
Role of Receptor Tyrosine Kinase Signaling in Renal Fibrosis

Abstract
:1. Introduction
2. Receptor Tyrosine Kinases
3. Platelet-Derived Growth Factor (PDGF) and Platelet-Derived Growth Factor Receptors (PDGFR)
4. The Vascular Endothelial Growth Factor (VEGF) and Vascular Endothelial Growth Factor Receptors (VEGFR)
5. Fibroblast Growth Factor (FGF) and Fibroblast Growth Factor Receptors (FGFR)
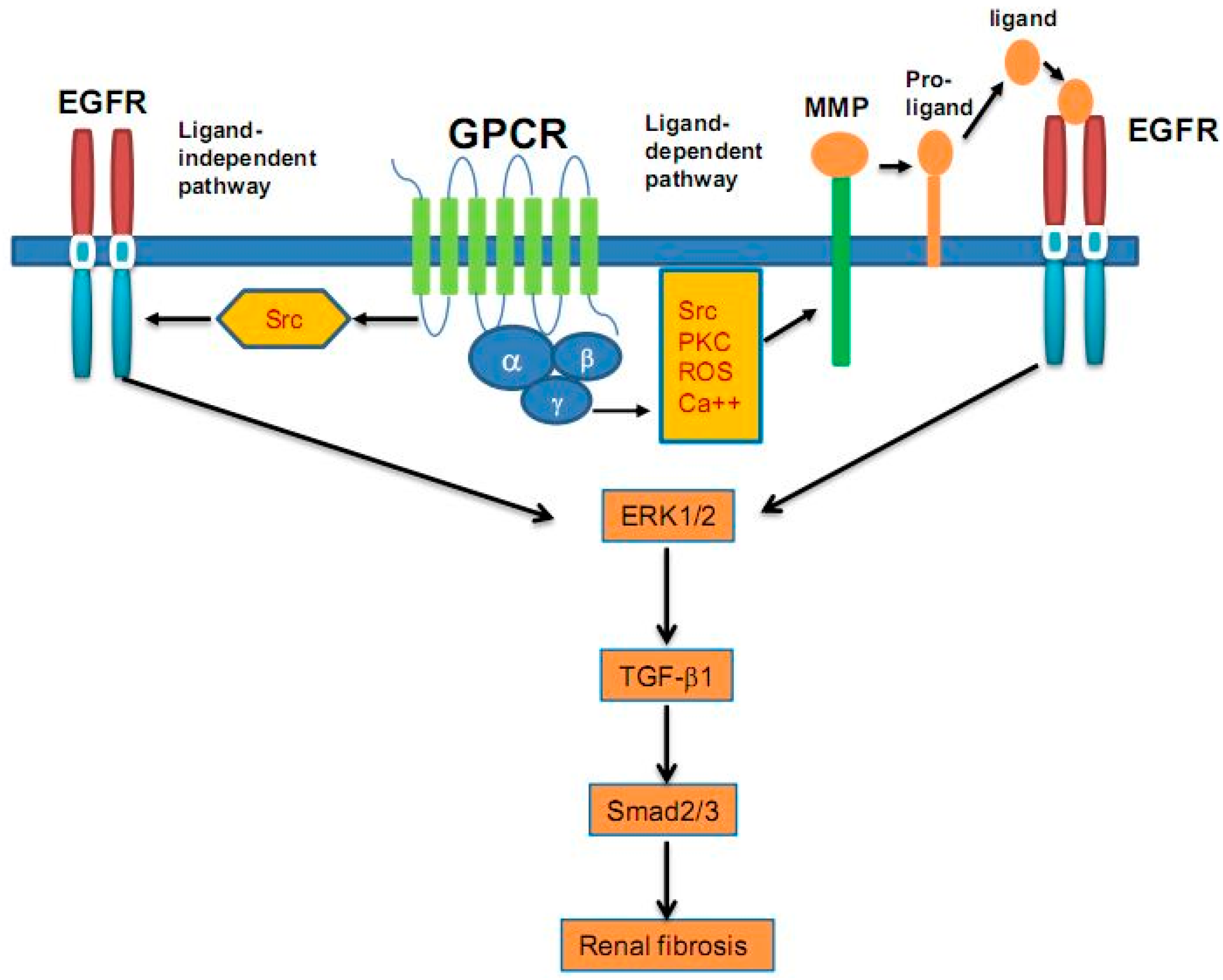
6. Epidermal Growth Factor (EGF) and Epidermal Growth Factor Receptor (EGFR)
7. Insulin-Like Growth Factors (IGF) and Insulin-Like Growth Factor Receptors (IGFR)
8. Discoidin Domain Receptor 1 (DDR1) and Discoidin Domain Receptor 2 (DDR2)
9. Growth Arrest-Specific Gene 6 (Gas6) and Axl Receptor
10. Tyrosine Kinase Inhibitors
11. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Negri, A.L. Prevention of progressive fibrosis in chronic renal diseases: Antifibrotic agents. J. Nephrol. 2004, 17, 496–503. [Google Scholar] [PubMed]
- Samuels, J.A.; Molony, D.A. Randomized controlled trials in nephrology: State of the evidence and critiquing the evidence. Adv. Chronic Kidney Dis. 2012, 19, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Tortora, G. EGFR antagonists in cancer treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef] [PubMed]
- Distler, J.H.; Distler, O. Intracellular tyrosine kinases as novel targets for anti-fibrotic therapy in systemic sclerosis. Rheumatology (Oxford, England) 2008, 47 (Suppl. 5), v10–v11. [Google Scholar] [CrossRef] [PubMed]
- Madhani, H.D. Accounting for specificity in receptor tyrosine kinase signaling. Cell 2001, 106, 9–11. [Google Scholar] [CrossRef]
- Miller, W.T. Determinants of substrate recognition in nonreceptor tyrosine kinases. Acc. Chem. Res. 2003, 36, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; El-Dahr, S.S.; Yosypiv, I.V. Receptor tyrosine kinases in kidney development. J. Signal Transduct. 2011, 2011, 869281. [Google Scholar] [CrossRef] [PubMed]
- Boor, P.; Ostendorf, T.; Floege, J. PDGF and the progression of renal disease. Nephrol. Dial. Transplant. 2014, 29 (Suppl. 1), i45–i54. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Inoue, H.; Sasahara, M. Platelet-derived growth factor and renal disease. Curr. Opin. Nephrol. Hypertens. 2012, 21, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Ostendorf, T.; Eitner, F.; Floege, J. The PDGF family in renal fibrosis. Pediatr. Nephrol. (Berlin, Germany) 2012, 27, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Floege, J.; Eitner, F.; Alpers, C.E. A new look at platelet-derived growth factor in renal disease. J. Am. Soc. Nephrol. 2008, 19, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Eitner, F.; Bucher, E.; van Roeyen, C.; Kunter, U.; Rong, S.; Seikrit, C.; Villa, L.; Boor, P.; Fredriksson, L.; Backstrom, G.; et al. PDGF-C is a proinflammatory cytokine that mediates renal interstitial fibrosis. J. Am. Soc. Nephrol. 2008, 19, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Tallquist, M.; Kazlauskas, A. PDGF signaling in cells and mice. Cytokine Growth Factor Rev. 2004, 15, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Olson, L.E.; Soriano, P. Increased PDGFRα activation disrupts connective tissue development and drives systemic fibrosis. Dev. Cell 2009, 16, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Tolbert, E.; Pang, M.; Ponnusamy, M.; Yan, H.; Zhuang, S. Suramin inhibits renal fibrosis in chronic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 1064–1075. [Google Scholar] [CrossRef] [PubMed]
- Ostendorf, T.; Kunter, U.; Grone, H.J.; Bahlmann, F.; Kawachi, H.; Shimizu, F.; Koch, K.M.; Janjic, N.; Floege, J. Specific antagonism of PDGF prevents renal scarring in experimental glomerulonephritis. J. Am. Soc. Nephrol. 2001, 12, 909–918. [Google Scholar] [PubMed]
- Ostendorf, T.; Rong, S.; Boor, P.; Wiedemann, S.; Kunter, U.; Haubold, U.; van Roeyen, C.R.; Eitner, F.; Kawachi, H.; Starling, G.; et al. Antagonism of PDGF-D by human antibody CR002 prevents renal scarring in experimental glomerulonephritis. J. Am. Soc. Nephrol. 2006, 17, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Shinkai, Y.; Cameron, J.S. Trial of platelet-derived growth factor antagonist, trapidil, in accelerated nephrotoxic nephritis in the rabbit. Br. J. Exp. Pathol. 1987, 68, 847–852. [Google Scholar] [PubMed]
- Nakagawa, T.; Sasahara, M.; Haneda, M.; Kataoka, H.; Nakagawa, H.; Yagi, M.; Kikkawa, R.; Hazama, F. Role of PDGF B-chain and PDGF receptors in rat tubular regeneration after acute injury. Am. J. Pathol. 1999, 155, 1689–1699. [Google Scholar] [CrossRef]
