
Human Aquaporin-4 and Molecular Modeling: Historical Perspective and View to the Future

 and
and 
Abstract
:
1. Introduction
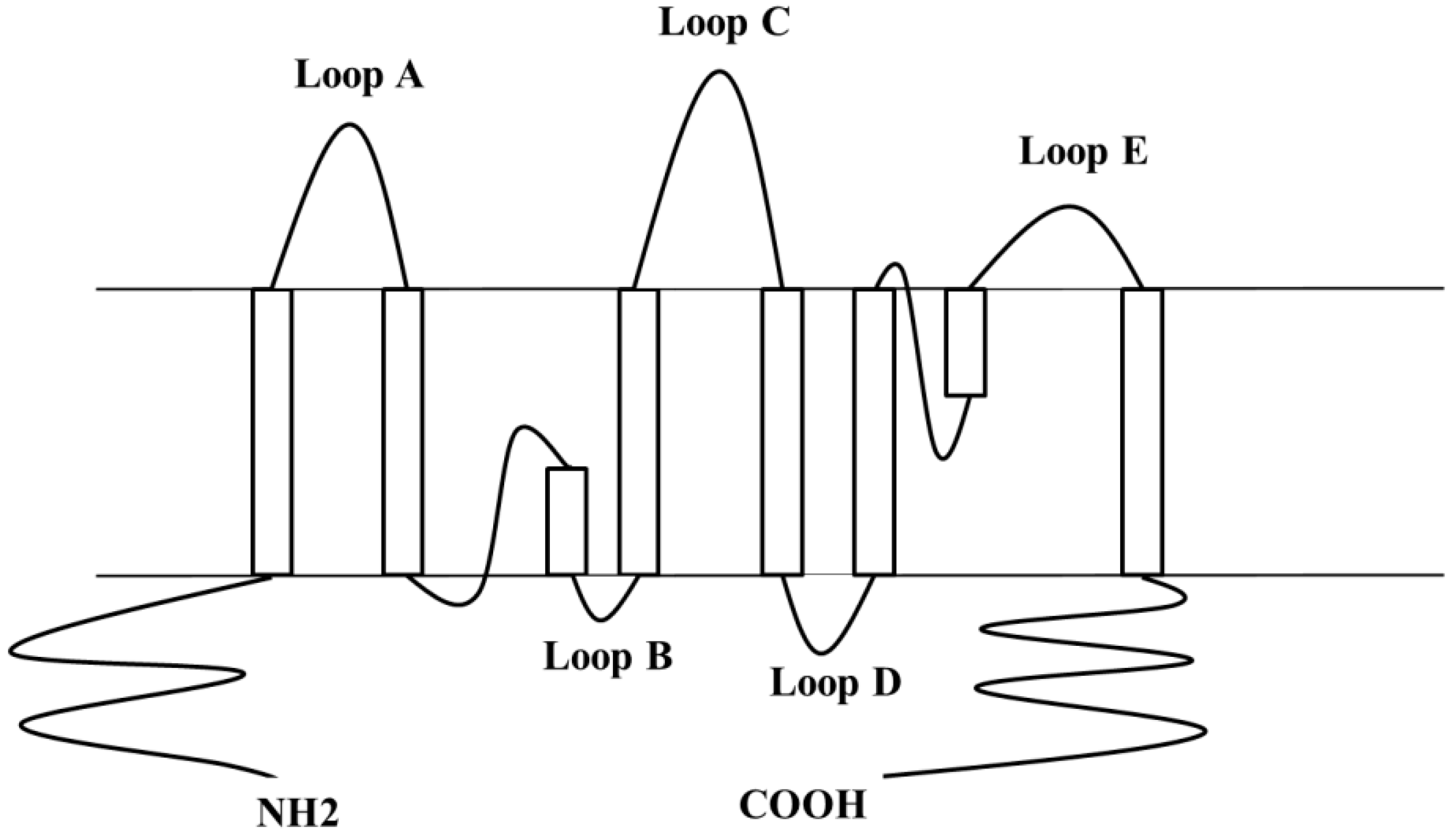
2. Human Aquaporin-4 (hAQP4), the Predominant Water Channel in the Central Nervous System
3. Why Molecular Modeling?
3.1. Understanding the Water Permeation Mechanism
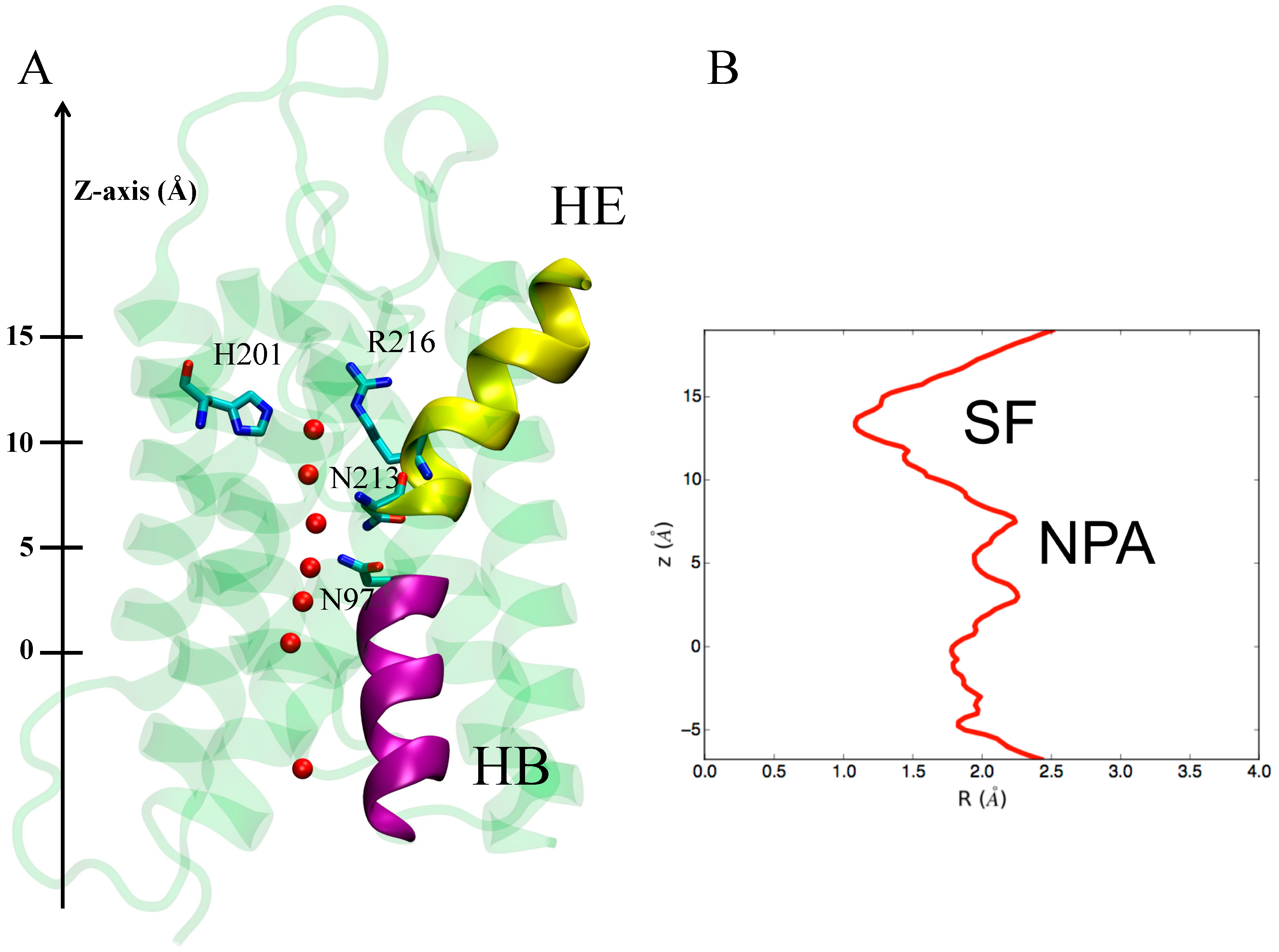
3.1.1. hAQP4: X-ray Data and First MD Simulations
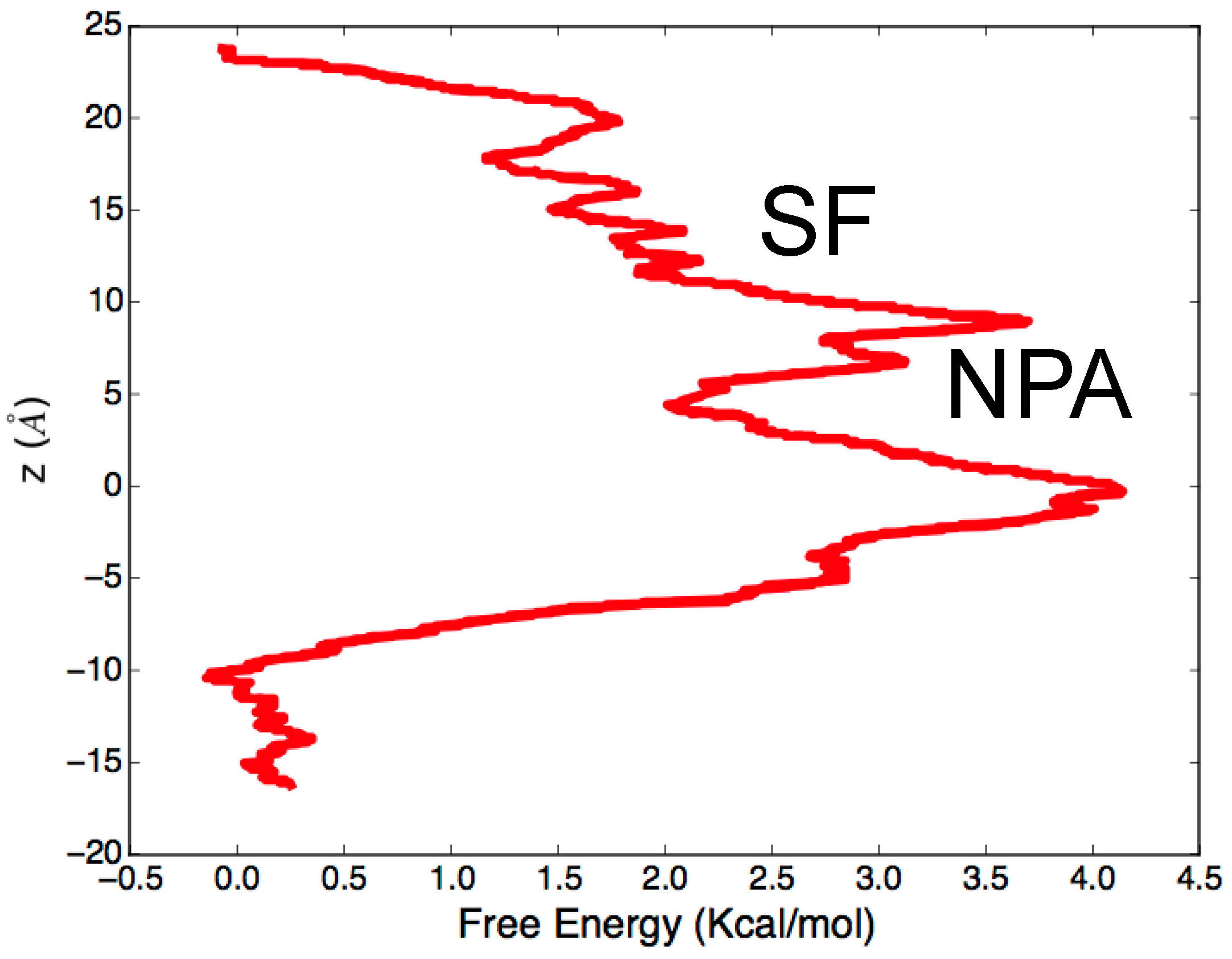
3.2. Computing Free-Energy Profile of Water Conduction
3.3. Identifying Gating Sites
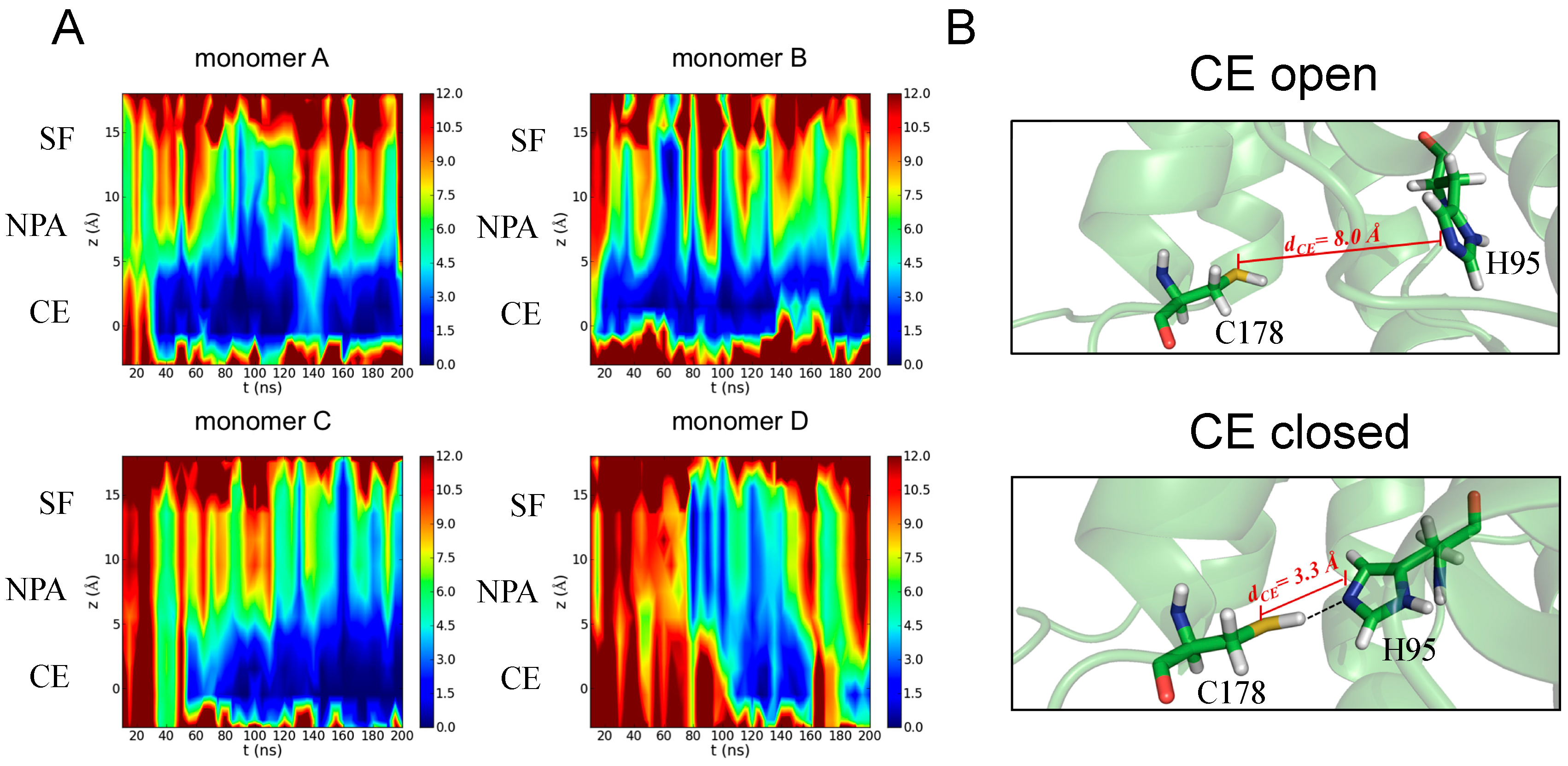
3.3.1. Identification of H95 as a New Gating Residue in hAQP4
3.4. Rationalizing Pathological Mutations
3.5. Characterizing Epitope Structural Features
3.6. Assessing Druggability
4. Future Challenges
4.1. Investigating Orthogonal Array of Particles (OAPs) Aggregation
4.2. Structure-Based Virtual Screening
4.3. Drug-Repurposing Strategies
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| hAQP4 | Human Aquaporin-4 |
| NMO | Neuromyelitis optica |
| MD | Molecular Dynamics |
| SMD | Steered Molecular Dynamics |
| BD-FDT | Brownian dynamics fluctuation-dissipation theorems |
| VS | Virtual Screening |
References
- Nielsen, S.; Nagelhus, E.A.; Amiry-Moghaddam, M.; Bourque, C.; Agre, P.; Ottersen, O.P. Specialized membrane domains for water transport in glial cells: High-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J. Neurosci. Off. J. Soc. Neurosci. 1997, 17, 171–180. [Google Scholar]
- Borgnia, M.; Nielsen, S.; Engel, A.; Agre, P. Cellular and molecular biology of the aquaporin water channels. Annu. Rev. Biochem. 1999, 68, 425–458. [Google Scholar] [CrossRef] [PubMed]
- Sui, H.; Han, B.G.; Lee, J.K.; Walian, P.; Jap, B.K. Structural basis of water-specific transport through the AQP1 water channel. Nature 2001, 414, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, R.; Singh, B. Insights into structural mechanisms of gating induced regulation of aquaporins. Prog. Biophys. Mol. Biol. 2014, 114, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Ilan, B.; Tajkhorshid, E.; Schulten, K.; Voth, G.A. The mechanism of proton exclusion in aquaporin channels. Proteins 2004, 55, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, H.; Steinbronn, C.; Wu, B.; Beitz, E.; Zeuthen, T.; Voth, G.A. Enhancement of proton conductance by mutations of the selectivity filter of aquaporin-1. J. Mol. Biol. 2011, 407, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Hub, J.S.; de Groot, B.L. Mechanism of selectivity in aquaporins and aquaglyceroporins. Proc. Natl. Acad. Sci. USA 2008, 105, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Zeidel, M.L.; Ambudkar, S.V.; Smith, B.L.; Agre, P. Reconstitution of functional water channels in liposomes containing purified red cell CHIP28 protein. Biochemistry 1992, 31, 7436–7440. [Google Scholar] [CrossRef] [PubMed]
