
Neuroprotective Effects of Inhibiting Fyn S-Nitrosylation on Cerebral Ischemia/Reperfusion-Induced Damage to CA1 Hippocampal Neurons

Abstract
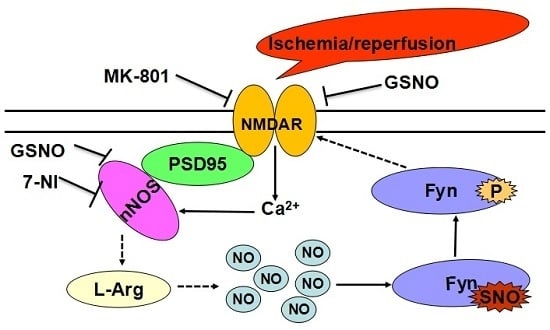
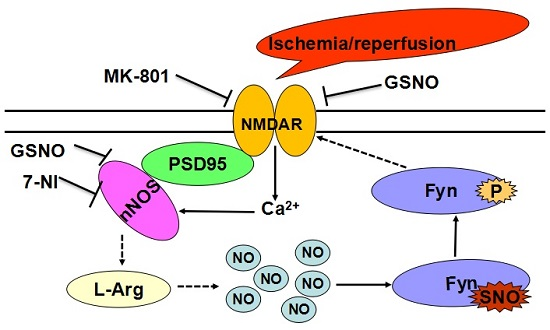
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
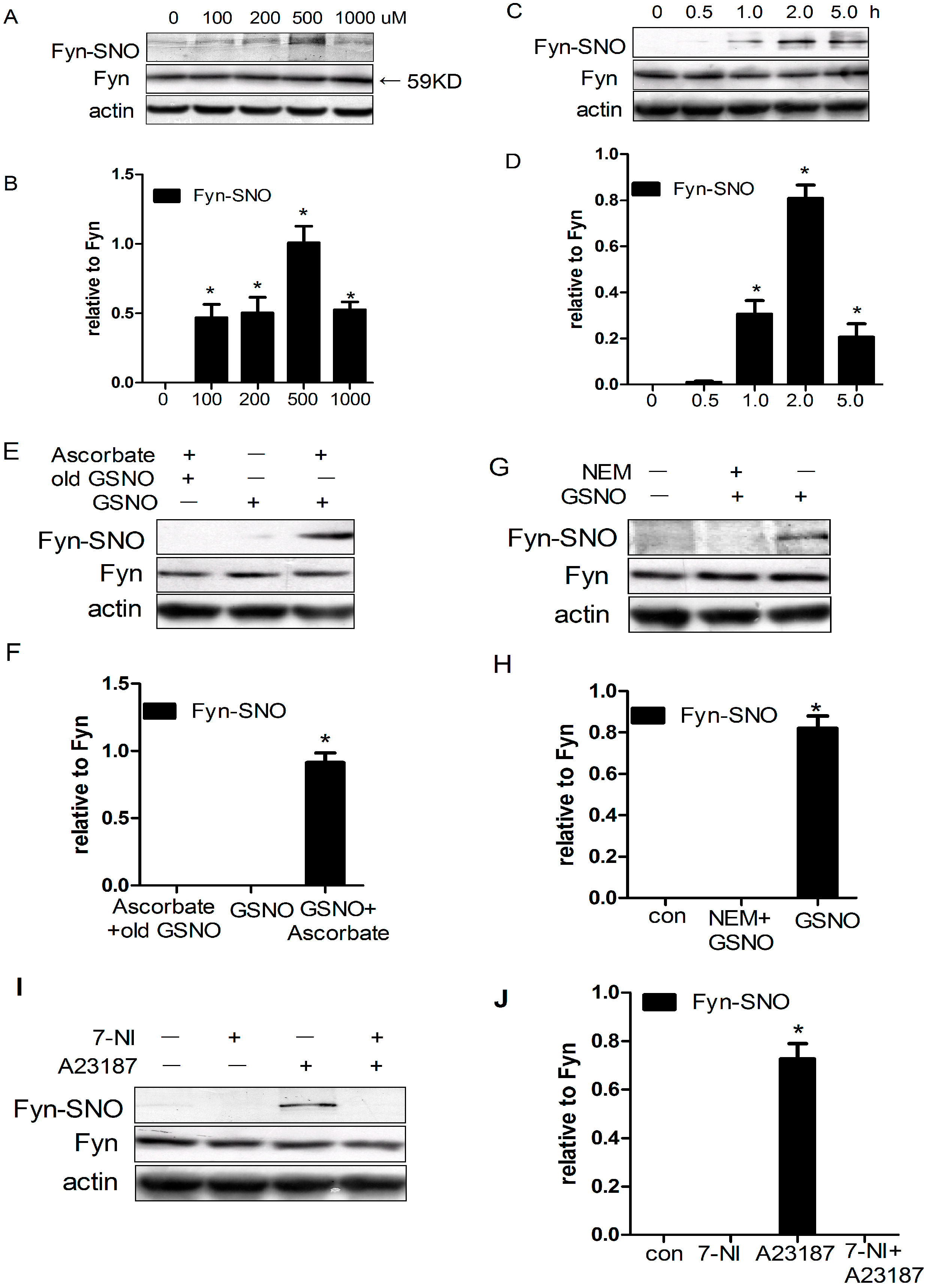
2.1. Fyn Was S-Nitrosylated by S-Nitrosoglutathione (GSNO) in HEK293 Cells
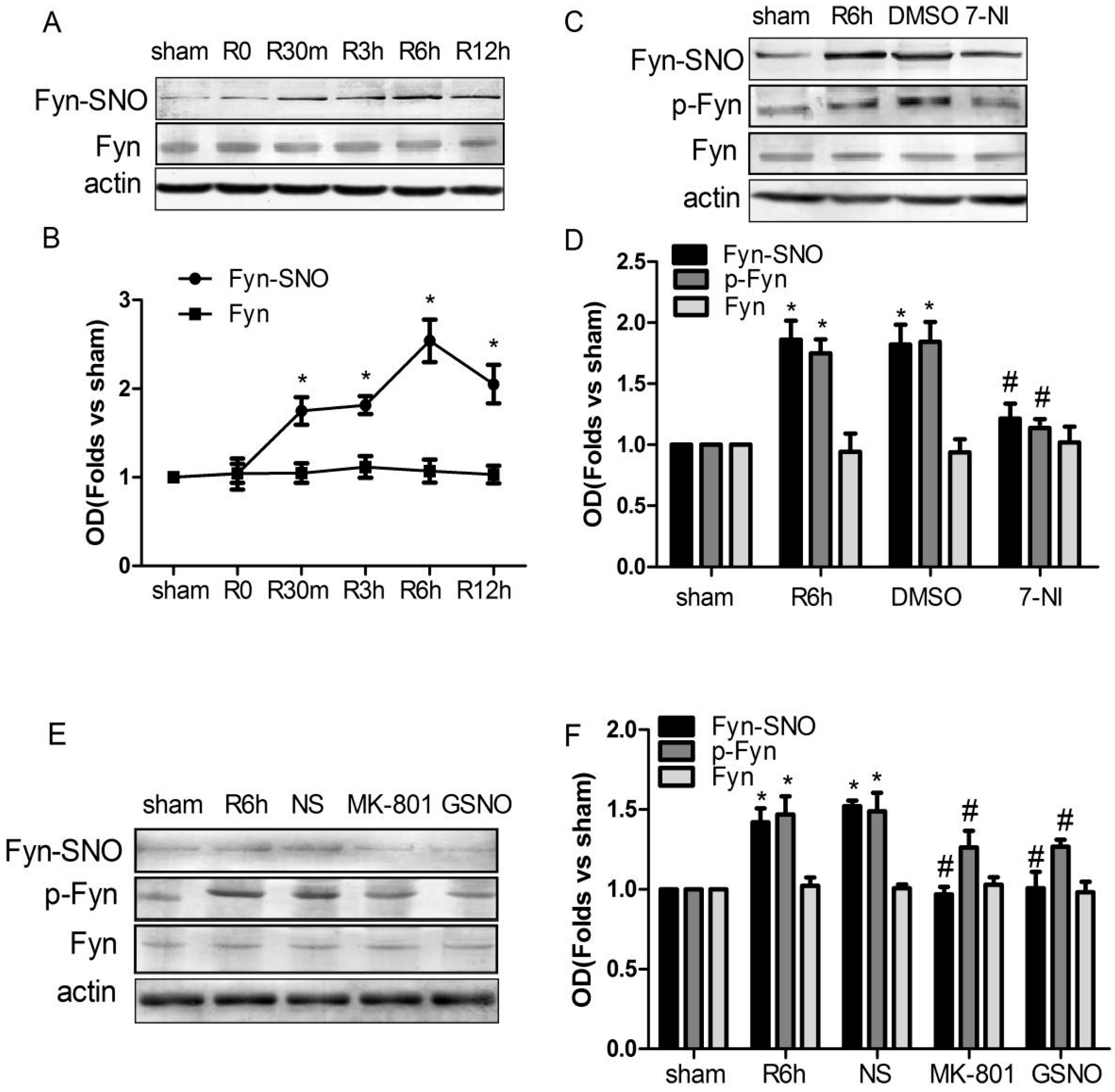
2.2. S-Nitrosylation and Phosphorylation of Fyn Are Mediated by Nitric Oxide Synthase (nNOS), with Potential Involvement of N-Methyl-d-aspartate Receptors (NMDARs) in Vivo
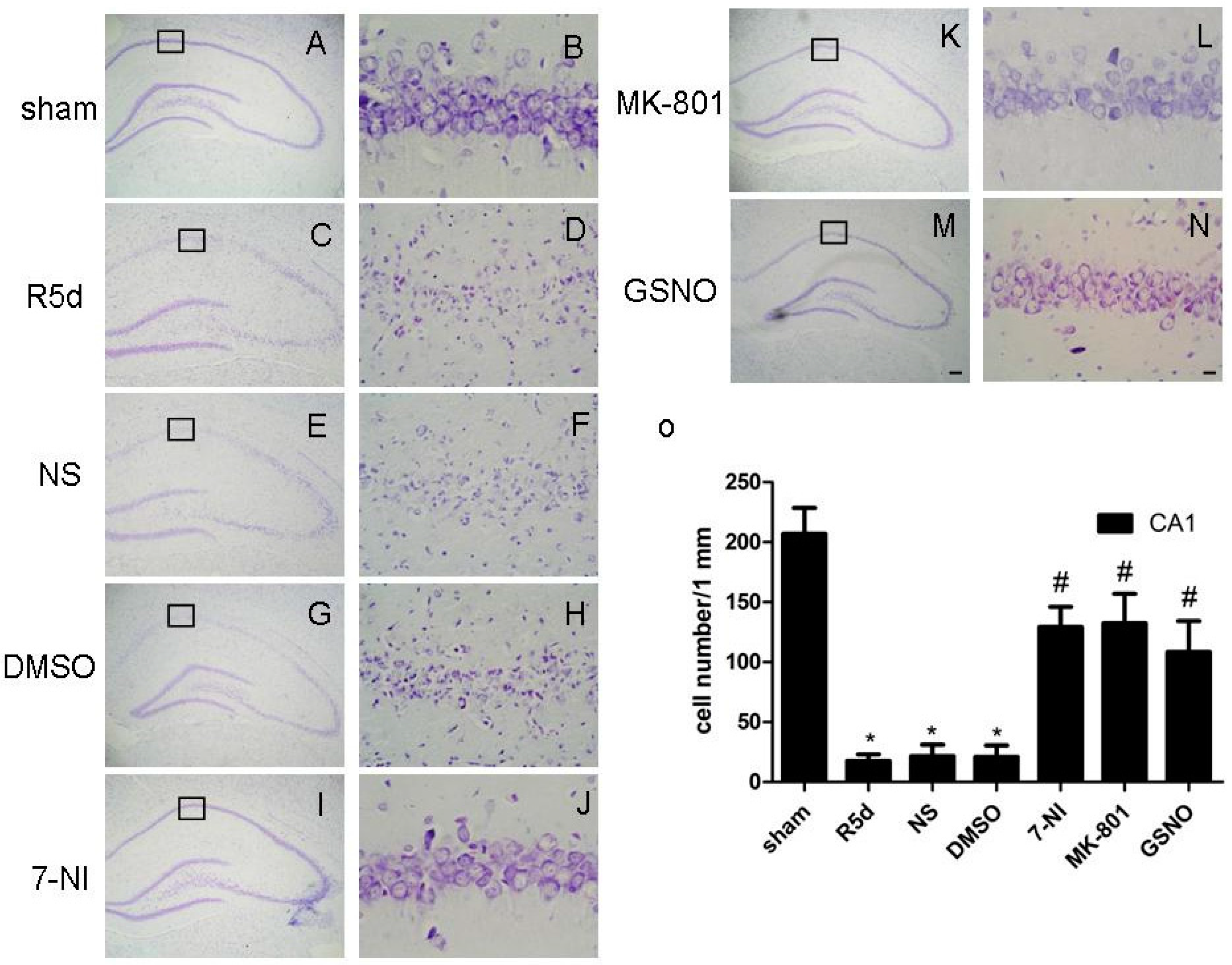
2.3. Treatment with Drugs that Attenuate Cerebral Ischemia/Reperfusion (I/R)-Induced Fyn S-Nitrosylation Results in Neuroprotection
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Culture and Plasmid Transfection Analysis
4.3. Experimental Animals
4.4. Induction of Transient Cerebral Ischemia
4.5. Administration of Drugs
4.6. Sample Preparation
4.7. S-Nitrosylation Assay
4.8. Immunoblotting Assay
4.9. Histological Analysis
4.10. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DMSO | dimethyl sulfoxide |
| DTT | dithiothreitol |
| EDTA | ethylenediamine tetraacetic acid |
| EGTA | ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid |
| JNK | c-Jun N-terminal kinase |
| NMDAR | N-methyl-d-aspartate receptor |
| nNOS | neuronal nitric oxide synthase |
| GSNO | S-nitrosoglutathione |
| TBST | tris-buffered saline with 0.1% Tween 20 |
| 7-NI | 7-nitroindazole |
| NBT/BCIP | nitro blue tetrazolium/5-bromo-4-chloro-3-indolyl-phosphate |
References
- Davidson, D.; Fournel, M.; Veillette, A. Oncogenic activation of p59fyn tyrosine protein kinase by mutation of its carboxyl-terminal site of tyrosine phosphorylation, tyrosine 528. J. Biol. Chem. 1994, 269, 10956–10963. [Google Scholar] [PubMed]
