
Exploring the Molecular Basis for Binding of Inhibitors by Threonyl-tRNA Synthetase from Brucella abortus: A Virtual Screening Study

 , ,
, ,
Abstract
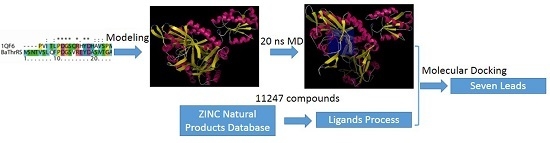
:
1. Introduction
2. Results and Discussion
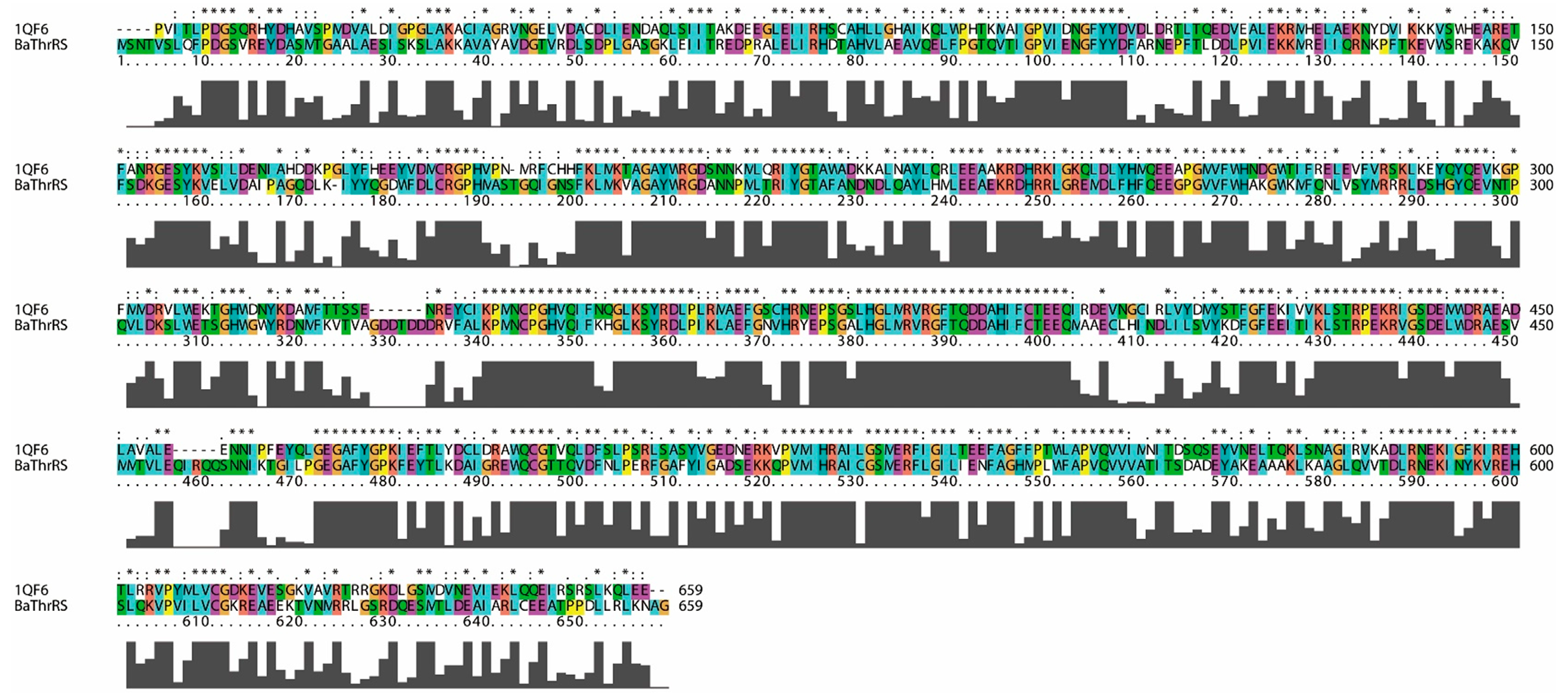
2.1. Sequence Alignments and Molecular Modeling
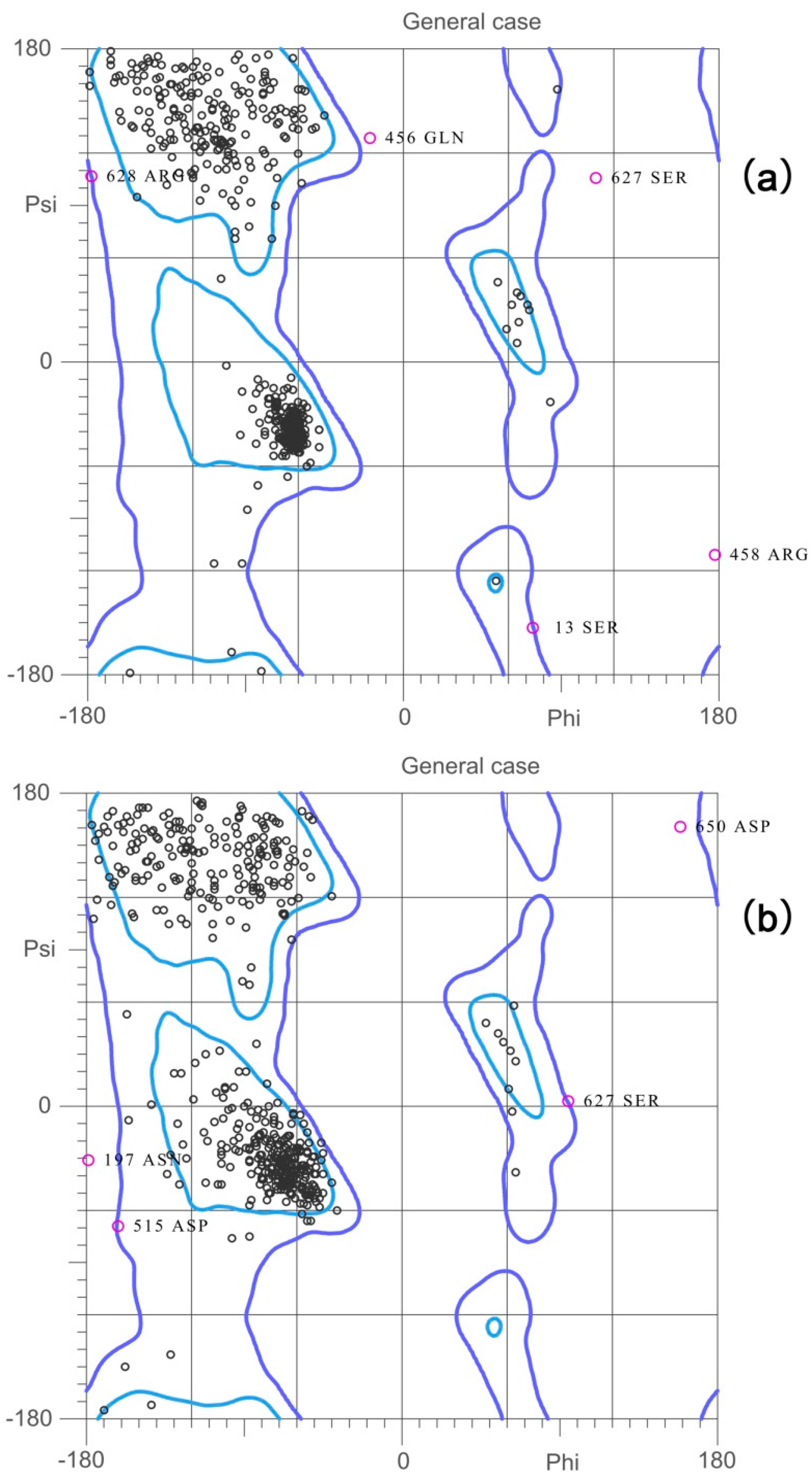
2.2. Validation of the Homology Model
2.3. Identification of Substrate-Binding Region in BaThrRS
2.4. Docking Study
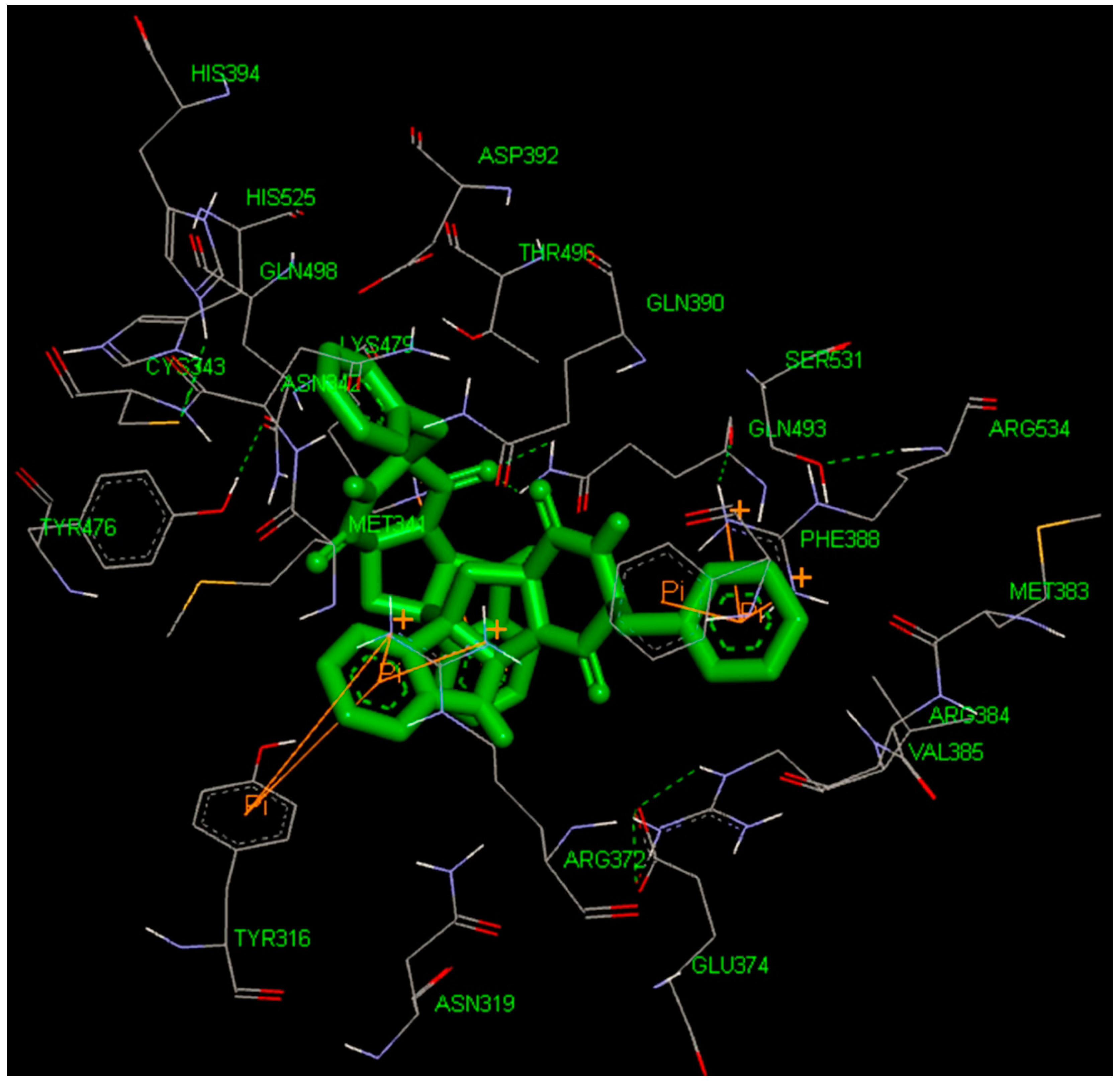
2.5. High-Throughput Virtual Screening Procedure and Docking of the Best Inhibitor to BaThrRS
3. Materials and Methods
3.1. Molecular Modeling
3.2. Assessment of the Homology Model
3.3. Binding Pocket Analyses
3.4. Validation of the Model by Docking Analysis
3.5. Virtual Screening
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ThrRS | threonyl-tRNA synthetase |
| BaThrRS | Brucella abortus threonyl-tRNA synthetase |
| PDB | Protein Data Bank |
| ATP | adenosine triphosphate |
| tRNA | transfer RNA |
| aaRS | aminoacyl-tRNA synthetase |
| EThrRS | Escherichia coli threonyl-tRNA synthetase |
| NPD | Natural Products Database |
References
- Olsen, S.C.; Carlson, S.A. In vitro bactericidal activity of aminoglycosides, including the next-generation drug plazomicin, against Brucella spp. Int. J. Antimicrob. Agents 2015, 45, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Joseph, P.; Turtaut, F.; Ouahrani-Bettache, S.; Montero, J.L.; Nishimori, I.; Minakuchi, T.; Vullo, D.; Scozzafava, A.; Kohler, S.; Winum, J.Y.; et al. Cloning, characterization, and inhibition studies of a beta-carbonic anhydrase from Brucella suis. J. Med. Chem. 2010, 53, 2277–2285. [Google Scholar] [CrossRef] [PubMed]
- Wareth, G.; Eravci, M.; Weise, C.; Roesler, U.; Melzer, F.; Sprague, L.D.; Neubauer, H.; Murugaiyan, J. Comprehensive identification of immunodominant proteins of Brucella abortus and Brucella melitensis using antibodies in the sera from naturally infected hosts. Int. J. Mol. Sci. 2016, 17, 659. [Google Scholar] [CrossRef] [PubMed]
