
Fourier Transform Mass Spectrometry and Nuclear Magnetic Resonance Analysis for the Rapid and Accurate Characterization of Hexacosanoylceramide

Abstract
:
1. Introduction
2. Results and Discussion
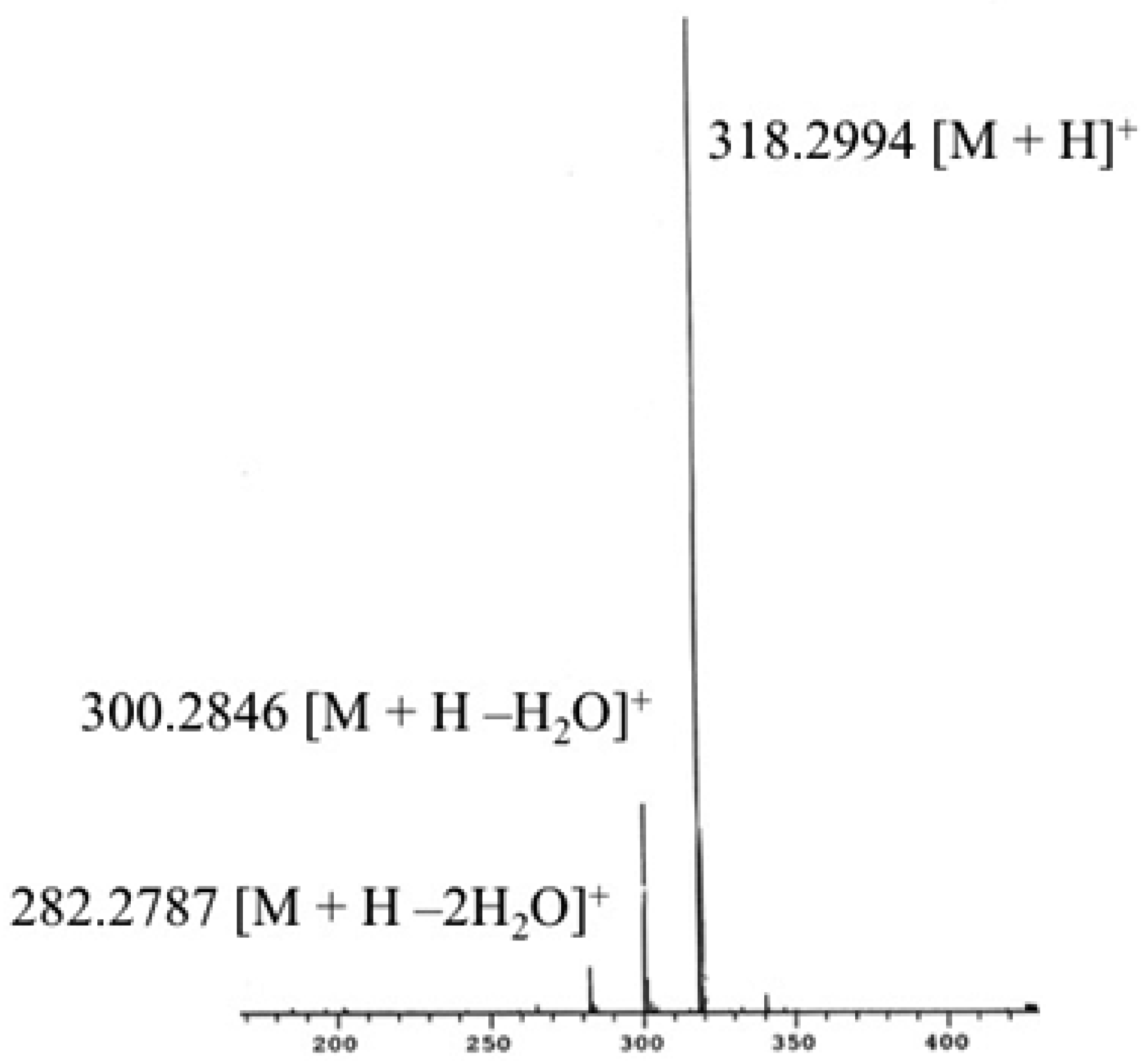
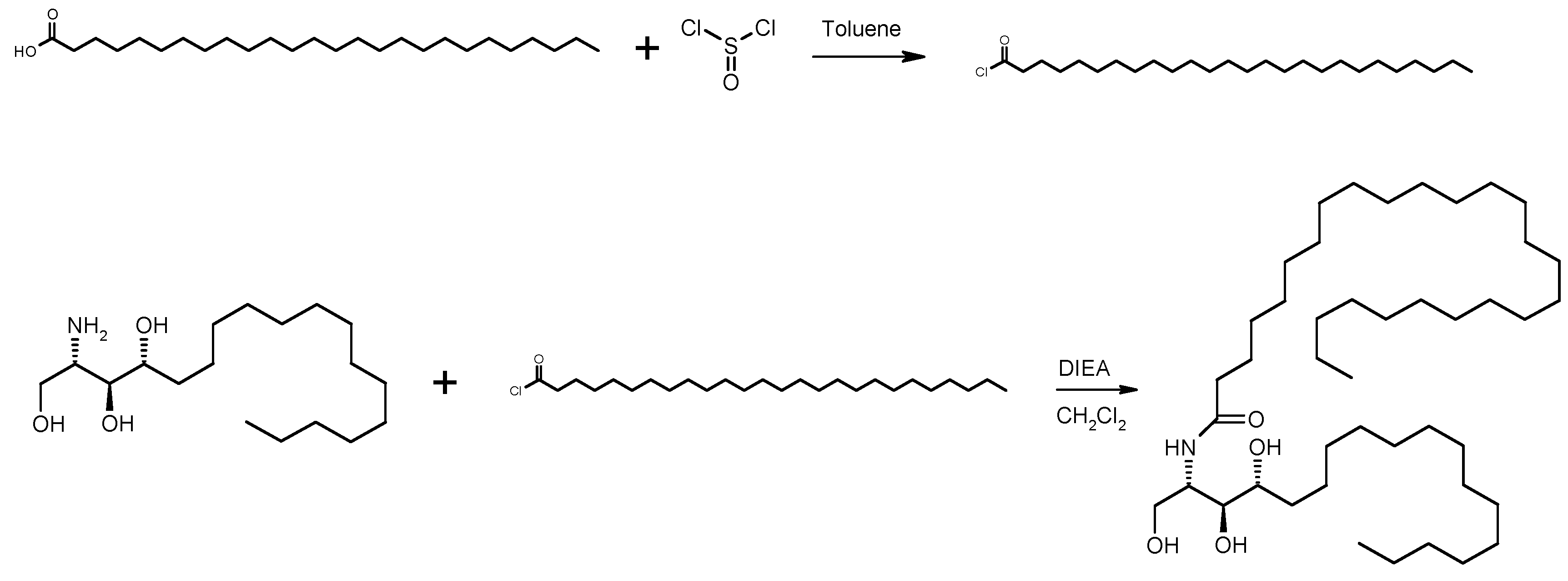
2.1. Analysis of Starting Materials
2.2. Analysis of Reaction Products
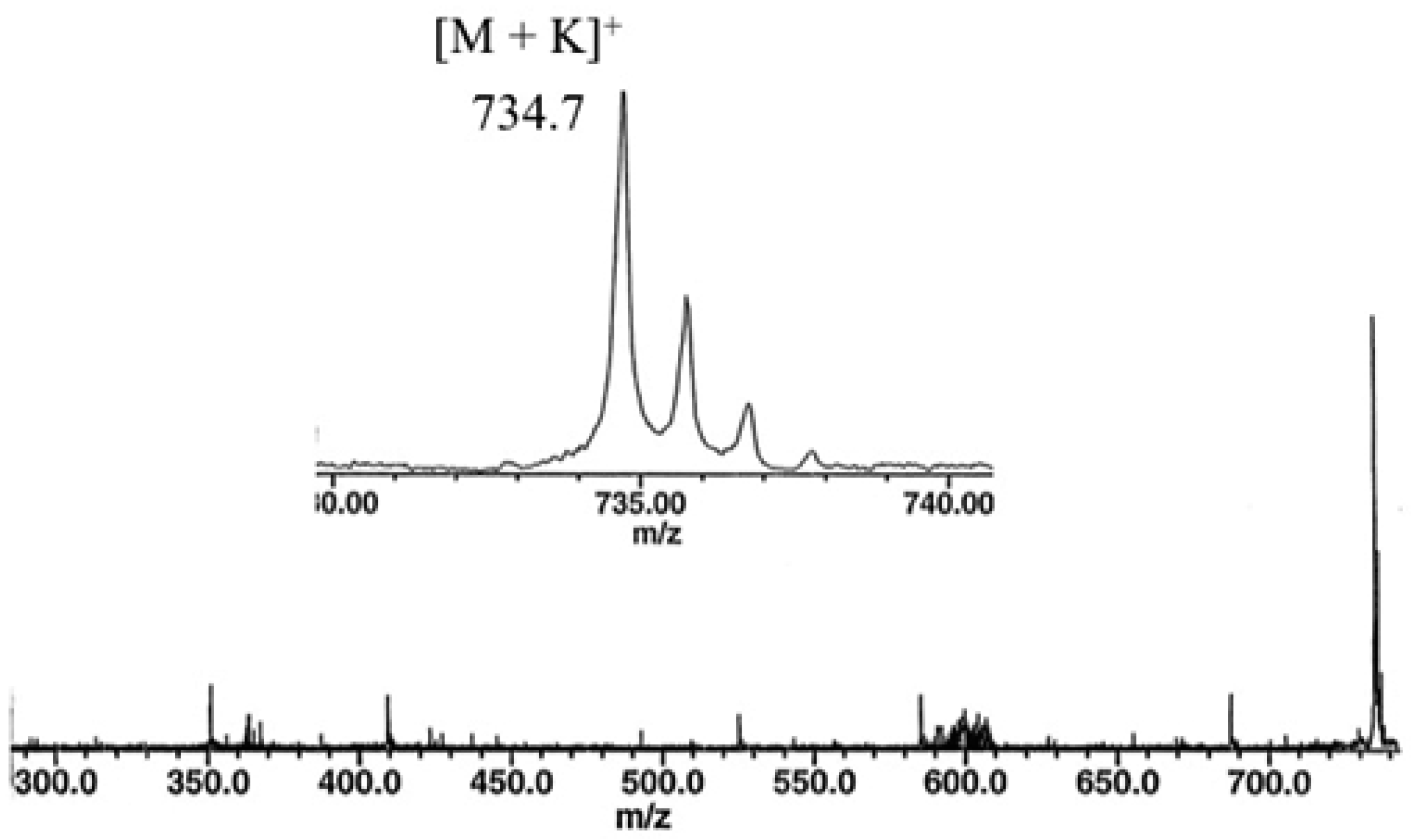
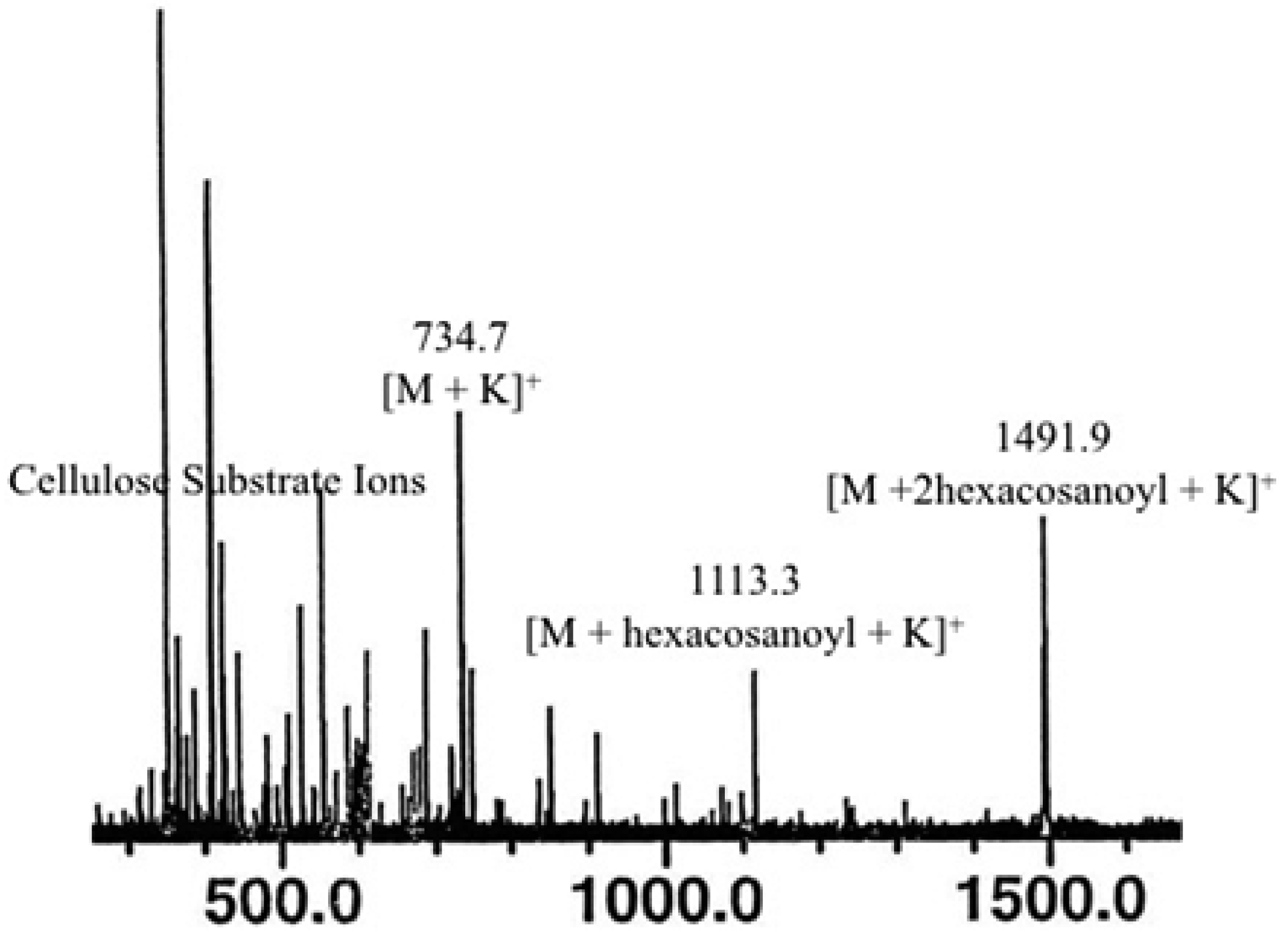
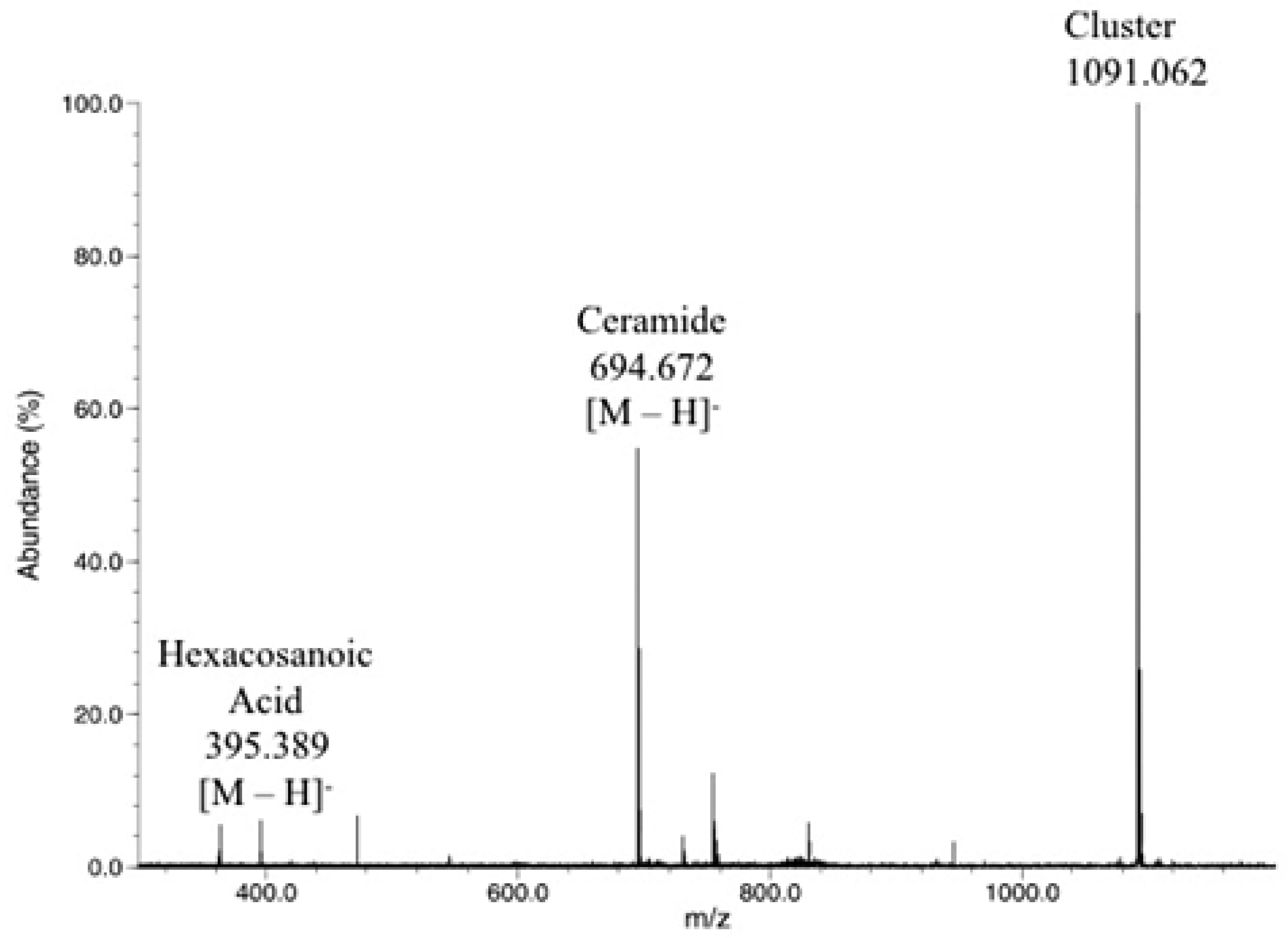
2.3. Direct Laser Desorption Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry (FTICR MS) Analysis
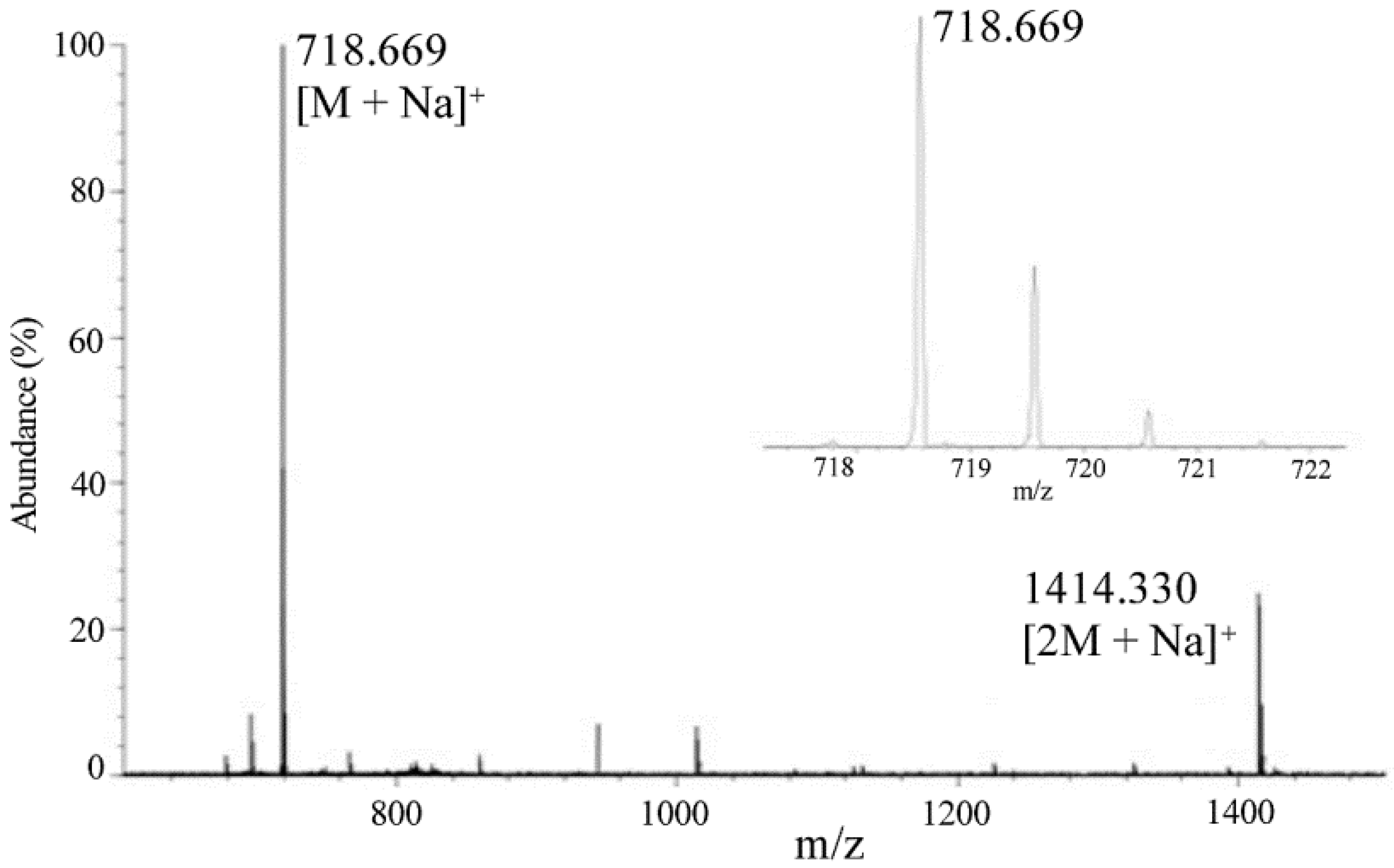
2.4. Sheath Flow Electrospray Ionization
2.5. Final Analyses
3. Materials and Methods
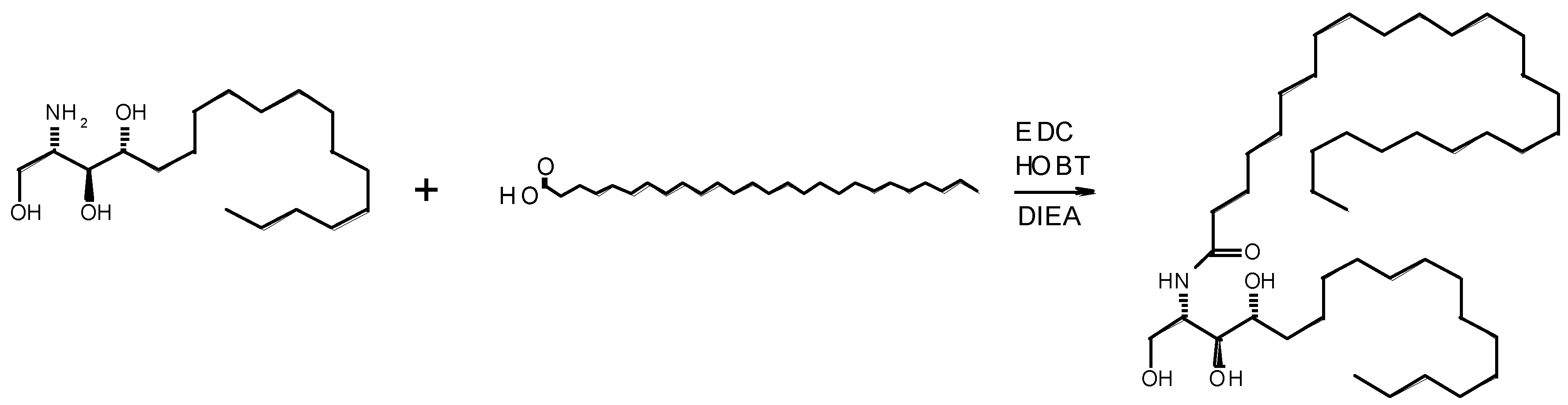
3.1. Synthesis of N-[(1S,2S,3R)-2,3-Dihydroxy-1-(hydroxymethyl)heptadecyl] Hexacosanamide
3.1.1. Samples via EDC/HOBT Coupling
Starting Materials and Products
- Phytosphingosine (C18H39NO3, 317.293 Da)
- Hexacosanoic acid (C26H52O2, 396.396 Da)
- Hexacosanoylceramide (C44H89NO4, 695.679 Da)
3.1.2. Via the Acid Chloride
3.2. Instrumentation
3.2.1. 7T Bruker BioApex II™ FTICR MS
3.2.2. Finnigan FTICR MS NewStar
3.2.3. Nuclear Magnetic Resonance (NMR)
3.3. MS Ionization Methods
3.3.1. Standard Monospray Electrospray Ionization
3.3.2. Direct Laser Desorption Ionization (DLDI)
3.3.3. Sheath Flow Electrospray Ionization
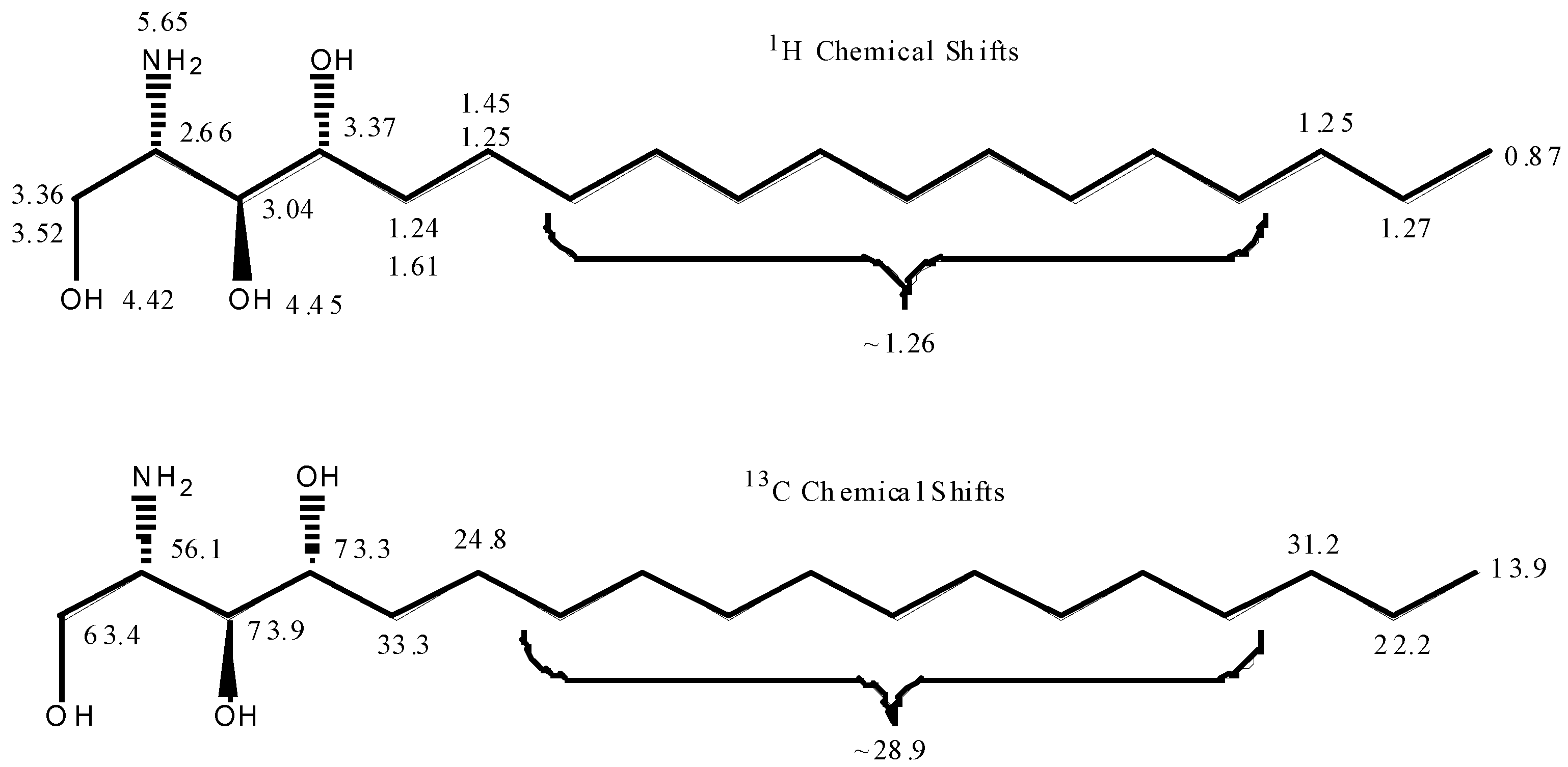
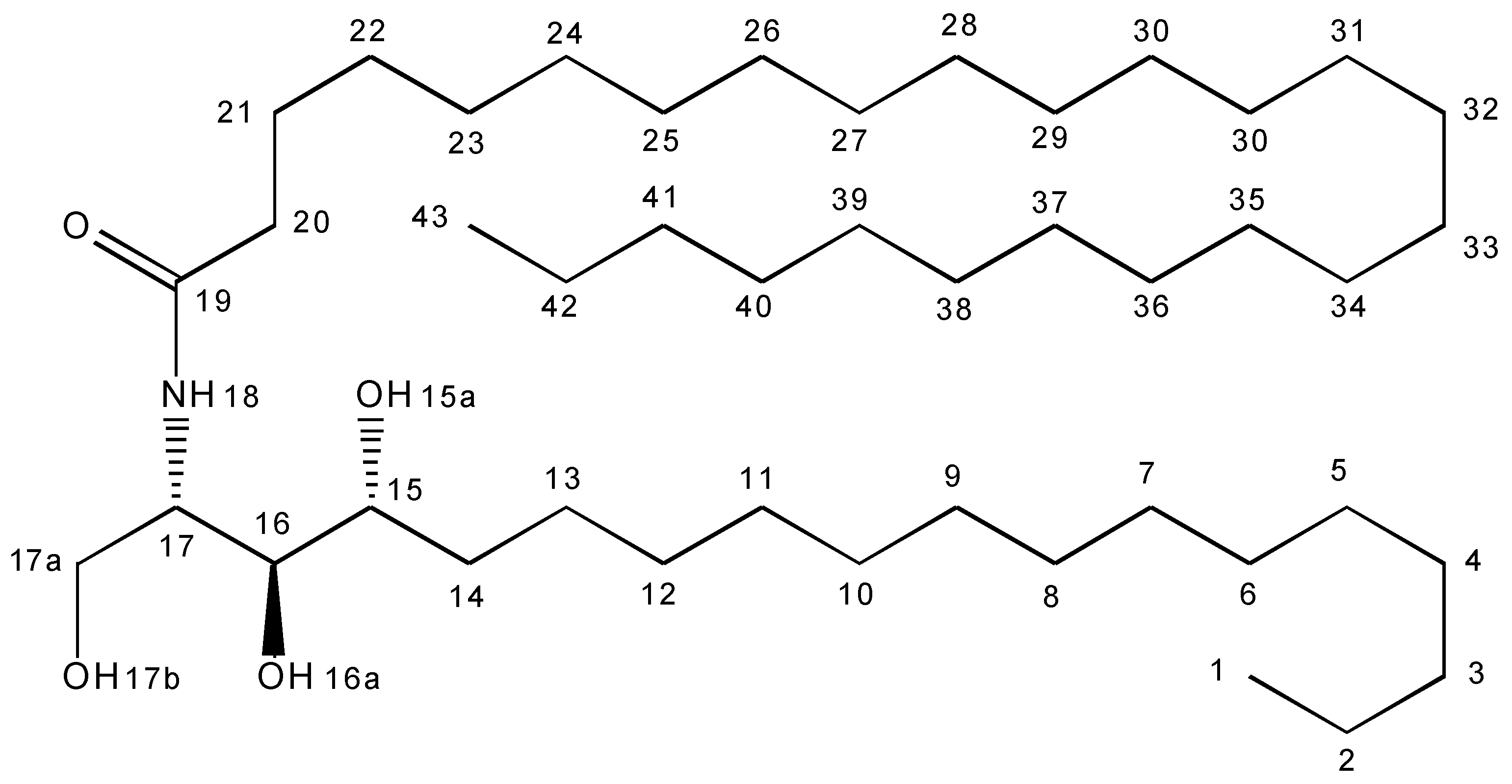
3.4. NMR Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DLDI | Direct laser desorption ionization |
| ESI | Electrospray ionization |
| FTICR MS | Fourier transform ion cyclotron resonance mass spectrometry |
| HPLC | High performance liquid chromatography |
| TLC | Thin layer chromatography |
| NMR | Nuclear magnetic resonance |
| EDC | N-[3-(dimethylamino)propyl]-N′-ethylcarbodiimide hydrochloride |
| HOBT | 1H–1,2,3-benzotriazol-1-ol hydrate |
