Lee, B.; Dill, K.A.; Vlachy, V. Protein aggregation in salt solutions. Proc. Natl. Acad. Sci. USA 2015, 112, 6766–6770. [Google Scholar] [CrossRef] [PubMed]
- Doig, A.J.; Derreumaux, P. Inhibition of protein aggregation and amyloid formation by small molecules. Curr. Opin. Struct. Biol. 2015, 30, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, S.; Kita, Y.; Arakawa, T. Interactions of formulation excipients with proteins in solution and in the dried state. Adv. Drug Deliv. Rev. 2011, 63, 1053–1073. [Google Scholar] [CrossRef] [PubMed]
- Kamerzell, T.J.; Esfandiary, R.; Joshi, S.B.; Middaugh, C.R.; Volkin, D.B. Protein–excipient interactions: Mechanisms and biophysical characterization applied to protein formulation development. Adv. Drug Deliv. Rev. 2011, 63, 1118–1159. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.R.; Sternberg, M.J. Prediction of protein–protein interactions by docking methods. Curr. Opin. Struct. Biol. 2002, 12, 28–35. [Google Scholar] [CrossRef]
- Novikov, F.N.; Chilov, G.G. Molecular docking: Theoretical background, practical applications and perspectives. Mendeleev Commun. 2009, 19, 237–242. [Google Scholar] [CrossRef]
- Yuriev, E.; Holien, J.; Ramsland, P.A. Improvements, trends, and new ideas in molecular docking: 2012–2013 in review. J. Mol. Recognit. 2015, 28, 581–604. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Grinter, S.Z.; Zou, X. Scoring functions and their evaluation methods for protein–ligand docking: Recent advances and future directions. Phys. Chem. Chem. Phys. 2010, 12, 12899–12908. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N. Scoring functions for protein–ligand docking. Curr. Protein Pept. Sci. 2006, 7, 407–420. [Google Scholar] [PubMed]
- Wang, R.; Lu, Y.; Wang, S. Comparative evaluation of 11 scoring functions for molecular docking. J. Med. Chem. 2003, 46, 2287–2303. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Kalyanaraman, C.; Irwin, J.J.; Jacobson, M.P. Physics-based scoring of protein–ligand complexes: Enrichment of known inhibitors in large-scale virtual screening. J. Chem. Inf. Model. 2006, 46, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Bohm, H.J. The development of a simple empirical scoring function to estimate the binding constant for a protein–ligand complex of known three-dimensional structure. J. Comput. Aided Mol. Des. 1994, 8, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lai, L.; Wang, S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comput. Aided Mol. Des. 2002, 16, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Sippl, M.J. Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins. J. Mol. Biol. 1990, 213, 859–883. [Google Scholar] [CrossRef]
- Arakawa, T.; Bhat, R.; Timasheff, S.N. Preferential interactions determine protein solubility in three-component solutions: The magnesium chloride system. Biochemistry 1990, 29, 1914–1923. [Google Scholar] [CrossRef] [PubMed]
- Baynes, B.M.; Trout, B.L. Proteins in mixed solvents: A molecular-level perspective. J. Phys. Chem. B 2003, 107, 14058–14067. [Google Scholar] [CrossRef]
- Shulgin, I.L.; Ruckenstein, E. Relationship between preferential interaction of a protein in an aqueous mixed solvent and its solubility. Biophys. Chem. 2005, 118, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Goodford, P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-M.; Chen, C.-C. Gemdock: A generic evolutionary method for molecular docking. Proteins Struct. Funct. Bioinform. 2004, 55, 288–304. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.J.; Kumar, S.; Wang, X.; Helk, B.; Singh, S.K.; Trout, B.L. Aggregation in protein-based biotherapeutics: Computational studies and tools to identify aggregation-prone regions. J. Pharm. Sci. 2011, 100, 5081–5095. [Google Scholar] [CrossRef] [PubMed]
- Morriss-Andrews, A.; Shea, J.-E. Computational studies of protein aggregation: Methods and applications. Annu. Rev. Phys. Chem. 2015, 66, 643–666. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Magnero, K.H.; Valiente, P.A.; Ruiz-Peña, M.; Pérez-Gramatges, A.; Pons, T. Unraveling the binding mechanism of polyoxyethylene sorbitan esters with bovine serum albumin: A novel theoretical model based on molecular dynamic simulations. Colloids Surfaces B 2014, 116, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Antoniw, P.; Farnsworth, A.P.; Turner, A.; Haines, A.M.; Mountain, A.; Mackintosh, J.; Shochat, D.; Humm, J.; Welt, S.; Old, L.J.; et al. Radioimmunotherapy of colorectal carcinoma xenografts in nude mice with yttrium-90 a33 IGG and tri-fab (TFM). Br. J. Cancer 1996, 74, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, C.; Shumway, J.; Castano, J.; Wagner, M.; Waghmare, R. Challenges and strategies for the downstream purification and formulation of fab antibody fragments. Biopharm. Int. 2014, 27, 42–44. [Google Scholar]