- Agre, P.; King, L.S.; Yasui, M.; Guggino, W.B.; Ottersen, O.P.; Fujiyoshi, Y.; Engel, A.; Nielsen, S. Aquaporin water channels—From atomic structure to clinical medicine. J. Physiol. 2002, 542, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Day, R.E.; Kitchen, P.; Owen, D.S.; Bland, C.; Marshall, L.; Conner, A.C.; Bill, R.M.; Conner, M.T. Human aquaporins: Regulators of transcellular water flow. Biochim. Biophys. Acta 2014, 1840, 1492–1506. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W. Aquaporin-related disorders of water homeostasis. Drug News Perspect. 2007, 20, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Agre, P.; Kozono, D. Aquaporin water channels: Molecular mechanisms for human diseases 1. FEBS Lett. 2003, 555, 72–78. [Google Scholar] [CrossRef]
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S.; Hinson, S.R. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005, 202, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Castle, N.A. Aquaporins as targets for drug discovery. Drug Discov. Today 2005, 10, 485–493. [Google Scholar] [CrossRef]
- De Groot, B.L.; Frigato, T.; Helms, V.; Grubmüller, H. The mechanism of proton exclusion in the aquaporin-1 water channel. J. Mol. Biol. 2003, 333, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Hub, J.S.; Grubmüller, H.; de Groot, B.L. Dynamics and energetics of permeation through aquaporins. What do we learn from molecular dynamics simulations? Handb. Exp. Pharmacol. 2009, 190, 57–76. [Google Scholar]
- Hub, J.S.; Aponte-Santamaría, C.; Grubmüller, H.; de Groot, B.L. Voltage-regulated water flux through aquaporin channels in silico. Biophys. J. 2010, 99, L97–L99. [Google Scholar] [CrossRef] [PubMed]
- Aponte-Santamaría, C.; Briones, R.; Schenk, A.D.; Walz, T.; de Groot, B.L. Molecular driving forces defining lipid positions around aquaporin-0. Proc. Natl. Acad. Sci. USA 2012, 109, 9887–9892. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, D.; Zapater, C.; Krenc, D.; Haddoub, R.; Flitsch, S.; Beitz, E.; Cerdà, J.; de Groot, B.L. Discovery of novel human aquaporin-1 blockers. ACS Chem. Biol. 2013, 8, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Assentoft, M.; Kaptan, S.; Fenton, R.A.; Hua, S.Z.; de Groot, B.L.; MacAulay, N. Phosphorylation of rat aquaporin-4 at Ser111 is not required for channel gating. Glia 2013, 61, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.D.; Yeh, R.; Sandstrom, A.; Chorny, I.; Harries, W.E.C.; Robbins, R.A.; Miercke, L.J.W.; Stroud, R.M. Crystal structure of human aquaporin 4 at 1.8 A and its mechanism of conductance. Proc. Natl. Acad. Sci. USA 2009, 106, 7437–7442. [Google Scholar] [CrossRef] [PubMed]
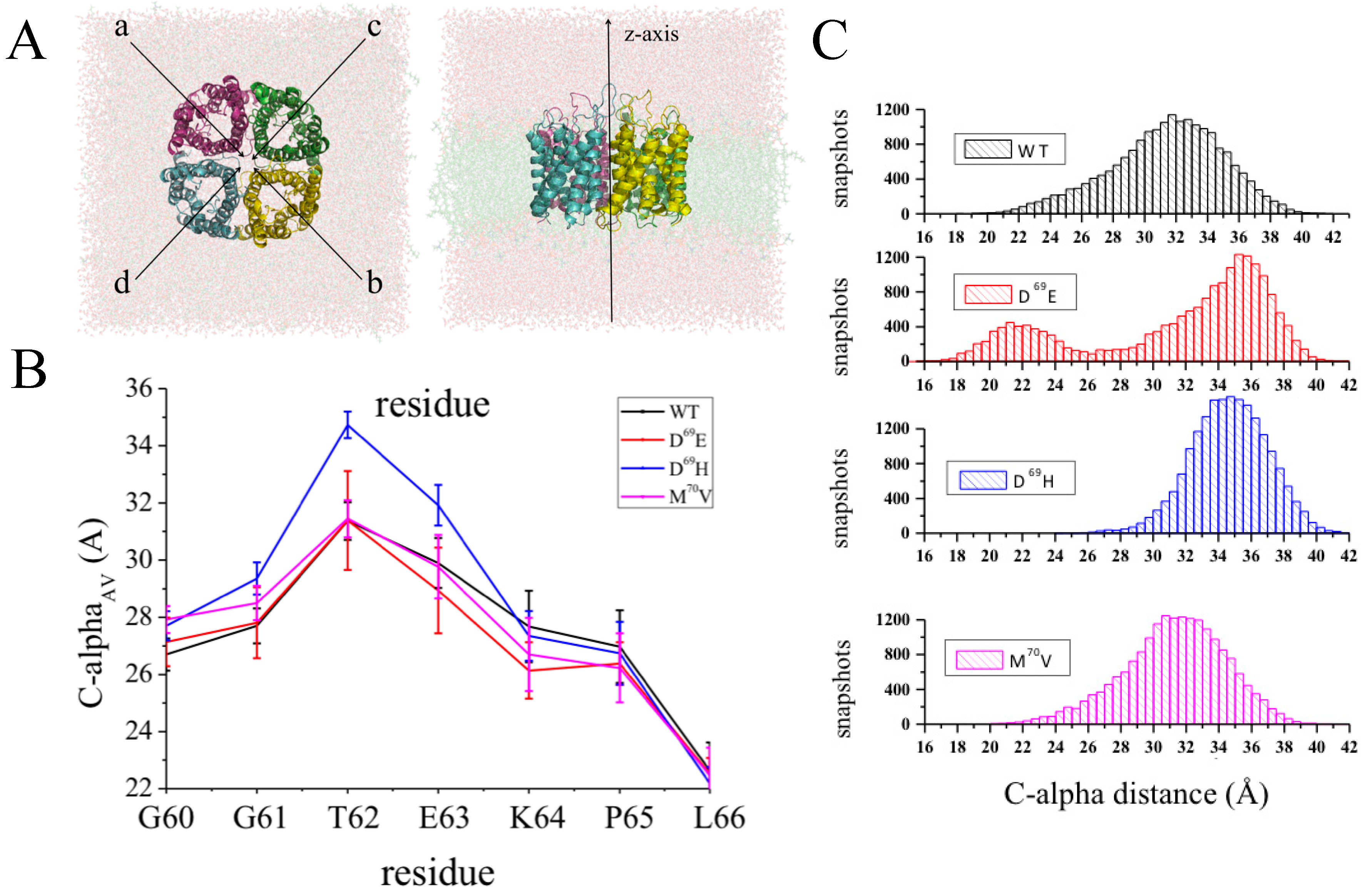
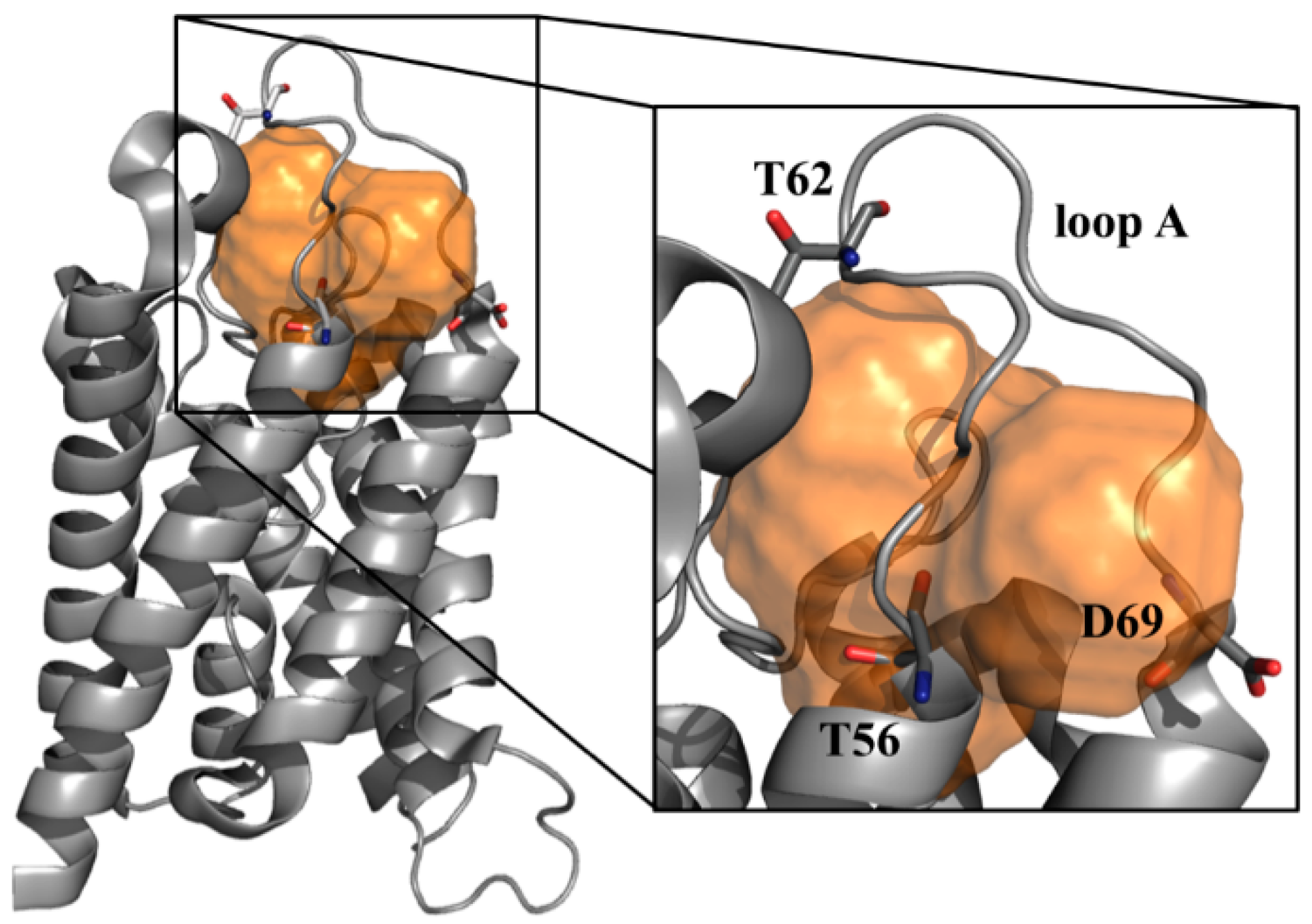
- Alberga, D.; Nicolotti, O.; Lattanzi, G.; Nicchia, G.P.; Frigeri, A.; Pisani, F.; Benfenati, V.; Mangiatordi, G.F. A new gating site in human aquaporin-4: Insights from molecular dynamics simulations. Biochim. Biophys. Acta 2014, 1838, 3052–3060. [Google Scholar] [CrossRef] [PubMed]