| MALDI | Matrix-assisted laser desorption ionization |
| PFG-HMBC | Pulsed field gradient heteronuclear multiple bond correlation spectroscopy |
| HMQC | 2D heteronuclear multiple quantum coherence spectroscopy |
| HMBC | 2D heteronuclear multiple bond correlation spectroscopy |
| COSY | 2D homonuclear correlation spectroscopy |
| GARP | Globally optimized alternating phase rectangular pulse |
References
- Gamard, C.J.; Dbaibo, G.S.; Liu, B.; Obeid, L.; Hannun, Y. Selective involvement of ceramide in cytokine-induced apoptosis. Ceramide inhibits phorbol ester activation of nuclear factor κB. J. Biol. Chem. 1997, 272, 16474–16481. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Kondo, T.; Kitano, T.; Tashima, M. Diversity and complexity of ceramide signalling in apoptosis. Cell Signal. 1998, 10, 685–692. [Google Scholar] [CrossRef]
- Hannun, Y. Functions of ceramide in coordinating cellular responses to stress. Science 1996, 272, 1855–1858. [Google Scholar] [CrossRef]
- Obeid, L.M.; Hannun, Y. Ceramide: A stress signal and mediator of growth suppression and apoptosis. J. Cell. Biochem. 1995, 58, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Miller, H.C.; Esselman, W.S. Modulation of the immune response by antigen-reactive lymphocytes after cultivation with gangliosides. J. Immunol. 1975, 115, 839–843. [Google Scholar] [PubMed]
- Miller, H.C.; Chaney, W.G.; Klinhan, N.R.; Essalman, W.S. Regulation of B cell tolerance by murine gangliosides. Cell. Immunol. 1982, 67, 390–395. [Google Scholar] [CrossRef]
- Constantino, V.; Fattorusso, E.; Mangoni, A. Glycolipids from Sponges. IV. Immunomodulating glycosyl ceramide from the marine sponge. Tetrahedron 1996, 52, 1573–1578. [Google Scholar] [CrossRef]
- Motoki, K.; Kobayashi, E.; Uchida, T.; Fukushima, H.; Koezuka, Y. Radioprotective effects of α-galactosylceramides. Bioorg. Med. Chem. Lett. 1995, 5, 705–710. [Google Scholar] [CrossRef]