- Nelson, A.L.; Reichert, J.M. Development trends for therapeutic antibody fragments. Nat. Biotechnol. 2009, 27, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.S.; Gussow, D.; Griffiths, A.D.; Jones, P.T.; Winter, G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia-coli. Nature 1989, 341, 544–546. [Google Scholar] [CrossRef] [PubMed]
- Kudou, M.; Shiraki, K.; Fujiwara, S.; Imanaka, T.; Takagi, M. Prevention of thermal inactivation and aggregation of lysozyme by polyamines. Eur. J. Biochem. 2003, 270, 4547–4554. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Lyophilization and development of solid protein pharmaceuticals. Int. J. Pharm. 2000, 203, 1–60. [Google Scholar] [CrossRef]
- Kasraian, K.; Spitznagel, T.M.; Juneau, J.A.; Yim, K. Characterization of the sucrose/glycine/water system by differential scanning calorimetry and freeze-drying microscopy. Pharm. Dev. Technol. 1998, 3, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Chongprasert, S.; Knopp, S.A.; Nail, S.L. Characterization of frozen solutions of glycine. J. Pharm. Sci. 2001, 90, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Nomura, M.; Tanaka, K.; Kataoka, N.; Oshitani, J.; Imanaka, H.; Nakanishi, K. Impacts of compression on crystallization behavior of freeze-dried amorphous sucrose. J. Pharm. Sci. 2010, 99, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.E.; Kirchhoff, C.F.; Gaud, H.T. Mannitol-sucrose mixtures—Versatile formulations for protein lyophilization. J. Pharm. Sci. 2002, 91, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, S.; Schebor, C.; Palecek, S.P.; de Pablo, J.J. Phase behavior of freeze-dried phospholipid–cholesterol mixtures stabilized with trehalose. Biochim. Biophys. Acta Biomembr. 2005, 1713, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, S.; Wang, Y.J. Trehalose: Current use and future applications. J. Pharm. Sci. 2011, 100, 2020–2053. [Google Scholar] [CrossRef] [PubMed]
- Pyne, A.; Surana, R.; Suryanarayanan, R. Crystallization of mannitol below TG’ during freeze-drying in binary and ternary aqueous systems. Pharm. Res. 2002, 19, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Krishnamurthy, R.; Suryanarayanan, R. Influence of processing conditions on the physical state of mannitol—Implications in freeze-drying. Pharm. Res. 2007, 24, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.-H.; Brown, M.; Quader, A.; Martin, G. Protective mechanism of stabilizing excipients against dehydration in the freeze-drying of proteins. Pharm. Res. 2002, 19, 1854–1861. [Google Scholar] [CrossRef] [PubMed]
- Nema, S.; Washkuhn, R.J.; Brendel, R.J. Excipients and their use in injectable products. PDA J. Pharm. Sci. Technol. 1997, 51, 166–171. [Google Scholar] [PubMed]
- Brader, M.L.; Estey, T.; Bai, S.; Alston, R.W.; Lucas, K.K.; Lantz, S.; Landsman, P.; Maloney, K.M. Examination of thermal unfolding and aggregation profiles of a series of developable therapeutic monoclonal antibodies. Mol. Pharm. 2015, 12, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Fedoroff, O.Y.; Townson, S.A.; Golovanov, A.P.; Baron, M.; Avis, J.M. The structure and dynamics of tandem WW domains in a negative regulator of notch signaling, suppressor of deltex. J. Biol. Chem. 2004, 279, 34991–35000. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.W.; Kemp, G.J. Protein docking using spherical polar fourier correlations. Proteins 2000, 39, 178–194. [Google Scholar] [CrossRef]
- Macindoe, G.; Mavridis, L.; Venkatraman, V.; Devignes, M.D.; Ritchie, D.W. Hexserver: An FFT-based protein docking server powered by graphics processors. Nucleic Acids Res. 2010, 38, W445–W449. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D. Hex 5.0 User Manual; Hex: Vavdoeuvre-les-Nancy, France, 1996. [Google Scholar]
- Tovchigrechko, A.; Vakser, I.A. Gramm-x public web server for protein–protein docking. Nucleic Acids Res. 2006, 34, W310–W314. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-M. Development and evaluation of a generic evolutionary method for protein–ligand docking. J. Comput. Chem. 2004, 25, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Lab, B. Igemdock Scoring Function. Available online: http://gemdock.life.nctu.edu.tw/dock/method_sf.php (accessed on 18 February 2016).