- Ludewig, D.; Kosmehl, H.; Sommer, M.; Bohmer, F.D.; Stein, G. PDGF receptor kinase blocker AG1295 attenuates interstitial fibrosis in rat kidney after unilateral obstruction. Cell Tissue Res. 2000, 299, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, B.F.; Flyvbjerg, A.; de Vriese, A.S. The role of vascular endothelial growth factor (VEGF) in renal pathophysiology. Kidney Int. 2004, 65, 2003–2017. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P. The role of vascular endothelial growth factor in angiogenesis. Acta Haematol. 2001, 106, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Tsurumi, Y.; Murohara, T.; Krasinski, K.; Chen, D.; Witzenbichler, B.; Kearney, M.; Couffinhal, T.; Isner, J.M. Reciprocal relation between VEGF and NO in the regulation of endothelial integrity. Nat. Med. 1997, 3, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Eremina, V.; Sood, M.; Haigh, J.; Nagy, A.; Lajoie, G.; Ferrara, N.; Gerber, H.P.; Kikkawa, Y.; Miner, J.H.; Quaggin, S.E. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Investig. 2003, 111, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Foster, R.R.; Hole, R.; Anderson, K.; Satchell, S.C.; Coward, R.J.; Mathieson, P.W.; Gillatt, D.A.; Saleem, M.A.; Bates, D.O.; Harper, S.J. Functional evidence that vascular endothelial growth factor may act as an autocrine factor on human podocytes. Am. J. Physiol. Ren. Physiol. 2003, 284, F1263–F1273. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.J.; Downs, L.; Tomson, C.R.; Dwight, J.S.; Bolton, C. Elevated plasma vascular endothelial growth factor levels in non-diabetic predialysis uraemia. Nephron 2002, 90, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Chou, E.; Suzuma, I.; Way, K.J.; Opland, D.; Clermont, A.C.; Naruse, K.; Suzuma, K.; Bowling, N.L.; Vlahos, C.J.; Aiello, L.P.; et al. Decreased cardiac expression of vascular endothelial growth factor and its receptors in insulin-resistant and diabetic States: A possible explanation for impaired collateral formation in cardiac tissue. Circulation 2002, 105, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Suga, S.I.; Kang, D.H.; Jefferson, J.A.; Mazzali, M.; Gordon, K.L.; Matsui, K.; Breiteneder-Geleff, S.; Shankland, S.J.; Hughes, J.; et al. Vascular endothelial growth factor accelerates renal recovery in experimental thrombotic microangiopathy. Kidney Int. 2000, 58, 2390–2399. [Google Scholar] [CrossRef] [PubMed]
- Suga, S.; Kim, Y.G.; Joly, A.; Puchacz, E.; Kang, D.H.; Jefferson, J.A.; Abraham, J.A.; Hughes, J.; Johnson, R.J.; Schreiner, G.F. Vascular endothelial growth factor (VEGF121) protects rats from renal infarction in thrombotic microangiopathy. Kidney Int. 2001, 60, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Malmstrom, N.K.; Kallio, E.A.; Rintala, J.M.; Nykanen, A.I.; Raisanen-Sokolowski, A.K.; Paavonen, T.; Lemstrom, K.B.; Koskinen, P.K. Vascular endothelial growth factor in chronic rat allograft nephropathy. Transpl. Immunol. 2008, 19, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Shihab, F.S.; Bennett, W.M.; Yi, H.; Andoh, T.F. Expression of vascular endothelial growth factor and its receptors Flt-1 and KDR/Flk-1 in chronic cyclosporine nephrotoxicity. Transplantation 2001, 72, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Shihab, F.S.; Bennett, W.M.; Isaac, J.; Yi, H.; Andoh, T.F. Angiotensin II regulation of vascular endothelial growth factor and receptors Flt-1 and KDR/Flk-1 in cyclosporine nephrotoxicity. Kidney Int. 2002, 62, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Shihab, F.S.; Bennett, W.M.; Isaac, J.; Yi, H.; Andoh, T.F. Nitric oxide modulates vascular endothelial growth factor and receptors in chronic cyclosporine nephrotoxicity. Kidney Int. 2003, 63, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Flyvbjerg, A.; Dagnaes-Hansen, F.; de Vriese, A.S.; Schrijvers, B.F.; Tilton, R.G.; Rasch, R. Amelioration of long-term renal changes in obese type 2 diabetic mice by a neutralizing vascular endothelial growth factor antibody. Diabetes 2002, 51, 3090–3094. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, B.F.; Rasch, R.; Tilton, R.G.; Flyvbjerg, A. High protein-induced glomerular hypertrophy is vascular endothelial growth factor-dependent. Kidney Int. 2002, 61, 1600–1604. [Google Scholar] [CrossRef] [PubMed]
- Pillebout, E.; Burtin, M.; Yuan, H.T.; Briand, P.; Woolf, A.S.; Friedlander, G.; Terzi, F. Proliferation and remodeling of the peritubular microcirculation after nephron reduction: Association with the progression of renal lesions. Am. J. Pathol. 2001, 159, 547–560. [Google Scholar] [CrossRef]