- Principi, N.; Esposito, S. Infectious discitis and spondylodiscitis in children. Int. J. Mol. Sci. 2016, 17, 539. [Google Scholar] [CrossRef] [PubMed]
- Ariza, J.; Bosilkovski, M.; Cascio, A.; Colmenero, J.D.; Corbel, M.J.; Falagas, M.E.; Memish, Z.A.; Roushan, M.R.; Rubinstein, E.; Sipsas, N.V.; et al. Perspectives for the treatment of brucellosis in the 21st century: The Ioannina recommendations. PLoS Med. 2007, 4, e317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnhill, A.E.; Brewer, M.T.; Carlson, S.A. Adverse effects of antimicrobials via predictable or idiosyncratic inhibition of host mitochondrial components. Antimicrob. Agents Chemother. 2012, 56, 4046–4051. [Google Scholar] [CrossRef] [PubMed]
- Van de Vijver, P.; Ostrowski, T.; Sproat, B.; Goebels, J.; Rutgeerts, O.; van Aerschot, A.; Waer, M.; Herdewijn, P. Aminoacyl-tRNA synthetase inhibitors as potent and synergistic immunosuppressants. J. Med. Chem. 2008, 51, 3020–3029. [Google Scholar] [CrossRef] [PubMed]
- Havrylenko, S.; Mirande, M. Aminoacyl-tRNA synthetase complexes in evolution. Int. J. Mol. Sci. 2015, 16, 6571–6594. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Macnamara, L.M.; Leuchter, J.D.; Alexander, R.W.; Cho, S.S. MD simulations of tRNA and aminoacyl-tRNA synthetases: Dynamics, folding, binding, and allostery. Int. J. Mol. Sci. 2015, 16, 15872–15902. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Nair, S.K. Aminoacyl tRNA synthetases as targets for antibiotic development. Med. Chem. Commun. 2012, 3, 887–898. [Google Scholar] [CrossRef]
- Zhao, Y.; Meng, Q.; Bai, L.; Zhou, H. In silico discovery of aminoacyl-tRNA synthetase inhibitors. Int. J. Mol. Sci. 2014, 15, 1358–1373. [Google Scholar] [CrossRef] [PubMed]
- Ravishankar, S.; Ambady, A.; Swetha, R.G.; Anbarasu, A.; Ramaiah, S.; Sambandamurthy, V.K. Essentiality assessment of cysteinyl and lysyl-tRNA synthetases of Mycobacterium smegmatis. PLoS ONE 2016, 11, e0147188. [Google Scholar] [CrossRef] [PubMed]