- Goldsmith, J.F.; Hall, C.G.; Atkinson, T.P. Identification of an alternatively spliced isoform of the fyn tyrosine kinase. Biochem. Biophys. Res. Commun. 2002, 298, 501–504. [Google Scholar] [CrossRef]
- Cooke, M.P.; Perlmutter, R.M. Expression of a novel form of the fyn proto-oncogene in hematopoietic cells. New Biol. 1989, 1, 66–74. [Google Scholar] [PubMed]
- Ingley, E. Src family kinases: Regulation of their activities, levels and identification of new pathways. Biochim. Biophys. Acta 2008, 1784, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Schenone, S.; Brullo, C.; Musumeci, F.; Biava, M.; Falchi, F.; Botta, M. Fyn kinase in brain diseases and cancer: The search for inhibitors. Curr. Med. Chem. 2011, 18, 2921–2942. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Senga, T.; Ito, S.; Hyodo, T.; Hasegawa, H.; Hamaguchi, M. S-nitrosylation at cysteine 498 of c-Src tyrosine kinase regulates nitric oxide-mediated cell invasion. J. Biol. Chem. 2010, 285, 3806–3814. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.J.; Li, C.; Hu, S.Q.; Wu, Y.P.; Zong, Y.Y.; Sun, C.C.; Zhang, F.; Zhang, G.Y. S-nitrosylation of c-Src via NMDAR-nNOS module promotes c-Src activation and NR2A phosphorylation in cerebral ischemia/reperfusion. Mol. Cell. Biochem. 2012, 365, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Christopherson, K.S.; Hillier, B.J.; Lim, W.A.; Bredt, D.S. PSD-95 assembles a ternary complex with the N-methyl-d-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J. Biol. Chem. 1999, 274, 27467–27473. [Google Scholar] [CrossRef] [PubMed]
- Sattler, R.; Xiong, Z.; Lu, W.Y.; Hafner, M.; MacDonald, J.F.; Tymianski, M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science 1999, 284, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- Szabadits, E.; Cserep, C.; Szonyi, A.; Fukazawa, Y.; Shigemoto, R.; Watanabe, M.; Itohara, S.; Freund, T.F.; Nyiri, G. NMDA receptors in hippocampal GABAergic synapses and their role in nitric oxide signaling. J. Neurosci. 