- Kang, D.H.; Joly, A.H.; Oh, S.W.; Hugo, C.; Kerjaschki, D.; Gordon, K.L.; Mazzali, M.; Jefferson, J.A.; Hughes, J.; Madsen, K.M.; et al. Impaired angiogenesis in the remnant kidney model: I. Potential role of vascular endothelial growth factor and thrombospondin-1. J. Am. Soc. Nephrol. 2001, 12, 1434–1447. [Google Scholar] [PubMed]
- Pupilli, C.; Lasagni, L.; Romagnani, P.; Bellini, F.; Mannelli, M.; Misciglia, N.; Mavilia, C.; Vellei, U.; Villari, D.; Serio, M. Angiotensin II stimulates the synthesis and secretion of vascular permeability factor/vascular endothelial growth factor in human mesangial cells. J. Am. Soc. Nephrol. 1999, 10, 245–255. [Google Scholar] [PubMed]
- Hara, A.; Wada, T.; Furuichi, K.; Sakai, N.; Kawachi, H.; Shimizu, F.; Shibuya, M.; Matsushima, K.; Yokoyama, H.; Egashira, K.; et al. Blockade of VEGF accelerates proteinuria, via decrease in nephrin expression in rat crescentic glomerulonephritis. Kidney Int. 2006, 69, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Lian, Y.G.; Zhou, Q.G.; Zhang, Y.J.; Zheng, F.L. VEGF ameliorates tubulointerstitial fibrosis in unilateral ureteral obstruction mice via inhibition of epithelial-mesenchymal transition. Acta Pharmacol. Sin. 2011, 32, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Feng, H.; Kan, T.; Huang, B.; Zhang, M.; Li, Y.; Shi, C.; Wu, M.; Luo, Y.; Yang, J.; et al. Bevacizumab attenuates hepatic fibrosis in rats by inhibiting activation of hepatic stellate cells. PLoS ONE 2013, 8, e73492. [Google Scholar] [CrossRef] [PubMed]
- Ada, S.; Ersan, S.; Sifil, A.; Unlu, M.; Kolatan, E.; Sert, M.; Sarioglu, S.; Yilmaz, O.; Camsari, T. Effect of bevacizumab, a vascular endothelial growth factor inhibitor, on a rat model of peritoneal sclerosis. Int. Urol. Nephrol. 2015, 47, 2047–2051. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chu, S.; Zeng, F.; Xu, H. Bevacizumab modulates the process of fibrosis in vitro. Clin. Exp. Ophthalmol. 2015, 43, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Cancilla, B.; Davies, A.; Cauchi, J.A.; Risbridger, G.P.; Bertram, J.F. Fibroblast growth factor receptors and their ligands in the adult rat kidney. Kidney Int. 2001, 60, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Powers, C.J.; McLeskey, S.W.; Wellstein, A. Fibroblast growth factors, their receptors and signaling. Endocr. Relat. Cancer 2000, 7, 165–197. [Google Scholar] [CrossRef] [PubMed]
- Boulet, A.M.; Moon, A.M.; Arenkiel, B.R.; Capecchi, M.R. The roles of Fgf4 and Fgf8 in limb bud initiation and outgrowth. Dev. Biol. 2004, 273, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Sil, A.K.; Maeda, S.; Sano, Y.; Roop, D.R.; Karin, M. IκB kinase-α acts in the epidermis to control skeletal and craniofacial morphogenesis. Nature 2004, 428, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Strutz, F.; Zeisberg, M.; Hemmerlein, B.; Sattler, B.; Hummel, K.; Becker, V.; Muller, G.A. Basic fibroblast growth factor expression is increased in human renal fibrogenesis and may mediate autocrine fibroblast proliferation. Kidney Int. 2000, 57, 1521–1538. [Google Scholar] [CrossRef] [PubMed]
- Mas, V.; Maluf, D.; Archer, K.; Yanek, K.; Mas, L.; King, A.; Gibney, E.; Massey, D.; Cotterell, A.; Fisher, R.; et al. Establishing the molecular pathways involved in chronic allograft nephropathy for testing new noninvasive diagnostic markers. Transplantation 2007, 83, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Floege, J.; Hudkins, K.L.; Eitner, F.; Cui, Y.; Morrison, R.S.; Schelling, M.A.; Alpers, C.E. Localization of fibroblast growth factor-2 (basic FGF) and FGF receptor-1 in adult human kidney. Kidney Int. 1999, 56, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Floege, J.; Eng, E.; Young, B.A.; Alpers, C.E.; Barrett, T.B.; Bowen-Pope, D.F.; Johnson, R.J. Infusion of platelet-derived growth factor or basic fibroblast growth factor induces selective glomerular mesangial cell proliferation and matrix accumulation in rats. J. Clin. Investig. 1993, 92, 2952–2962. [Google Scholar] [CrossRef] [PubMed]