- Nagamitsu, T.; Takano, D.; Marumoto, K.; Fukuda, T.; Furuya, K.; Otoguro, K.; Takeda, K.; Kuwajima, I.; Harigaya, Y.; Omura, S. Total synthesis of borrelidin. J. Org. Chem. 2007, 72, 2744–2756. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.L.; Yin, Y.W.; Carter, C.W., Jr. Selective inhibition of bacterial tryptophanyl-tRNA synthetases by indolmycin is mechanism-based. J. Biol. Chem. 2016, 291, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.; Hilgers, M.T.; Cunningham, M.L.; Borchardt, A.; Locke, J.B.; Abraham, S.; Haley, G.; Kwan, B.P.; Hall, C.; Hough, G.W.; et al. Identification of bacteria-selective threonyl-tRNA synthetase substrate inhibitors by structure-based design. J. Med. Chem. 2013, 56, 1748–1760. [Google Scholar] [CrossRef] [PubMed]
- Pavadai, E.; El Mazouni, F.; Wittlin, S.; de Kock, C.; Phillips, M.A.; Chibale, K. Identification of new human malaria parasite Plasmodium falciparum dihydroorotate dehydrogenase inhibitors by pharmacophore and structure-based virtual screening. J. Chem. Inf. Model. 2016, 56, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Cele, F.N.; Ramesh, M.; Soliman, M.E. Per-residue energy decomposition pharmacophore model to enhance virtual screening in drug discovery: A study for identification of reverse transcriptase inhibitors as potential anti-HIV agents. Drug Des. Dev. Ther. 2016, 10, 1365–1377. [Google Scholar]
- Zhou, X.; Yu, S.; Su, J.; Sun, L. Computational study on new natural compound inhibitors of pyruvate dehydrogenase kinases. Int. J. Mol. Sci. 2016, 17, 340. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Y.; Liu, Q.; Ai, Z.; Xiang, Y.; Qiao, Y. Discovery of dual ETA/ETB receptor antagonists from traditional Chinese herbs through in silico and in vitro screening. Int. J. Mol. Sci. 2016, 17, 389. [Google Scholar] [CrossRef] [PubMed]
- Billones, J.B.; Carrillo, M.C.; Organo, V.G.; Macalino, S.J.; Sy, J.B.; Emnacen, I.A.; Clavio, N.A.; Concepcion, G.P. Toward antituberculosis drugs: In silico screening of synthetic compounds against Mycobacterium tuberculosis l,d-transpeptidase 2. Drug Des. Dev. Ther. 2016, 10, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Wójcikowski, M.; Zielenkiewicz, P.; Siedlecki, P. Open drug discovery toolkit (ODDT): A new open-source player in the drug discovery field. J. Cheminform. 2015, 7, 26. [Google Scholar] [CrossRef]