2011, 31, 5893–5904. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Murphy, E. Protein S-nitrosylation and cardioprotection. Circ. Res. 2010, 106, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Seth, D.; Stamler, J.S. The SNO-proteome: Causation and classifications. Curr. Opin. Chem. Biol. 2011, 15, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Bolanos, J.P. Nitric oxide, cell bioenergetics and neurodegeneration. J. Neurochem. 2006, 97, 1676–1689. [Google Scholar] [CrossRef] [PubMed]
- Shahani, N.; Sawa, A. Protein S-nitrosylation: Role for nitric oxide signaling in neuronal death. Biochim. Biophys. Acta 2012, 1820, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Trepanier, C.H.; Jackson, M.F.; MacDonald, J.F. Regulation of NMDA receptors by the tyrosine kinase Fyn. FEBS J. 2012, 279, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, Y.; Liu, Y.; Hou, X.Y.; Zhang, Q.G.; Meng, F.J.; Zhang, G.Y. Possible mechanisms underlying the protective effects of SY-21, an extract of a traditional Chinese herb, on transient brain ischemia/reperfusion-induced neuronal death in rat hippocampus. Brain Res. 2003, 989, 180–186. [Google Scholar] [CrossRef]
- Yu, H.M.; Xu, J.; Li, C.; Zhou, C.; Zhang, F.; Han, D.; Zhang, G.Y. Coupling between neuronal nitric oxide synthase and glutamate receptor 6-mediated c-Jun N-terminal kinase signaling pathway via S-nitrosylation contributes to ischemia neuronal death. Neuroscience 2008, 155, 1120–1132. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.Q.; Ye, J.S.; Zong, Y.Y.; Sun, C.C.; Liu, D.H.; Wu, Y.P.; Song, T.; Zhang, G.Y. S-nitrosylation of mixed lineage kinase 3 contributes to its activation after cerebral ischemia. J. Biol. Chem. 2012, 287, 2364–2377. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.H.; Hao, L.Y.; Yue, J.; Zong, Y.Y.; Zhang, G.Y. Exogenous nitric oxide negatively regulates the S-nitrosylation p38 mitogen-activated protein kinase activation during cerebral ischaemia and reperfusion. Neuropathol. Appl. Neurobiol. 2013, 39, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.W.; Hao, L.Y.; Qi, S.H. Inhibition on the S-nitrosylation of MKK4 can protect hippocampal CA1 neurons in rat cerebral ischemia/reperfusion. Brain Res. Bull. 2016, 124, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Cornelius, C.; Rizzarelli, E.; Owen, J.B.; Dinkova-Kostova, A.T.; Butterfield, D.A. Nitric oxide in cell survival: A Janus molecule. Antioxid. Redox Signal. 2009, 11, 2717–2739. [Google Scholar] [CrossRef] [PubMed]
- Knott, A.B.; Bossy-Wetzel, E. Nitric oxide in health and disease of the nervous system. Antioxid. Redox Signal. 2009, 11, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.C.; Andriantsitohaina, R. Reactive nitrogen species: Molecular mechanisms and potential significance in health and disease. Antioxid. Redox Signal. 2009, 11, 669–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamler, J.S.; Simon, D.I.; Osborne, J.A.; Mullins, M.E.; Jaraki, O.; Michel, T.; Singel, D.J.; Loscalzo, J. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc. Natl. Acad. Sci. USA 1992, 89, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Jaffrey, S.R. Detection and characterization of protein nitrosothiols. Methods Enzymol. 2005, 396, 105–118. [Google Scholar] [PubMed]
- Wei, G.; Dawson, V.L.; Zweier, J.L. Role of neuronal and endothelial nitric oxide synthase in nitric oxide generation in the brain following cerebral ischemia. Biochim. Biophys. Acta 1999, 1455, 23–34. [Google Scholar] [CrossRef]