- Floege, J.; Kriz, W.; Schulze, M.; Susani, M.; Kerjaschki, D.; Mooney, A.; Couser, W.G.; Koch, K.M. Basic fibroblast growth factor augments podocyte injury and induces glomerulosclerosis in rats with experimental membranous nephropathy. J. Clin. Investig. 1995, 96, 2809–2819. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W.; Hahnel, B.; Rosener, S.; Elger, M. Long-term treatment of rats with FGF-2 results in focal segmental glomerulosclerosis. Kidney Int. 1995, 48, 1435–1450. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.E.; Bruggeman, L.A.; Weeks, B.S.; Kopp, J.B.; Bryant, J.L.; Owens, J.W.; Notkins, A.L.; Klotman, P.E. bFGF and its low affinity receptors in the pathogenesis of HIV-associated nephropathy in transgenic mice. Kidney Int. 1994, 46, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Strutz, F.; Zeisberg, M.; Renziehausen, A.; Raschke, B.; Becker, V.; van Kooten, C.; Muller, G. TGF-β 1 induces proliferation in human renal fibroblasts via induction of basic fibroblast growth factor (FGF-2). Kidney Int. 2001, 59, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Holbro, T.; Hynes, N.E. ErbB receptors: Directing key signaling networks throughout life. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 195–217. [Google Scholar] [CrossRef] [PubMed]
- Makki, N.; Thiel, K.W.; Miller, F.J., Jr. The epidermal growth factor receptor and its ligands in cardiovascular disease. Int. J. Mol. Sci. 2013, 14, 20597–20613. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S.; Liu, N. EGFR signaling in renal fibrosis. Kidney Int. Suppl. 2014, 4, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, W.E.; Chen, Y.; Nakanishi, K.; Frost, P.; Avner, E.D. Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int. 2000, 57, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Francois, H.; Placier, S.; Flamant, M.; Tharaux, P.L.; Chansel, D.; Dussaule, J.C.; Chatziantoniou, C. Prevention of renal vascular and glomerular fibrosis by epidermal growth factor receptor inhibition. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 926–928. [Google Scholar] [CrossRef] [PubMed]
- Bollee, G.; Flamant, M.; Schordan, S.; Fligny, C.; Rumpel, E.; Milon, M.; Schordan, E.; Sabaa, N.; Vandermeersch, S.; Galaup, A.; et al. Epidermal growth factor receptor promotes glomerular injury and renal failure in rapidly progressive crescentic glomerulonephritis. Nat. Med. 2011, 17, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Berk, B.C. Transactivation: A novel signaling pathway from angiotensin II to tyrosine kinase receptors. J. Mol. Cell. Cardiol. 2001, 33, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, J.K.; Nagai, K.; Plieth, D.; Tan, M.; Lee, T.C.; Threadgill, D.W.; Neilson, E.G.; Harris, R.C. EGFR signaling promotes TGFβ-dependent renal fibrosis. J. Am. Soc. Nephrol. 2012, 23, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; He, S.; Tolbert, E.; Gong, R.; Bayliss, G.; Zhuang, S. Suramin alleviates glomerular injury and inflammation in the remnant kidney. PLoS ONE 2012, 7, e36194. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Liu, N.; Tolbert, E.; Ponnusamy, M.; Ma, L.; Gong, R.; Bayliss, G.; Yan, H.; Zhuang, S. Sustained activation of EGFR triggers renal fibrogenesis after acute kidney injury. Am. J. Pathol. 2013, 183, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Laouari, D.; Burtin, M.; Phelep, A.; Martino, C.; Pillebout, E.; Montagutelli, X.; Friedlander, G.; Terzi, F. TGF-α mediates genetic susceptibility to chronic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Sureshbabu, A.; Muhsin, S.A.; Choi, M.E. TGF-β Signaling in the Kidney: Pro-fibrotic and Protective Effects. Am. J. Physiol. Ren. Physiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Ni, H.F.; Pan, M.M.; Liu, H.; Xu, M.; Zhang, M.H.; Liu, B.C. Pirfenidone inhibits macrophage infiltration in 5/6 nephrectomized rats. Am. J. Physiol. Ren. Physiol. 2013, 304, F676–F685. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Iglesias-de la Cruz, M.C.; Jim, B.; Hong, S.W.; Isono, M.; Ziyadeh, F.N. Reversibility of established diabetic glomerulopathy by anti-TGF-β antibodies in db/db mice. Biochem. Biophys. Res. Commun. 2003, 300, 16–22. [Google Scholar] [CrossRef]
- Lautrette, A.; Li, S.; Alili, R.; Sunnarborg, S.W.; Burtin, M.; Lee, D.C.; Friedlander, G.; Terzi, F. Angiotensin II and EGF receptor cross-talk in chronic kidney diseases: A new therapeutic approach. Nat. Med. 2005, 11, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Mondorf, U.F.; Geiger, H.; Herrero, M.; Zeuzem, S.; Piiper, A. Involvement of the platelet-derived growth factor receptor in angiotensin II-induced activation of extracellular regulated kinases 1 and 2 in human mesangial cells. FEBS Lett. 2000, 472, 129–132. [Google Scholar] [CrossRef]