- Levy, R.M.; Gallicchio, E. Computer simulations with explicit solvent: Recent progress in the thermodynamic decomposition of free energies and in modeling electrostatic effects. Annu. Rev. Phys. Chem. 1998, 49, 531–567. [Google Scholar] [CrossRef] [PubMed]
- Leach, A. Molecular Modelling—Principles and Applications, 2nd ed.; Pearson: Harlow, UK, 2001. [Google Scholar]
- Chakroun, N.; Hilton, D.; Ahmad, S.S.; Platt, G.W.; Dalby, P.A. Mapping the aggregation kinetics of a therapeutic antibody fragment. Mol. Pharm. 2015, 13, 307–319. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Arg1 | Arg2 | Arg3 | Arg4 | Arg5 | Gly1 | Gly2 | Gly3 |
|---|---|---|---|---|---|---|---|---|
| Ebinding kcal·mol−1 | −25.029 | −22.737 | −19.848 | −15.913 | −15.583 | −27.304 | −26.352 | −25.334 |
| Spot | 3 | 1 | 3 | 2 | 2 | 1 | 2 | 1 |
| Molecule | Tre1 | Tre2 | Tre3 | Tre4 | Tre5 | Suc1 | Suc2 | Suc3 | Suc4 | Suc5 |
|---|---|---|---|---|---|---|---|---|---|---|
| Ebinding kcal·mol−1 | −16.638 | −13.783 | −13.023 | −12.262 | −11.762 | −20.233 | −16.780 | −16.334 | −13.868 | −13.286 |
| Spot | 2 | 1 | 2 | 1 | 1 | 2 | 1 | 1 | 3 | 4 |
| Molecule | Man1 | Man2 | Man3 | Man4 | Man5 | Sor1 | Sor2 | Sor3 | Sor4 | Sor5 |
|---|---|---|---|---|---|---|---|---|---|---|
| Ebinding kcal·mol−1 | −11.901 | −10.67 | −8.537 | −8.415 | −7.852 | −9.879 | −9.372 | −8.904 | −8.524 | −8.329 |
| Spot | 1 | 1 | 2 | 3 | 1 | 2 | 1 | 1 | 1 | 4 |
| Molecule | P20 | P20 | P20 | P20 | P80 | P80 | P80 |
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 1 | 2 | 3 | |
| Ebinding kcal·mol−1 | −21.3 | −18.3 | −15.6 | −14.4 | −20.6 | −20.1 | −18.9 |
| Spot | 2 | 3 | 1 | 1 | 2 | 3 | 1 |
| Excipient | Ebinding kcal·mol−1 | Hotspot in Target |
|---|---|---|
| Trehalose_1 | −13.8 | Hotspot 1 Heavy Chain: Lys43-Trp47; Arg67; Glu89 Light Chain: Ile1-Trp5; Thr97-Gln100 |
| Trehalose_2 | −12.3 | |
| Sucrose_1 | −16.9 | |
| Sucrose_2 | −13.9 | |
| Arginine_1 | −22.7 | Hotspot 2 Heavy Chain: Ser131-Ser136 Light Chain: Ala112-Ile117; Leu201-Asn210; Glu213-Cys214 |
| Glycine_1 | −27.3 | |
| Glycine_2 | −25.3 | |
| Mannitol_1 | −11.9 | |
| Mannitol_2 | −10.7 | |
| Mannitol_3 | −7.8 | |
| Sorbitol_1 | −9.4 | |
| Sorbitol_2 | −8.9 | |
| Sorbitol_3 | −8.5 | |
| Tween20_1 | −14.4 | Hotspot 3 Heavy Chain: Val2-Leu4; Ala105-Trp107; Gln109 Light Chain: Lys39; Lys42-Thr46; His55-Val58; Pro80-Phe83; Gln166-Ser168 |
| Tween20_2 | −15.6 | |
| Tween80_1 | −18.9 |
| Solution 1 | Solution 2 | Solution 3 | |
|---|---|---|---|
| Chain with higher number of interactions | Heavy | Light | Light |
| Matching hotspots | 3 (light and heavy chain) | 1 (light and heavy chain) and 2 (light chain) | 1 (light and heavy chain) |
| Sample | Concentration | Tm (°C) | Average Ebinding kcal·mol−1 |
|---|---|---|---|
| A33 | 1 mg/mL | 79.37 | - |
| A33 + trehalose | 5% w/v | 80.4 | −13.05 |
| A33 + sucrose | 5% w/v | 80.16 | −15.4 |
| A33 + mannitol | 4% w/v | 80.06 | −10.13 |
| A33 + sorbitol | 4% w/v | 80.3 | −8.93 |
| A33 + Tween 20 | 0.4% w/v | 80.5 | −15.00 |
| A33 + Tween 80 | 0.4% w/v | 80.1 | −18.9 |
| A33 + Gly | 2% w/v | 81.6 | −26.3 |
| A33 + Arg | 2% w/v | 71.6 | −22.7 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barata, T.S.; Zhang, C.; Dalby, P.A.; Brocchini, S.; Zloh, M. Identification of Protein–Excipient Interaction Hotspots Using Computational Approaches. Int. J. Mol. Sci. 2016, 17, 853. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17060853
Barata TS, Zhang C, Dalby PA, Brocchini S, Zloh M. Identification of Protein–Excipient Interaction Hotspots Using Computational Approaches. International Journal of Molecular Sciences. 2016; 17(6):853. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17060853
Chicago/Turabian StyleBarata, Teresa S., Cheng Zhang, Paul A. Dalby, Steve Brocchini, and Mire Zloh. 2016. "Identification of Protein–Excipient Interaction Hotspots Using Computational Approaches" International Journal of Molecular Sciences 17, no. 6: 853. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17060853