- Plettenburg, O.; Bodmer-Narkevitch, V.; Wong, C. Bacterial glycolipids and analogs as antigens for CD1d-restricted NKT cells. J. Org. Chem. 2002, 67, 4559–4564. [Google Scholar] [CrossRef] [PubMed]
- Cremesti, A.E.; Fischl, A.S. Current methods for the identification and quantitation of ceramides: An overview. Lipids 2000, 35, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Blair, I.A. Analysis of etheno 2′-deoxyguanosine adducts as dosimeters of AKR mediated-oxidative stress (Chapter 10) in Aldo-keto reductases and toxicant metabolism. ACS Symp. Ser. 2004, 865, 139–152. [Google Scholar]
- Castegnaro, M.; Garren, L.; Gaucher, I.; Wild, C.P. Development of a new method for the analysis of sphinganine and sphingosine in urine and tissues. Nat. Toxins 1996, 4, 284–290. [Google Scholar] [CrossRef]
- Lagana, A.; Marino, A.; Fago, G.; Miccheli, A. Determination of free sphingosine in biological systems by HPLC. Ann. Chim. 1991, 81, 721–734. [Google Scholar]
- Ribar, S.; Mesaric, M.; Bauman, M. High-performance liquid chromatographic determination of sphinganine and sphingosine in serum and urine of subjects from an endemic nephropathy area in Croatia. J. Chromatogr. B Biomed. Sci. Appl. 2001, 754, 511–519. [Google Scholar] [CrossRef]
- Shephard, G.S.; van der Westhuizen, L. Liquid chromatographic determination of the sphinganine/sphingosine ratio in serum. J. Chromatogr. B Biomed. Sci. Appl. 1998, 710, 219–222. [Google Scholar] [CrossRef]
- Previati, M.; Bertolaso, L.; Tramarin, M.; Bertagnolo, V.; Capitani, S. Low nanogram range quantitation of diglycerides and ceramide by high performance liquid chromatography. Anal. Biochem. 1996, 233, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Gaudin, K.; Germain, D.P.; Baillet, A.; Prognon, P.; Chaminade, P. Optimisation of the separation of four major neutral glycosphingolipids: Application to a rapid and simple detection of urinary globotriaosylceramide in Fabry disease. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2004, 805, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Markello, T.C.; Guo, J.; Gahl, W.A. High-performance liquid chromatography of lipids for the identification of human metabolic disease. Anal. Biochem. 1991, 198, 368–374. [Google Scholar] [CrossRef]
- McNabb, T.J.; Cremesti, A.E.; Brown, P.R.; Fischl, A.S. The separation and direct detection of ceramides and sphingoid bases by normal-phase high-performance liquid chromatography and evaporative light-scattering detection. Anal. Biochem. 1999, 276, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Olsson, N.U.; Harding, A.J.; Harper, C.; Salem, N., Jr. High-performance liquid chromatography method with light-scattering detection for measurements of lipid class composition: Analysis of brains from alcoholics. J. Chromatogr. B Biomed. Appl. 1996, 681, 213–218. [Google Scholar] [CrossRef]
- Rubino, F.M.; Zecca, L.; Sonnino, S. Characterization of a complex mixture of ceramides by fast atom bombardment and precursor and fragment analysis tandem mass spectrometry. Biol. Mass Spectrom. 1994, 23, 82–90. [Google Scholar] [CrossRef]
- Rubino, F.M.; Sonnino, S. Characterization of ceramide mixtures by fast atom bombardment and tandem mass spectrometry. NATO ASI Ser. C Math. Phys. Sci. 1996, 475, 417–428. [Google Scholar]
- Ivanova, P.T.; Cerda, B.A.; Horn, D.M.; Cohen, J.S.; McLafferty, F.W.; Brown, H.A. Electrospray ionization mass spectrometry analysis of changes in phospholipids in RBL-2H3 mastocytoma cells during degranulation. Proc. Natl. Acad. Sci. USA 2001, 98, 7152–7157. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, R.P.; Blumer, E.N.; Emmett, M.R.; Marshall, A.G. Complete compositional monitoring of the weathering of transportation fuels based on elemental compositions from Fourier transform ion cyclotron resonance mass spectrometry. Environ. Sci. Technol. 2000, 34, 1671–1678. [Google Scholar] [CrossRef]