- Clemmons, D.R. Modifying IGF1 activity: An approach to treat endocrine disorders, atherosclerosis and cancer. Nat. Rev. Drug Discov. 2007, 6, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Bach, L.A.; Headey, S.J.; Norton, R.S. IGF-binding proteins—The pieces are falling into place. Trends Endocrinol. Metab. 2005, 16, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.A.; Ryan, G.; Hammerman, M.R. Insulin-like growth factors I and II are produced in the metanephros and are required for growth and development in vitro. J. Cell Biol. 1991, 113, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Bach, L.A.; Hale, L.J. Insulin-like growth factors and kidney disease. Am. J. Kidney Dis. 2015, 65, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Tonshoff, B.; Kaskel, F.J.; Moore, L.C. Effects of insulin-like growth factor I on the renal juxtamedullary microvasculature. Am. J. Physiol. 1998, 274, F120–F128. [Google Scholar] [PubMed]
- Kamenicky, P.; Mazziotti, G.; Lombes, M.; Giustina, A.; Chanson, P. Growth hormone, insulin-like growth factor-1, and the kidney: Pathophysiological and clinical implications. Endocr. Rev. 2014, 35, 234–281. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.P.; Urbonas, A.; Baddoo, A.; Baskin, S.; Malhotra, A.; Meggs, L.G. IGF-1 inhibits the mitochondrial apoptosis program in mesangial cells exposed to high glucose. Am. J. Physiol. Ren. Physiol. 2003, 285, F1013–F1024. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, J.A.; Mok, A.Y.; Parbtani, A.; Matsell, D.G. Increased expression of insulin-like growth factors in progressive glomerulonephritis of the MRL/lpr mouse. Lupus 2003, 12, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Tack, I.; Elliot, S.J.; Potier, M.; Rivera, A.; Striker, G.E.; Striker, L.J. Autocrine activation of the IGF-I signaling pathway in mesangial cells isolated from diabetic NOD mice. Diabetes 2002, 51, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Fujinaka, H.; Katsuyama, K.; Yamamoto, K.; Nameta, M.; Yoshida, Y.; Yaoita, E.; Tomizawa, S.; Yamamoto, T. Expression and localization of insulin-like growth factor binding proteins in normal and proteinuric kidney glomeruli. Nephrology 2010, 15, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Hale, L.J.; Welsh, G.I.; Perks, C.M.; Hurcombe, J.A.; Moore, S.; Hers, I.; Saleem, M.A.; Mathieson, P.W.; Murphy, A.J.; Jeansson, M.; et al. Insulin-like growth factor-II is produced by, signals to and is an important survival factor for the mature podocyte in man and mouse. J. Pathol. 2013, 230, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Kiepe, D.; Tonshoff, B. Insulin-like growth factors in normal and diseased kidney. Endocrinol. Metab. Clin. N. Am. 2012, 41, 351–374. [Google Scholar] [CrossRef] [PubMed]
- Bach, L.A. The insulin-like growth factor system in kidney disease and hypertension. Curr. Opin. Nephrol. Hypertens. 2012, 21, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Shinada, M.; Akdeniz, A.; Panagiotopoulos, S.; Jerums, G.; Bach, L.A. Proteolysis of insulin-like growth factor-binding protein-3 is increased in urine from patients with diabetic nephropathy. J. Clin. Endocrinol. Metab. 2000, 85, 1163–1169. [Google Scholar] [CrossRef] [PubMed]
- Gronbaek, H.; Nielsen, B.; Schrijvers, B.; Vogel, I.; Rasch, R.; Flyvbjerg, A. Inhibitory effects of octreotide on renal and glomerular growth in early experimental diabetes in mice. J. Endocrinol. 2002, 172, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Jia, T.; Gama Axelsson, T.; Heimburger, O.; Barany, P.; Lindholm, B.; Stenvinkel, P.; Qureshi, A.R. IGF-1 and survival in ESRD. Clin. J. Am. Soc. Nephrol. C 2014, 9, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Worthmann, K.; Peters, I.; Kumpers, P.; Saleem, M.; Becker, J.U.; Agustian, P.A.; Achenbach, J.; Haller, H.; Schiffer, M. Urinary excretion of IGFBP-1 and -3 correlates with disease activity and differentiates focal segmental glomerulosclerosis and minimal change disease. Growth Factors 2010, 28, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, K.; Uto, H.; Takami, Y.; Mera, K.; Nishida, C.; Yoshimine, Y.; Fukumoto, M.; Oku, M.; Sogabe, A.; Nosaki, T.; et al. Insulin-like growth factor binding protein-1 levels are increased in patients with IgA nephropathy. Biochem. Biophys. Res. Commun. 2010, 399, 144–149. [Google Scholar] [CrossRef] [PubMed]
- New, D.D.; Block, K.; Bhandhari, B.; Gorin, Y.; Abboud, H.E. IGF-I increases the expression of fibronectin by Nox4-dependent Akt phosphorylation in renal tubular epithelial cells. Am. J. Physiol. Cell Physiol. 2012, 302, C122–C130. [Google Scholar] [CrossRef] [PubMed]