- Zhang, H.J.; Li, C.; Zhang, G.Y. ATPA induced GluR5-containing kainite receptor S-nitrosylation via activation of GluR5-Gq-PLC-IP3R pathway and signalling module GluR5·PSD-95·nNOS. Int. J. Biochem. Cell Biol. 2012, 44, 2261–2271. [Google Scholar] [CrossRef] [PubMed]
- Keynes, R.G.; Garthwaite, J. Nitric oxide and its role in ischaemic brain injury. Curr. Mol. Med. 2004, 4, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Samdani, A.F.; Dawson, T.M.; Dawson, V.L. Nitric oxide synthase in models of focal ischemia. Stroke 1997, 28, 1283–1288. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Xu, X.; Zhang, F.; el-Fakahany, E.E.; Ross, M.E. Marked induction of calcium-independent nitric oxide synthase activity after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1995, 15, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Otani, H.; Jesmin, S.; Togashi, H.; Sakuma, I.; Nakai, K.; Satoh, H.; Yoshioka, M.; Kitabatake, A. An S-nitrosylated hemoglobin derivative protects the rat hippocampus from ischemia-induced long-term potentiation impairment with a time window. J. Pharmacol. Sci. 2004, 96, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Stuehr, D.J. Mammalian nitric oxide synthases. Biochim. Biophys. Acta 1999, 1411, 217–230. [Google Scholar] [CrossRef]
- Pei, D.S.; Song, Y.J.; Yu, H.M.; Hu, W.W.; Du, Y.; Zhang, G.Y. Exogenous nitric oxide negatively regulates c-Jun N-terminal kinase activation via inhibiting endogenous NO-induced S-nitrosylation during cerebral ischemia and reperfusion in rat hippocampus. J. Neurochem. 2008, 106, 1952–1963. [Google Scholar] [CrossRef] [PubMed]
- Broniowska, K.A.; Diers, A.R.; Hogg, N. S-Nitrosoglutathione. Biochim. Biophys. Acta 2013, 1830, 3173–3181. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, N.; Root, P.; Jiang, X.M.; Hogg, P.J.; Mutus, B. Mechanism of transfer of NO from extracellular S-nitrosothiols into the cytosol by cell-surface protein disulfide isomerase. Proc. Natl. Acad. Sci. USA. 2001, 98, 9539–9544. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, N.; Jacob, S.; Zielinski, B.; Curatola, G.; Mazzanti, L.; Mutus, B. N-Dansyl-snitrosohomocysteine a fluorescent probe for intracellular thiols and S-nitrosothiols. Biochim. Biophys. Acta 1999, 1430, 149–154. [Google Scholar] [CrossRef]
- Kim, W.K.; Choi, Y.B.; Rayudu, P.V.; Das, P.; Asaad, W.; Arnelle, D.R.; Stamler, J.S.; Lipton, S.A. Attenuation of NMDA receptor activity and neurotoxicity by nitroxyl anion, NO. Neuron 1999, 24, 461–469. [Google Scholar] [CrossRef]
- Kiedrowski, L.; Costa, E.; Wroblewski, J.T. Glutamate receptor agonists stimulate nitric oxide synthase in primary cultures of cerebellar granule cells. J. Neurochem. 1992, 58, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Rameau, G.A.; Tukey, D.S.; Garcin-Hosfield, E.D.; Titcombe, R.F.; Misra, C.; Khatri, L.; Getzoff, E.D.; Ziff, E.B. Biphasic coupling of neuronal nitric oxide synthase phosphorylation to the NMDA receptor regulates AMPA receptor trafficking and neuronal cell death. J. Neurosci. 2007, 27, 3445–3455. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Dhammu, T.S.; Sakakima, H.; Shunmugavel, A.; Gilg, A.G.; Singh, A.K.; Singh, I. The inhibitory effect of S-nitrosoglutathione on blood–brain barrier disruption and peroxynitrite formation in a rat model of experimental stroke. J. Neurochem. 2012, 123, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Dhammu, T.S.; Matsuda, F.; Singh, A.K.; Singh, I. Blocking a vicious cycle nNOS/peroxynitrite/AMPK by S-nitrosoglutathione: implication for stroke therapy. BMC Neurosci. 2015, 16, 42. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.H.; Espino, P.C.; Marshall, J.; Harvey, R.; Merrill, J.; Smith, A.E. Structural elements that regulate pp59c-fyn catalytic activity, transforming potential, and ability to associate with polyomavirus middle-T antigen. J. Virol. 1991, 65, 170–179. [Google Scholar] [PubMed]