- Fridriksson, E.K.; Shipkova, P.A.; Sheets, E.D.; Holowka, D.; Baird, B.; McLafferty, F.W. Quantitative analysis of phospholipids in functionally important membrane domains from RBL-2H3 mast cells using tandem high-resolution mass spectrometry. Biochemistry 1999, 38, 8056–8063. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.G.; Rodgers, R.P.; Blumer, E.N.; Emmett, M.R. Efficacy of Bacterial bioremediation: demonstration of complete incorporation of hydrocarbons into membrane phospholipids from rhodococcus hydrocarbon degrading bacteria by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Environ. Sci. Technol. 2000, 34, 535–540. [Google Scholar]
- Byrdwell, W.C. Dual parallel mass spectrometers for analysis of sphingolipid, glycerophospholipid and plasmalogen molecular species. Rapid Commun. Mass Spectrom. 1998, 12, 256–272. [Google Scholar] [CrossRef]
- Sandhoff, R.; Hepbildikler, S.T.; Jennemann, R.; Geyer, R.; Gieselmann, V.; Proia, R.L.; Wiegandt, H.; Grone, H.-J. Kidney sulfatides in mouse models of inherited glycosphingolipid disorders: Determination by nano-electrospray ionization tandem mass spectrometry. J. Biol. Chem. 2002, 277, 20386–20398. [Google Scholar] [CrossRef] [PubMed]
- Levery, S.B.; Toledo, M.S.; Doong, R.L.; Straus, A.H.; Takahashi, H.K. Comparative analysis of ceramide structural modification found in fungal cerebrosides by electrospray tandem mass spectrometry with low energy collision-induced dissociation of Li+ adduct ions. Rapid Commun. Mass Spectrom. 2000, 14, 551–563. [Google Scholar] [CrossRef]