- Umenishi, F.; Verkman, A.S. Isolation and functional analysis of alternative promoters in the human aquaporin-4 water channel gene. Genomics 1998, 50, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Ma, T.; Verkman, A.S. cDNA cloning, gene organization, and chromosomal localization of a human mercurial insensitive water channel. J. Biol. Chem. 1995, 270, 22907–22913. [Google Scholar] [CrossRef] [PubMed]
- Amiry-Moghaddam, M.; Ottersen, O.P. The molecular basis of water transport in the brain. Nat. Rev. Neurosci. 2003, 4, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, K.; Yamamoto, N.; Asai, K.; Sobue, K.; Fujita, Y.; Fujita, M.; Mase, M.; Yamada, K.; Nakanishi, M.; Tada, T.; et al. Regulation of aquaporin-4 expression in astrocytes. Brain Res. Mol. Brain Res. 2001, 89, 94–102. [Google Scholar] [CrossRef]
- Nicchia, G.P.; Ficarella, R.; Rossi, A.; Giangreco, I.; Nicolotti, O.; Carotti, A.; Pisani, F.; Estivill, X.; Gasparini, P.; Svelto, M.; et al. D184E mutation in aquaporin-4 gene impairs water permeability and links to deafness. Neuroscience 2011, 197, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Verkman, A.S. Impaired hearing in mice lacking aquaporin-4 water channels. J. Biol. Chem. 2001, 276, 31233–31237. [Google Scholar] [CrossRef] [PubMed]
- Zador, Z.; Stiver, S.; Wang, V.; Manley, G.T. Role of aquaporin-4 in cerebral edema and stroke. Handb. Exp. Pharmacol. 2009, 190, 159–170. [Google Scholar] [PubMed]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin-4 and brain edema. Pediatr. Nephrol. Berl. Ger. 2007, 22, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Heuser, K.; Nagelhus, E.A.; Taubøll, E.; Indahl, U.; Berg, P.R.; Lien, S.; Nakken, S.; Gjerstad, L.; Ottersen, O.P. Variants of the genes encoding AQP4 and Kir4.1 are associated with subgroups of patients with temporal lobe epilepsy. Epilepsy Res. 2010, 88, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.J.; Bao, L.; Jacob, A.; Kraus, D.M.; Holers, V.M.; Quigg, R.J. Administration of the soluble complement inhibitor, Crry-Ig, reduces inflammation and aquaporin 4 expression in lupus cerebritis. Biochim. Biophys. Acta 2003, 1639, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Krampla, W.; Aboul-Enein, F.; Jecel, J.; Lang, W.; Fertl, E.; Hruby, W.; Kristoferitsch, W. Spinal cord lesions in patients with neuromyelitis optica: a retrospective long-term MRI follow-up study. Eur. Radiol. 2009, 19, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.J.; Tsujita, M.; Yamazaki, M.; Sakimura, K.; Nakada, T. Identification of arylsulfonamides as Aquaporin 4 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 1270–1273. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, Y.; Hiroaki, Y.; Fujiyoshi, Y. Acetazolamide reversibly inhibits water conduction by aquaporin-4. J. Struct. Biol. 2009, 166, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, H.; Huber, V.J.; Tsujita, M.; Nakada, T. Pretreatment with a novel aquaporin 4 inhibitor, TGN-020, significantly reduces ischemic cerebral edema. Neurol. Sci. 2011, 32, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.J.; Tsujita, M.; Nakada, T. Identification of aquaporin 4 inhibitors using in vitro and in silico methods. Bioorg. Med. Chem. 2009, 17, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Tradtrantip, L.; Zhang, H.; Anderson, M.O.; Saadoun, S.; Phuan, P.-W.; Papadopoulos, M.C.; Bennett, J.L.; Verkman, A.S. Small-molecule inhibitors of NMO-IgG binding to aquaporin-4 reduce astrocyte cytotoxicity in neuromyelitis optica. FASEB J. 2012, 26, 2197–2208. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Bennett, J.L.; Verkman, A.S. Treatment of neuromyelitis optica: State-of-the-art and emerging therapies. Nat. Rev. Neurol. 2014, 10, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Carbrey, J.M.; Agre, P. Discovery of the aquaporins and development of the field. In Handbook of Experimental Pharmacology; Beitz, P.D.E., Ed.; Springer: Berlin/Heidelberg, Germany, 2009; Volume 190, pp. 3–28. [Google Scholar]
- Verkman, A.S.; Mitra, A.K. Structure and function of aquaporin water channels. Am. J. Physiol. 2000, 278, F13–F28. [Google Scholar]
- Agmon, N. The Grotthuss mechanism. Chem. Phys. Lett. 1995, 244, 456–462. [Google Scholar] [CrossRef]
- De Groot, B.L.; Engel, A.; Grubmüller, H. A refined structure of human aquaporin-1. FEBS Lett. 2001, 504, 206–211. [Google Scholar] [CrossRef]
- De Groot, B.L.; Grubmüller, H. Water permeation across biological membranes: Mechanism and dynamics of aquaporin-1 and GlpF. Science 2001, 294, 2353–2357. [Google Scholar] [CrossRef] [PubMed]
- Tajkhorshid, E.; Nollert, P.; Jensen, M.Ø.; Miercke, L.J.W.; O’Connell, J.; Stroud, R.M.; Schulten, K. Control of the selectivity of the aquaporin water channel family by global orientational tuning. Science 2002, 296, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, N.; Roux, B.; Pomès, R. Structural determinants of proton blockage in aquaporins. J. Mol. Biol. 2004, 343, 493–510. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wu, Y.; Voth, G.A. Origins of proton transport behavior from selectivity domain mutations of the aquaporin-1 channel. Biophys. J. 2006, 90, L73–L75. [Google Scholar] [CrossRef] [PubMed]
- Kreida, S.; Törnroth-Horsefield, S. Structural insights into aquaporin selectivity and regulation. Curr. Opin. Struct. Biol. 2015, 33, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Kosinska Eriksson, U.; Fischer, G.; Friemann, R.; Enkavi, G.; Tajkhorshid, E.; Neutze, R. Subangstrom resolution X-ray structure details aquaporin-water interactions. Science 2013, 340, 1346–1349. [Google Scholar] [CrossRef] [PubMed]