- Damm-Ganamet, K.L.; Bembenek, S.D.; Venable, J.W.; Castro, G.G.; Mangelschots, L.; Peeters, D.C.; McAllister, H.M.; Edwards, J.P.; Disepio, D.; Mirzadegan, T. A prospective virtual screening study: Enriching hit rates and designing focus libraries to find inhibitors of PI3Kδ and PI3Kγ. J. Med. Chem. 2016, 59, 4302–4313. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wong, C.F. Inexpensive method for selecting receptor structures for virtual screening. J. Chem. Inf. Model. 2016, 56, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Xu, Y.; Liu, X.; Wang, Y.; Peng, J.; Luo, X.; Zheng, M.; Chen, K.; Jiang, H. Combinatorial pharmacophore-based 3D-QSAR analysis and virtual screening of FGFR1 inhibitors. Int. J. Mol. Sci. 2015, 16, 13407–13426. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Chen, L.; Zhang, J.; Xie, X.; Qiu, K.; Fu, J. A combined pharmacophore modeling, 3D QSAR and virtual screening studies on imidazopyridines as B-Raf inhibitors. Int. J. Mol. Sci. 2015, 16, 12307–12323. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, L.; Chen, Y.; Hu, J.; Liu, J.; Liu, Y.C.; Liu, R.; Zhang, Y.; Meng, F.; Zhu, K.; et al. Identification of novel disruptor of telomeric silencing 1-like (DOT1L) inhibitors through structure-based virtual screening and biological assays. J. Chem. Inf. Model. 2016, 56, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, J.; Chen, Z.; Wang, F.; Huang, W.; Hong, Z.; Lin, J. Multistage virtual screening and identification of novel HIV-1 protease inhibitors by integrating SVM, shape, pharmacophore and docking methods. Eur. J. Med. Chem. 2015, 101, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Veeramachaneni, G.K.; Raj, K.K.; Chalasani, L.M.; Bondili, J.S.; Talluri, V.R. High-throughput virtual screening with e-pharmacophore and molecular simulations study in the designing of pancreatic lipase inhibitors. Drug Des. Dev. Ther. 2015, 9, 4397–4412. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Ang, E.; Dai, L.; Yang, X.; Ye, D.; Chen, H.; Zhou, L.; Yang, M.; Teguh, D.; Tan, R.; et al. Natural germacrane sesquiterpenes inhibit osteoclast formation, bone resorption, RANKL-induced NF-κB activation, and IκBα degradation. Int. J. Mol. Sci. 2015, 16, 26599–26607. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.H.; Cheng, T.C.; Leu, Y.L.; Chuang, K.H.; Tzou, S.C.; Chen, C.S. Discovery of Akt kinase inhibitors through structure-based virtual screening and their evaluation as potential anticancer agents. Int. J. Mol. Sci. 2015, 16, 3202–3212. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Mishra, R.K.; Schiltz, G.E.; Makanji, Y.; Scheidt, K.A.; Mazar, A.P.; Woodruff, T.K. Virtual high-throughput screening to identify novel activin antagonists. J. Med. Chem. 2015, 58, 5637–5648. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.S.; Lin, S.Y.; Liao, F.Y.; Hsiao, W.C.; Lee, L.C.; Peng, Y.H.; Hsieh, C.L.; Wu, M.H.; Song, J.S.; Yueh, A.; et al. Identification of substituted naphthotriazolediones as novel tryptophan 2,3-dioxygenase (TDO) inhibitors through structure-based virtual screening. J. Med. Chem. 2015, 58, 7807–7819. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Narayanapillai, S.; Zhang, W.; Sham, Y.Y.; Xing, C. Rapid identification of Keap1-Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. J. Med. Chem. 2014, 57, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Chai, H.H.; Lim, D.; Lee, S.H.; Chai, H.Y.; Jung, E. Homology modeling study of bovine µ-calpain inhibitor-binding domains. Int. J. Mol. Sci. 2014, 15, 7897–7938. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; He, G.; Jiang, Q.; Han, B.; Peng, C. Novel hybrid virtual screening protocol based on molecular docking and structure-based pharmacophore for discovery of methionyl-tRNA synthetase inhibitors as antibacterial agents. Int. J. Mol. Sci. 2013, 14, 14225–14239. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, X.; Li, S.; Wang, Y.; Zhou, N.; Luo, C.; Luo, X.; Zheng, M.; Jiang, H.; Chen, K. Identification of novel small molecules as inhibitors of hepatitis C virus by structure-based virtual screening. Int. J. Mol. Sci. 2013, 14, 22845–22856. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2014, 47, 5601–5632. [Google Scholar]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2014, 1137, 1–15. [Google Scholar] [PubMed]
- Sankaranarayanan, R.; Dock-Bregeon, A.C.; Romby, P.; Caillet, J.; Springer, M.; Rees, B.; Ehresmann, C.; Ehresmann, B.; Moras, D. The structure of threonyl-tRNA synthetase-tRNAThr complex enlightens its repressor activity and reveals an essential zinc ion in the active site. Cell 1999, 97, 371–381. [Google Scholar] [CrossRef]
- Baker, D.; Sali, A. Protein structure prediction and structural genomics. Science 2001, 294, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Luthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Bowie, J.; Luthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006, 34, W116–W118. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, J.; Liu, C.; Fang, B.; Wang, X.; Xiang, W. Identification of borrelidin binding site on threonyl-tRNA synthetase. Biochem. Biophys. Res. Commun. 2014, 451, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Zhan, D.; Wang, D.; Min, W.; Han, W. Exploring the molecular basis for selective binding of homoserine dehydrogenase from Mycobacterium leprae TN toward inhibitors: A virtual screening study. Int. J. Mol. Sci. 2014, 15, 1826–1841. [Google Scholar] [CrossRef] [PubMed]
- Minogue, T.D.; Daligault, H.A.; Davenport, K.W.; Bishop-Lilly, K.A.; Broomall, S.M.; Bruce, D.C.; Chain, P.S.; Chertkov, O.; Coyne, S.R.; Frey, K.G.; et al. Whole-genome sequences of 24 Brucella strains. Genome Announc. 2014, 2, e00915-14. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Morgulis, A.; Coulouris, G.; Raytselis, Y.; Madden, T.L.; Agarwala, R.; Schäffer, A.A. Database indexing for production MegaBLAST searches. Bioinformatics 2008, 24, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. Gromacs 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Validation | BaThrRS (0 ns) | BaThrRS (20 ns) | 1QF6 |