- Bielawski, J.; Pierce, J.S.; Snider, J.; Rembiesa, B.; Szulc, Z.M.; Bielawski, A. Sphingolipid Analysis by High Performance Liquid Chromatography-Tandem Mass Spectrometry (HPLC-MS/MS). In Sphingolipids as Signaling and Regulatory Molecules; Landes Bioscience and Spring Science: New York, NY, USA, 2010; pp. 46–59. [Google Scholar]
- Selvam, R.; Radin, N.S. Quantitation of lipids by charring on thin-layer plates and scintillation quenching: Application to ceramide determination. Anal. Biochem. 1981, 112, 338–345. [Google Scholar] [CrossRef]
- Pettus, B.J.; Kroesen, B.-J.; Szulc, Z.M.; Bielawska, A.; Bielawski, J.; Hannun, Y.A.; Busman, M. Quantitative measurement of different ceramide species from crude cellular extracts by normal-phase high-performance liquid chromatography coupled to atmospheric pressure ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2004, 18, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Ramjit, H.G.; Newton, R.; Guare, J.P. A novel coaxial electrospray ionization method for characterizing hexacosanoylceramides by Fourier transform ion cyclotron resonance mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 1257–1262. [Google Scholar] [CrossRef] [PubMed]
- Aaserud, D.J.; Prokai, L.; Simonsick, W.J., Jr. Gel permeation chromatography coupled to Fourier transform mass spectrometry for polymer characterization. Anal. Chem. 1999, 71, 4793–4799. [Google Scholar] [CrossRef] [PubMed]
- Simonsick, W.J.; Ross, C.W., III. The characterization of novel dispersants, fluorinated surfactants, and modified natural oils by laser desorption Fourier transform ion cyclotron resonance mass spectrometry (LD-FTICR-MS). Int. J. Mass Spectrom. Ion Process. 1996, 158, 379–390. [Google Scholar] [CrossRef]
- Aaserud, D.J.; Simonsick, W.J. Modern mass spectrometry for coatings. Prog. Org. Coat. 1998, 34, 206–213. [Google Scholar] [CrossRef]