- Beitz, E.; Wu, B.; Holm, L.M.; Schultz, J.E.; Zeuthen, T. Point mutations in the aromatic/arginine region in aquaporin 1 allow passage of urea, glycerol, ammonia, and protons. Proc. Natl. Acad. Sci. USA 2006, 103, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Smart, O.S.; Goodfellow, J.M.; Wallace, B.A. The pore dimensions of gramicidin A. Biophys. J. 1993, 65, 2455–2460. [Google Scholar] [CrossRef]
- Cui, Y.; Bastien, D.A. Water transport in human aquaporin-4: Molecular dynamics (MD) simulations. Biochem. Biophys. Res. Commun. 2011, 412, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Izrailev, S.; Stepaniants, S.; Isralewitz, B.; Kosztin, D.; Lu, H.; Molnar, F.; Wriggers, W.; Schulten, K. Steered molecular dynamics. In Computational Molecular Dynamics: Challenges, Methods, Ideas; Deuflhard, P., Hermans, J., Leimkuhler, B., Mark, A.E., Reich, S., Skeel, R.D., Eds.; Springer: Berlin/Heidelberg, Germany, 1999; pp. 39–65. [Google Scholar]
- Bossis, G.; Quentrec, B.; Boon, J.P. Brownian dynamics and the fluctuation-dissipation theorem. Mol. Phys. 1982, 45, 191–196. [Google Scholar] [CrossRef]
- Kutzner, C.; Grubmüller, H.; de Groot, B.L.; Zachariae, U. Computational electrophysiology: the molecular dynamics of ion channel permeation and selectivity in atomistic detail. Biophys. J. 2011, 101, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Németh-Cahalan, K.L.; Kalman, K.; Hall, J.E. Molecular basis of pH and Ca2+ regulation of aquaporin water permeability. J. Gen. Physiol. 2004, 123, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Zelenina, M.; Bondar, A.A.; Zelenin, S.; Aperia, A. Nickel and extracellular acidification inhibit the water permeability of human aquaporin-3 in lung epithelial cells. J. Biol. Chem. 2003, 278, 30037–30043. [Google Scholar] [CrossRef] [PubMed]
- Zelenina, M.; Tritto, S.; Bondar, A.A.; Zelenin, S.; Aperia, A. Copper inhibits the water and glycerol permeability of aquaporin-3. J. Biol. Chem. 2004, 279, 51939–51943. [Google Scholar] [CrossRef] [PubMed]
- Tamás, M.J.; Karlgren, S.; Bill, R.M.; Hedfalk, K.; Allegri, L.; Ferreira, M.; Thevelein, J.M.; Rydström, J.; Mullins, J.G.L.; Hohmann, S. A short regulatory domain restricts glycerol transport through yeast Fps1p. J. Biol. Chem. 2003, 278, 6337–6345. [Google Scholar] [CrossRef] [PubMed]
- Johansson, I.; Larsson, C.; Ek, B.; Kjellbom, P. The major integral proteins of spinach leaf plasma membranes are putative aquaporins and are phosphorylated in response to Ca2+ and apoplastic water potential. Plant. Cell 1996, 8, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Tournaire-Roux, C.; Sutka, M.; Javot, H.; Gout, E.; Gerbeau, P.; Luu, D.-T.; Bligny, R.; Maurel, C. Cytosolic pH regulates root water transport during anoxic stress through gating of aquaporins. Nature 2003, 425, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Zelenina, M.; Zelenin, S.; Bondar, A.A.; Brismar, H.; Aperia, A. Water permeability of aquaporin-4 is decreased by protein kinase C and dopamine. Am. J. Physiol. Ren. Physiol. 2002, 283, F309–F318. [Google Scholar] [CrossRef] [PubMed]