- Luque, V.; Escribano, J.; Grote, V.; Ferre, N.; Koletzko, B.; Gruszfeld, D.; Socha, P.; Langhendries, J.P.; Goyens, P.; Closa-Monasterolo, R. Does insulin-like growth factor-1 mediate protein-induced kidney growth in infants? A secondary analysis from a randomized controlled trial. Pediatr. Res. 2013, 74, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Pricci, F.; Pugliese, G.; Romano, G.; Romeo, G.; Locuratolo, N.; Pugliese, F.; Mene, P.; Galli, G.; Casini, A.; Rotella, C.M.; et al. Insulin-like growth factors I and II stimulate extracellular matrix production in human glomerular mesangial cells. Comparison with transforming growth factor-β. Endocrinology 1996, 137, 879–885. [Google Scholar] [PubMed]
- Wang, S.; Denichilo, M.; Brubaker, C.; Hirschberg, R. Connective tissue growth factor in tubulointerstitial injury of diabetic nephropathy. Kidney Int. 2001, 60, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Oldroyd, S.D.; Miyamoto, Y.; Moir, A.; Johnson, T.S.; El Nahas, A.M.; Haylor, J.L. An IGF-I antagonist does not inhibit renal fibrosis in the rat following subtotal nephrectomy. Am. J. Physiol. Ren. Physiol. 2006, 290, F695–F702. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, R.L.; Goyal, S.; Kim, A.; Chang, A.Y.; Landau, D.; LeRoith, D. Renal tubulointerstitial injury from ureteral obstruction in the neonatal rat is attenuated by IGF-1. Kidney Int. 2000, 57, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Eidman, K.E.; Kren, S.M.; Hostetter, T.H.; Segal, Y. Localization of discoidin domain receptors in rat kidney. Nephron. Exp. Nephrol. 2004, 97, e62–e70. [Google Scholar] [CrossRef] [PubMed]
- Flamant, M.; Placier, S.; Rodenas, A.; Curat, C.A.; Vogel, W.F.; Chatziantoniou, C.; Dussaule, J.C. Discoidin domain receptor 1 null mice are protected against hypertension-induced renal disease. J. Am. Soc. Nephrol. 2006, 17, 3374–3381. [Google Scholar] [CrossRef] [PubMed]
- Guerrot, D.; Kerroch, M.; Placier, S.; Vandermeersch, S.; Trivin, C.; Mael-Ainin, M.; Chatziantoniou, C.; Dussaule, J.C. Discoidin domain receptor 1 is a major mediator of inflammation and fibrosis in obstructive nephropathy. Am. J. Pathol. 2011, 179, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Kerroch, M.; Guerrot, D.; Vandermeersch, S.; Placier, S.; Mesnard, L.; Jouanneau, C.; Rondeau, E.; Ronco, P.; Boffa, J.J.; Chatziantoniou, C.; et al. Genetic inhibition of discoidin domain receptor 1 protects mice against crescentic glomerulonephritis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 4079–4091. [Google Scholar] [CrossRef] [PubMed]
- Wallace, E.; Gewin, L. Imatinib: Novel treatment of immune-mediated kidney injury. J. Am. Soc. Nephrol. 2013, 24, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, V.A. Axl-dependent signalling: A clinical update. Clin. Sci. 2012, 122, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Hyde, G.D.; Taylor, R.F.; Ashton, N.; Borland, S.J.; Wu, H.S.; Gilmore, A.P.; Canfield, A.E. Axl tyrosine kinase protects against tubulo-interstitial apoptosis and progression of renal failure in a murine model of chronic kidney disease and hyperphosphataemia. PLoS ONE 2014, 9, e102096. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.J.; Hilliard, B.; Swami, A.; Madara, J.C.; Rao, S.; Patel, T.; Gaughan, J.P.; Lee, J.; Gadegbeku, C.A.; Choi, E.T.; et al. Growth arrest-specific gene 6 (Gas6) levels are elevated in patients with chronic renal failure. Nephrol. Dial. Transplant. 2012, 27, 4166–4172. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Nagai, K.; Doi, T. Role of growth arrest-specific gene 6 in diabetic nephropathy. Vitam. Horm. 2008, 78, 375–392. [Google Scholar] [PubMed]
- Lee, C.H.; Shieh, Y.S.; Hsiao, F.C.; Kuo, F.C.; Lin, C.Y.; Hsieh, C.H.; Hung, Y.J. High glucose induces human endothelial dysfunction through an Axl-dependent mechanism. Cardiovasc. Diabetol. 2014, 13, 53. [Google Scholar] [CrossRef] [PubMed]
- Cavet, M.E.; Smolock, E.M.; Ozturk, O.H.; World, C.; Pang, J.; Konishi, A.; Berk, B.C. Gas6–Axl receptor signaling is regulated by glucose in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Nagai, K.; Miyoshi, M.; Kake, T.; Fukushima, N.; Matsuura, M.; Shibata, E.; Yamada, S.; Yoshikawa, K.; Kanayama, H.O.; Fukawa, T.; et al. Dual involvement of growth arrest-specific gene 6 in the early phase of human IgA nephropathy. PLoS ONE 2013, 8, e66759. [Google Scholar]
- Yin, J.L.; Hambly, B.D.; Bao, S.S.; Painter, D.; Bishop, G.A.; Eris, J.M. Expression of growth arrest-specific gene 6 and its receptors in dysfunctional human renal allografts. Transpl. Int. Off. J. Eur. Soc. Org. Transpl. 2003, 16, 681–688. [Google Scholar]