- Marto, J.A.; White, F.M.; Seldomridge, S.; Marshall, A.G. Structural characterization of phospholipids by matrix-assisted laser desorption/ionization Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 1995, 67, 3979–3984. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Wahl, M.C.; Wood, T.D.; Marshall, A.G. Enhanced mass resolving power, sensitivity, and selectivity in laser desorption Fourier transform ion cyclotron resonance mass spectrometry by ion axialization and cooling. Anal. Chem. 1993, 65, 1753–1757. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.D.; Ross, C.W., III; Marshall, A.G. Selective parent ion axialization for improved efficiency of collision-induced dissociation in laser desorption/ionization Fourier Transform ion cyclotron resonance mass spectrometry/mass spectrometry. J. Am. Soc. Mass Spectrom. 1994, 5, 900–907. [Google Scholar] [CrossRef]
- Corless, S.; Tetler, L.W.; Parr, V.; Wood, D. The usefulness of MALDI and electrospray in the characterisation of synthetic polymers. In Proceedings of the 42nd Annual Conference on Mass Spectrometry and Allied Topics, Chicago, IL, USA, 29 May–3 June 1994; American Society for Mass Spectrometry: Santa Fe, NM, USA, 1994; p. 515. [Google Scholar]
- Nielen, M.W.F. Characterization of synthetic polymers by size-exclusion chromatography/electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 1996, 10, 1652–1660. [Google Scholar] [CrossRef]
- Simonsick, W.J.; Prokai, L. Gel permeation chromatography coupled to mass spectrometry and tandem mass spectrometry for oligomer analysis. Adv. Chem. Ser. 1995, 247, 41–56. [Google Scholar]
- Shaka, A.J.; Barker, P.B.; Freeman, R. Computer-optimized decoupling scheme for wideband applications and low-level operation. J. Magn. Reson. 1985, 64, 547–552. [Google Scholar] [CrossRef]
- Hurd, R.E. Gradient-enhanced spectroscopy. J. Magn. Reson. 1990, 87, 422–428. [Google Scholar] [CrossRef]
- Bax, A.; Subramanian, S. Sensitivity-enhanced two-dimensional heteronuclear shift correlation NMR spectroscopy. J. Magn. Reson. 1986, 67, 565–569. [Google Scholar] [CrossRef]