- Gunnarson, E.; Zelenina, M.; Axehult, G.; Song, Y.; Bondar, A.; Krieger, P.; Brismar, H.; Zelenin, S.; Aperia, A. Identification of a molecular target for glutamate regulation of astrocyte water permeability. Glia 2008, 56, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, P.; Conner, A.C. Control of the aquaporin-4 channel water permeability by structural dynamics of aromatic/arginine selectivity filter residues. Biochemistry 2015, 54, 6753–6755. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yool, A.J.; Schulten, K.; Tajkhorshid, E. Mechanism of gating and ion conductivity of a possible tetrameric pore in aquaporin-1. Structure 2006, 14, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Nyblom, M.; Frick, A.; Wang, Y.; Ekvall, M.; Hallgren, K.; Hedfalk, K.; Neutze, R.; Tajkhorshid, E.; Törnroth-Horsefield, S. Structural and Functional Analysis of SoPIP2;1 Mutants Adds Insight into Plant Aquaporin Gating. J. Mol. Biol. 2009, 387, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Janosi, L.; Ceccarelli, M. The gating mechanism of the human aquaporin 5 revealed by molecular dynamics simulations. PLoS ONE 2013, 8, e59897. [Google Scholar] [CrossRef] [PubMed]
- Hashido, M.; Kidera, A.; Ikeguchi, M. Water transport in aquaporins: Osmotic permeability matrix analysis of molecular dynamics simulations. Biophys. J. 2007, 93, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Nicolotti, O.; Miscioscia, T.F.; Leonetti, F.; Muncipinto, G.; Carotti, A. Screening of matrix metalloproteinases available from the protein data bank: Insights into biological functions, domain organization, and zinc binding groups. J. Chem. Inf. Model. 2007, 47, 2439–2448. [Google Scholar] [CrossRef] [PubMed]
- Zong, X.; Stieber, J.; Ludwig, A.; Hofmann, F.; Biel, M. A single histidine residue determines the pH sensitivity of the pacemaker channel HCN2. J. Biol. Chem. 2001, 276, 6313–6319. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda, F.V.; Pablo Cid, L.; Teulon, J.; Niemeyer, M.I. Molecular aspects of structure, gating, and physiology of pH-sensitive background K2P and Kir K+-transport channels. Physiol. Rev. 2015, 95, 179–217. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.N.; Posson, D.J.; Parsa, P.V.; Nimigean, C.M. Molecular mechanism of pH sensing in KcsA potassium channels. Proc. Natl. Acad. Sci. USA 2008, 105, 6900–6905. [Google Scholar] [CrossRef] [PubMed]
- Kaptan, S.; Assentoft, M.; Schneider, H.P.; Fenton, R.A.; Deitmer, J.W.; MacAulay, N.; de Groot, B.L. H95 is a pH-dependent gate in aquaporin 4. Structures 2015, 23, 2309–2318. [Google Scholar] [CrossRef] [PubMed]
- Krivobokova, T.; Briones, R.; Hub, J.S.; Munk, A.; de Groot, B.L. Partial least-squares functional mode analysis: Application to the membrane proteins AQP1, Aqy1, and CLC-ec1. Biophys. J. 2012, 103, 786–796. [Google Scholar] [CrossRef] [PubMed]
- Henzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Studer, R.A.; Dessailly, B.H.; Orengo, C.A. Residue mutations and their impact on protein structure and function: detecting beneficial and pathogenic changes. Biochem. J. 2013, 449, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Benet-Pagès, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Gsponer, J.; Ferrara, P.; Caflisch, A. Flexibility of the murine prion protein and its Asp178Asn mutant investigated by molecular dynamics simulations. J. Mol. Graph. Model. 2001, 20, 169–182. [Google Scholar] [CrossRef]
- Shamsir, M.S.; Dalby, A.R. One gene, two diseases and three conformations: Molecular dynamics simulations of mutants of human prion protein at room temperature and elevated temperatures. Proteins Struct. Funct. Bioinform. 2005, 59, 275–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doss, C.G.P.; Rajith, B.; Garwasis, N.; Mathew, P.R.; Raju, A.S.; Apoorva, K.; William, D.; Sadhana, N.R.; Himani, T.; Dike, I. Screening of mutations affecting protein stability and dynamics of FGFR1—A simulation analysis. Appl. Transl. Genom. 2012, 1, 37–43. [Google Scholar] [CrossRef]
- Elmore, D.E.; Dougherty, D.A. Molecular dynamics simulations of wild-type and mutant forms of the Mycobacterium tuberculosis MscL channel. Biophys. J. 2001, 81, 1345–1359. [Google Scholar] [CrossRef]
- Pirolli, D.; Carelli Alinovi, C.; Capoluongo, E.; Satta, M.A.; Concolino, P.; Giardina, B.; de Rosa, M.C. Insight into a novel p53 single point mutation (G389E) by molecular dynamics simulations. Int. J. Mol. Sci. 2010, 12, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Imbrici, P.; Maggi, L.; Mangiatordi, G.F.; Dinardo, M.M.; Altamura, C.; Brugnoni, R.; Alberga, D.; Pinter, G.L.; Ricci, G.; Siciliano, G.; et al. ClC-1 mutations in myotonia congenita patients: insights into molecular gating mechanisms and genotype–phenotype correlation. J. Physiol. 2015, 593, 4181–4199. [Google Scholar] [CrossRef] [PubMed]
- Horsten, U.; Müller-Newen, G.; Gerhartz, C.; Wollmer, A.; Wijdenes, J.; Heinrich, P.C.; Grötzinger, J. Molecular modeling-guided mutagenesis of the extracellular part of gp130 leads to the identification of contact sites in the interleukin-6 (IL-6)·IL-6 receptor·gp130 Complex. J. Biol. Chem. 1997, 272, 23748–23757. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, C.; Oostenbrink, C.; Keizers, P.H.; van Vugt-Lussenburg, B.M.; van Waterschoot, R.A.; Tschirret-Guth, R.A.; Commandeur, J.N.; Vermeulen, N.P. Molecular modeling-guided site-directed mutagenesis of cytochrome P450 2D6. Curr. Drug Metab. 2007, 8, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Schushan, M.; Xiang, M.; Bogomiakov, P.; Padan, E.; Rao, R.; Ben-Tal, N. Model-guided mutagenesis drives functional studies of human NHA2, implicated in hypertension. J. Mol. Biol. 2010, 396, 1181–1196. [Google Scholar] [CrossRef] [PubMed]