- Braithwaite, S.P.; Paul, S.; Nairn, A.C.; Lombroso, P.J. Synaptic plasticity: One STEP at a time. Trends Neurosci. 2006, 29, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.; Liu, J.; Lombroso, P.J. Striatal enriched phosphatase 61 dephosphorylates Fyn at phosphotyrosine 420. J. Biol. Chem. 2002, 277, 24274–24279. [Google Scholar] [CrossRef] [PubMed]
- Neill, S.; Bright, J.; Desikan, R.; Hancock, J.; Harrison, J.; Wilson, I. Nitric oxide evolution and perception. J. Exp. Bot. 2008, 59, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Daaka, Y. S-nitrosylation-regulated GPCR signaling. Biochim. Biophys. Acta 2012, 1820, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci. 1997, 20, 132–139. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Pulsinelli, W.A.; Brierley, J.B. A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke 1979, 10, 267–272. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, L.; Wei, X.; Guo, P.; Zhang, G.; Qi, S. Neuroprotective Effects of Inhibiting Fyn S-Nitrosylation on Cerebral Ischemia/Reperfusion-Induced Damage to CA1 Hippocampal Neurons. Int. J. Mol. Sci. 2016, 17, 1100. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17071100
Hao L, Wei X, Guo P, Zhang G, Qi S. Neuroprotective Effects of Inhibiting Fyn S-Nitrosylation on Cerebral Ischemia/Reperfusion-Induced Damage to CA1 Hippocampal Neurons. International Journal of Molecular Sciences. 2016; 17(7):1100. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17071100
Chicago/Turabian StyleHao, Lingyun, Xuewen Wei, Peng Guo, Guangyi Zhang, and Suhua Qi. 2016. "Neuroprotective Effects of Inhibiting Fyn S-Nitrosylation on Cerebral Ischemia/Reperfusion-Induced Damage to CA1 Hippocampal Neurons" International Journal of Molecular Sciences 17, no. 7: 1100. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17071100