|---|---|---|---|
| Favored regions | 95.1% (623/655) | 92.2% (604/655) | 87.8% (561/639) |
| Allowed regions | 98.9% (648/655) | 99.2% (650/655) | 98.7% (631/639) |
| Ramachandran outliers | 1.07% (7/655) | 0.76% (5/655) | 1.25% (8/639) |
| Verify3D | 90.27% | 93.76% | 99.53% |
| ERRAT | 76.425 | 85.440 | 90.047 |
| 1QF6 | BaThrRS |
|---|---|
| Cys334 | Cys343 |
| Arg363 | Arg372 |
| Glu365 | Glu374 |
| Met374 | Met383 |
| Arg375 | Arg384 |
| Val376 | Val385 |
| Phe379 | Phe388 |
| Gln381 | Gln390 |
| His385 | His394 |
| Gln479 | Gln493 |
| Cys480 | Cys494 |
| Thr482 | Thr496 |
| His511 | His525 |
| Gly516 | Gly530 |
| Ser517 | Ser531 |
| Arg520 | Arg534 |
| ZINC ID | Score (kcal/mol) | Structure |
|---|---|---|
| ZINC27215482 | −12.6 |  |
| ZINC67910544 | −12.4 |  |
| ZINC42805205 | −12.4 |  |
| ZINC72320615 | −12.3 |  |
| ZINC72320626 | −12.2 |  |
| ZINC35270978 | −12.1 |  |
| ZINC35458951 | −12.1 |  |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Wen, F.; Zhao, S.; Wang, P.; Li, S.; Zhang, Y.; Zheng, N.; Wang, J. Exploring the Molecular Basis for Binding of Inhibitors by Threonyl-tRNA Synthetase from Brucella abortus: A Virtual Screening Study. Int. J. Mol. Sci. 2016, 17, 1078. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17071078
Li M, Wen F, Zhao S, Wang P, Li S, Zhang Y, Zheng N, Wang J. Exploring the Molecular Basis for Binding of Inhibitors by Threonyl-tRNA Synthetase from Brucella abortus: A Virtual Screening Study. International Journal of Molecular Sciences. 2016; 17(7):1078. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17071078
Chicago/Turabian StyleLi, Ming, Fang Wen, Shengguo Zhao, Pengpeng Wang, Songli Li, Yangdong Zhang, Nan Zheng, and Jiaqi Wang. 2016. "Exploring the Molecular Basis for Binding of Inhibitors by Threonyl-tRNA Synthetase from Brucella abortus: A Virtual Screening Study" International Journal of Molecular Sciences 17, no. 7: 1078. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17071078