- Bax, A.; Summers, M.F. Proton and carbon-13 assignments from sensitivity-enhanced detection of heteronuclear multiple-bond connectivity by 2D multiple quantum NMR. J. Am. Chem. Soc. 1986, 108, 2093–2094. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 13C (ppm) | 1H (ppm) |
|---|---|---|
| 1 | 14.5 | 0.89 |
| 2 | 23.7 | 1.32 |
| 3 | 32.9 | 1.3 |
| 4–12 | ~30.7 | ~1.31 |
| 13 | 26.7 | 1.32, 1.53 |
| 14 | 34.3 | 1.33, 1.71 |
| 15 | 73.2 | 3.44 |
| 15a | 3.77 | |
| 16 | 77.2 | 3.41 |
| 16a | 4.21 | |
| 17 | 53.9 | 4.01 |
| 17a | 62.5 | 3.61, 3.71 |
| 17b | 4.12 | |
| 18 | 6.93 | |
| 19 | 173.3 | |
| 20 | 36.7 | 2.12 |
| 21 | 26.6 | 1.59 |
| 22–40 | ~30.3 | ~1.31 |
| 41 | 32.9 | 1.3 |
| 42 | 23.7 | 1.32 |
| 43 | 14.5 | 0.89 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ross, C.W.; Simonsick, W.J.; Bogusky, M.J.; Celikay, R.W.; Guare, J.P.; Newton, R.C. Fourier Transform Mass Spectrometry and Nuclear Magnetic Resonance Analysis for the Rapid and Accurate Characterization of Hexacosanoylceramide. Int. J. Mol. Sci. 2016, 17, 1024. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17071024
Ross CW, Simonsick WJ, Bogusky MJ, Celikay RW, Guare JP, Newton RC. Fourier Transform Mass Spectrometry and Nuclear Magnetic Resonance Analysis for the Rapid and Accurate Characterization of Hexacosanoylceramide. International Journal of Molecular Sciences. 2016; 17(7):1024. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17071024
Chicago/Turabian StyleRoss, Charles W., William J. Simonsick, Michael J. Bogusky, Recep W. Celikay, James P. Guare, and Randall C. Newton. 2016. "Fourier Transform Mass Spectrometry and Nuclear Magnetic Resonance Analysis for the Rapid and Accurate Characterization of Hexacosanoylceramide" International Journal of Molecular Sciences 17, no. 7: 1024. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17071024