- Guo, J.K.; Marlier, A.; Shi, H.; Shan, A.; Ardito, T.A.; Du, Z.P.; Kashgarian, M.; Krause, D.S.; Biemesderfer, D.; Cantley, L.G. Increased tubular proliferation as an adaptive response to glomerular albuminuria. J. Am. Soc. Nephrol. 2012, 23, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Yanagita, M. Gas6, warfarin, and kidney diseases. Clin. Exp. Nephrol. 2004, 8, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.E.; Kelly, D.J.; McKay, T.; Chadban, S.; Hill, P.A.; Cooper, M.E.; Atkins, R.C.; Nikolic-Paterson, D.J. PDGF signal transduction inhibition ameliorates experimental mesangial proliferative glomerulonephritis. Kidney Int. 2001, 59, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Hirai, T.; Masaki, T.; Kuratsune, M.; Yorioka, N.; Kohno, N. PDGF receptor tyrosine kinase inhibitor suppresses mesangial cell proliferation involving STAT3 activation. Clin. Exp. Immunol. 2006, 144, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Lassila, M.; Jandeleit-Dahm, K.; Seah, K.K.; Smith, C.M.; Calkin, A.C.; Allen, T.J.; Cooper, M.E. Imatinib attenuates diabetic nephropathy in apolipoprotein E-knockout mice. J. Am. Soc. Nephrol. 2005, 16, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Zoja, C.; Corna, D.; Rottoli, D.; Zanchi, C.; Abbate, M.; Remuzzi, G. Imatinib ameliorates renal disease and survival in murine lupus autoimmune disease. Kidney Int. 2006, 70, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Savikko, J.; Taskinen, E.; Von Willebrand, E. Chronic allograft nephropathy is prevented by inhibition of platelet-derived growth factor receptor: Tyrosine kinase inhibitors as a potential therapy. Transplantation 2003, 75, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Wang, L.; Yang, T.; Xiong, C.; Xu, L.; Shi, Y.; Bao, W.; Chin, Y.E.; Cheng, S.B.; Yan, H.; et al. EGF Receptor Inhibition Alleviates Hyperuricemic Nephropathy. J. Am. Soc. Nephrol. 2015, 26, 2716–2729. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Tolbert, E.; Ponnusamy, M.; Yan, H.; Zhuang, S. Delayed administration of suramin attenuates the progression of renal fibrosis in obstructive nephropathy. J. Pharmacol. Exp. Ther. 2011, 338, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Korrapati, M.C.; Shaner, B.E.; Neely, B.A.; Alge, J.L.; Arthur, J.M.; Schnellmann, R.G. Diabetes-induced renal injury in rats is attenuated by suramin. J. Pharmacol. Exp. Ther. 2012, 343, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Korrapati, M.C.; Howell, L.A.; Shaner, B.E.; Megyesi, J.K.; Siskind, L.J.; Schnellmann, R.G. Suramin: A potential therapy for diabetic nephropathy. PLoS ONE 2013, 8, e73655. [Google Scholar] [CrossRef] [PubMed]
- Santos, E.S.; Gomez, J.E.; Raez, L.E. Targeting angiogenesis from multiple pathways simultaneously: BIBF 1120, an investigational novel triple angiokinase inhibitor. Investig. New Drugs 2012, 30, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.J.; Binder, R.; Colbatzky, F.; Dallinger, C.; Schlenker-Herceg, R.; Hilberg, F.; Wollin, S.L.; Kaiser, R. Nintedanib: From discovery to the clinic. J. Med. Chem. 2015, 58, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Hostettler, K.E.; Zhong, J.; Papakonstantinou, E.; Karakiulakis, G.; Tamm, M.; Seidel, P.; Sun, Q.; Mandal, J.; Lardinois, D.; Lambers, C.; et al. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir. Res. 2014, 15, 157. [Google Scholar] [CrossRef] [PubMed]
- Wollin, L.; Maillet, I.; Quesniaux, V.; Holweg, A.; Ryffel, B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J. Pharmacol. Exp. Ther. 2014, 349, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, H.V.; Molyneaux, P.L.; Maher, T.M. Reducing lung function decline in patients with idiopathic pulmonary fibrosis: Potential of nintedanib. Drug Des. Dev. Ther. 2013, 7, 503–510. [Google Scholar]
- Richeldi, L.; Costabel, U.; Selman, M.; Kim, D.S.; Hansell, D.M.; Nicholson, A.G.; Brown, K.K.; Flaherty, K.R.; Noble, P.W.; Raghu, G.; et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2011, 365, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Antoniu, S.A. Nintedanib (BIBF 1120) for IPF: A tomorrow therapy? Multidiscip. Respire. Med. 2012, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Dimitroulis, I.A. Nintedanib: A novel therapeutic approach for idiopathic pulmonary fibrosis. Respir. Care 2014, 59, 1450–1455. [Google Scholar] [CrossRef] [PubMed]
- Ahmadieh, H.; Salti, I. Tyrosine kinase inhibitors induced thyroid dysfunction: A review of its incidence, pathophysiology, clinical relevance, and treatment. BioMed Res. Int. 2013, 2013, 725410. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Chinda, J.; Kuroshima, T.; Kabara, M.; Nakagawa, N.; Fujino, T.; Yamamoto, Y.; Ohsaki, Y.; Ogawa, Y.; Hasebe, N. Minimal change nephrotic syndrome associated with gefitinib and a successful switch to erlotinib. Intern. Med. 2015, 54, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Winn, S.K.; Ellis, S.; Savage, P.; Sampson, S.; Marsh, J.E. Biopsy-proven acute interstitial nephritis associated with the tyrosine kinase inhibitor sunitinib: A class effect? Nephrol. Dial. Transpl. 2009, 24, 673–675. [Google Scholar] [CrossRef] [PubMed]
- Masutani, K.; Fujisaki, K.; Maeda, H.; Toyonaga, J.; Inoshima, I.; Takayama, K.; Katafuchi, R.; Hirakata, H.; Tsuruya, K.; Iida, M. Tubulointerstitial nephritis and IgA nephropathy in a patient with advanced lung cancer treated with long-term gefitinib. Clin. Exp. Nephrol. 2008, 12, 398–402. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| RTK Superfamily | Members | Ligands |
|---|---|---|
| EGFR | EGFR (Erb1/Her1), Erb2/Her2, Erb3/Her3, Erb4/Her4 | TGF-α, EGF, HB-EGF, amphiregulin, betacellulin, epiregualtin, epigen |
| PDGFR | PDGFR-αα, PDGFR-αβ, PDGFR-ββ, M-CSFR, SCFR, FLT3L | PDGF-A, -B, -C, -D |
| VEGFR | VEGFR1 (Flt-1), VEGFR2 (KDR), VEGFR3 (Flt-4) | VEGF-A, -B, -C, -D, -E |
| FGFR | FGFR1, FGFR2, FGFR3, FGFR4 | FGF1, FGF2 |
| IGFR | Type I IGFR | IGF-1, IGF-2 |
| DDR | DDR1, DDR2 | collagen I–VI and VIII |
| Axl | Axl, MER, TYRO3 | Gas6, protein S |
| STYK | STYK1 | STYK |
| CCK | CCK4 | CCK, gastrin |
| NGFR | TRKA, TRKB, TRKC | NGF, BDNF, NT3, NT4 |
| HGFR | MET, RON | HGF, MSP |
| EPHR | EphA1-8, EphA10, EphB1-4, EphB6 | ephrin-A1-5, ephrin-B1-3 |
| TIE | TIE1, TIE2 | angiopoietin-1-4 |
| RYK | RYK | RYK |
| RET | RET | GDNF |
| ROS | ROS | orphan |
| LTK | LTK, ALK | Orphan/Pleiotrophin |
| ROR | ROR1, ROR2 | Wnt5A |
| MUSK | MUSK | Agrin, Dok-7 |
| LMR | LMR1, 2, 3 | (vestigial ECD) |
| Animal Models | Intervention | Targets | Effects | Reference |
|---|---|---|---|---|
| Murine unilateral ureteral obstruction | Suramin | PDGFR, EGFR | Attenuates glomerular and vascular injury and reduces inflammatory responses | [22] |
| Rat chronic anti-Thy 1.1 glomerulonephritis | B-specific oligonucleotideaptamer | PDGFR | Decreases proteinuria and improves renal function; Inhibits glomerulosclerosis and tubulointerstitial fibrosis | [23] |
| Rat chronic anti-Thy 1.1 glomerulonephritis | Neutralizing anti-PDGF-D IgG | PDGFR | Decreases proteinuria and improves renal function; Inhibits glomerulosclerosis and tubulointerstitial fibrosis and EMT | [24] |
| Rat acute anti–Thy 1.1 glomerulonephritis | Trapidil | PDGFR | Inhibits mesangial cells proliferation and matrix accumulation | [25] |
| Rat ischemia/reperfusion injury | Trapidil | PDGFR | Increases serum creatinine and mortality rate; Inhibits proliferation of tubular epithelial cells | [26] |
| Rat acute anti-Thy 1.1 GN | Imatinib | PDGFR | Inhibits mesangial cells proliferation and matrix accumulation | [16,122,123] |
| Murine streptozotocin-induced diabetes | Imatinib | PDGFR | Decreases albuminuria, glomerular and tubulointerstitial damage | [16] |
| Murine lupus | Imatinib | PDGFR | Improves survival, decreases proteinuria, glomerular and tubulointerstitial damage | [16] |
| Rat unilateral ureter obstruction | AG 1295 | PDGFR | Inhibits tubulointerstitial fibrosis | [27] |
| Rat crescentic glomerulonephritis | sFlt-1 | VEGFR | Accelerates proteinuria with massive ascites, glomerulosclerosis, and interstitial fibrosis | [51] |
| Rat model of 5/6 nephrectomy | Suramin | PDGFR,EGFR | Inhibits glomerulosclerosis and vascular sclerosis, as well as inflammation | [82] |
| Murine model of unilateral I/R model | Suramin | PDGFR,EGFR | Decreases tubular cell apoptosis, dedifferentiation and proliferation | [83] |
| Rat model of hyperuricemic nephropathy | Gefitinib | EGFR | Prevented renal dysfunction, reduced urine microalbumin, and inhibited activation of renal interstitial fibroblasts and expression of extracellular proteins | [127] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, F.; Zhuang, S. Role of Receptor Tyrosine Kinase Signaling in Renal Fibrosis. Int. J. Mol. Sci. 2016, 17, 972. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17060972
Liu F, Zhuang S. Role of Receptor Tyrosine Kinase Signaling in Renal Fibrosis. International Journal of Molecular Sciences. 2016; 17(6):972. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17060972
Chicago/Turabian StyleLiu, Feng, and Shougang Zhuang. 2016. "Role of Receptor Tyrosine Kinase Signaling in Renal Fibrosis" International Journal of Molecular Sciences 17, no. 6: 972. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17060972