- Shyue, S.-K.; Ruan, K.-H.; Wang, L.-H.; Wu, K.K. Prostacyclin synthase active sites identification by molecular modeling-guided site-directed mutagenesis. J. Biol. Chem. 1997, 272, 3657–3662. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-H.; Matijevic-Aleksic, N.; Hsu, P.-Y.; Ruan, K.-H.; Wu, K.K.; Kulmacz, R.J. Identification of thromboxane A2 synthase active site residues by molecular modeling-guided site-directed mutagenesis. J. Biol. Chem. 1996, 271, 19970–19975. [Google Scholar] [CrossRef] [PubMed]
- Lucchinetti, C.F.; Mandler, R.N.; McGavern, D.; Bruck, W.; Gleich, G.; Ransohoff, R.M.; Trebst, C.; Weinshenker, B.; Wingerchuk, D.; Parisi, J.E.; et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain 2002, 125, 1450–1461. [Google Scholar] [CrossRef] [PubMed]
- Kira, J. Autoimmunity in neuromyelitis optica and opticospinal multiple sclerosis: Astrocytopathy as a common denominator in demyelinating disorders. J. Neurol. Sci. 2011, 311, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012, 11, 535–544. [Google Scholar] [CrossRef]
- Pisani, F.; Mastrototaro, M.; Rossi, A.; Nicchia, G.P.; Tortorella, C.; Ruggieri, M.; Trojano, M.; Frigeri, A.; Svelto, M. Identification of two major conformational aquaporin-4 epitopes for neuromyelitis optica autoantibody binding. J. Biol. Chem. 2011, 286, 9216–9224. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Baumgart, F.; van Hoek, A.N.; Verkman, A.S. Post-golgi supramolecular assembly of aquaporin-4 in orthogonal arrays. Traffic 2012, 13, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Nicchia, G.P.; Mastrototaro, M.; Rossi, A.; Pisani, F.; Tortorella, C.; Ruggieri, M.; Lia, A.; Trojano, M.; Frigeri, A.; Svelto, M. Aquaporin-4 orthogonal arrays of particles are the target for neuromyelitis optica autoantibodies. Glia 2009, 57, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
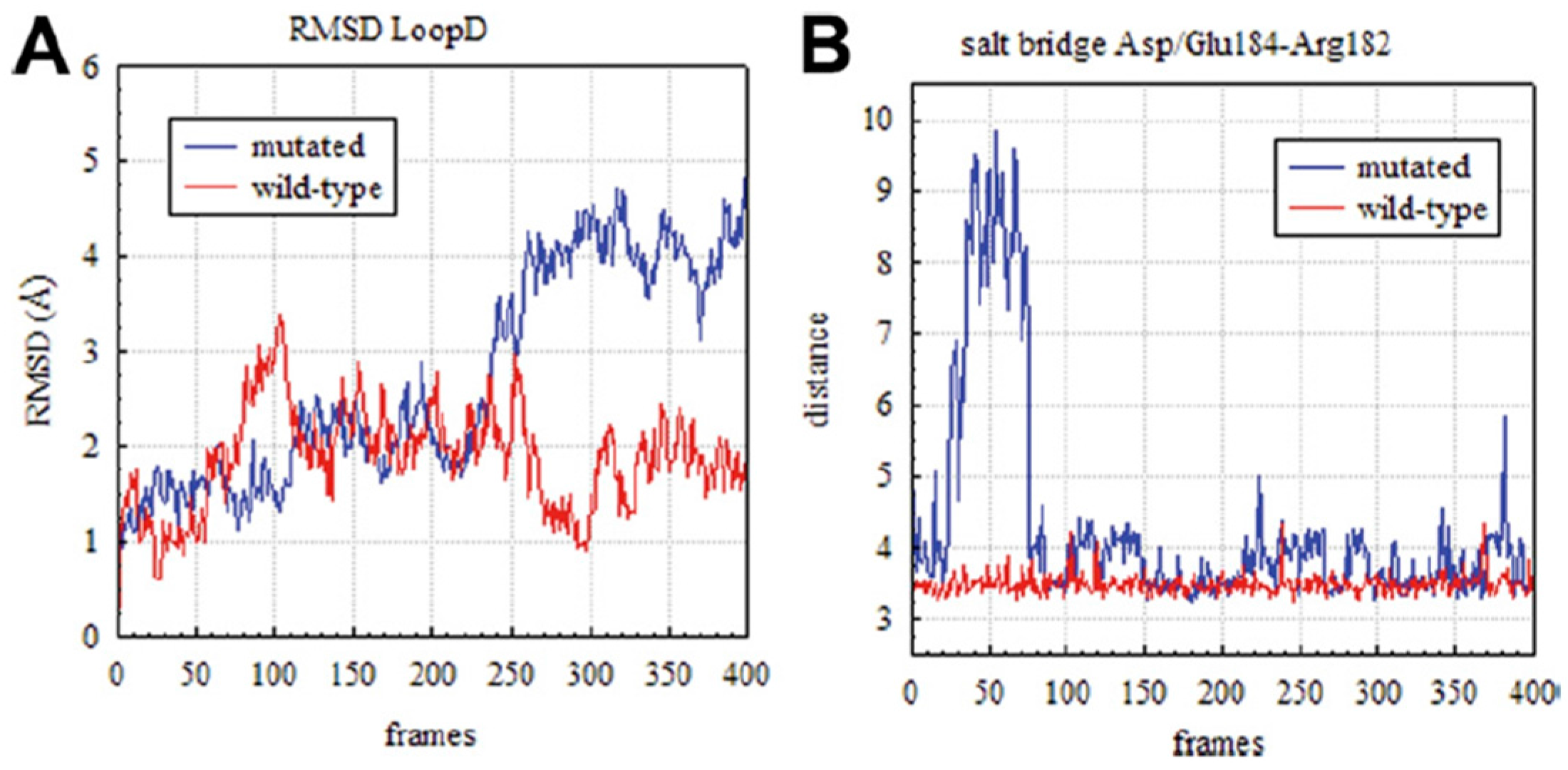
- Pisani, F.; Mola, M.G.; Simone, L.; Rosito, S.; Alberga, D.; Mangiatordi, G.F.; Lattanzi, G.; Nicolotti, O.; Frigeri, A.; Svelto, M.; et al. Identification of a point mutation impairing the binding between aquaporin-4 and neuromyelitis optica autoantibodies. J. Biol. Chem. 2014, 289, 30578–30589. [Google Scholar] [CrossRef] [PubMed]
- Mangiatordi, G.F.; Alberga, D.; Siragusa, L.; Goracci, L.; Lattanzi, G.; Nicolotti, O. Challenging AQP4 druggability for NMO-IgG antibody binding using molecular dynamics and molecular interaction fields. Biochim. Biophys. Acta 2015, 1848, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Grossfield, A.; Zuckerman, D.M. Quantifying uncertainty and sampling quality in biomolecular simulations. Annu. Rep. Comput. Chem. 2009, 5, 23–48. [Google Scholar] [PubMed]
- Keller, T.H.; Pichota, A.; Yin, Z. A practical view of ′druggability′. Curr. Opin. Chem. Biol. 2006, 10, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S.; Anderson, M.O.; Papadopoulos, M.C. Aquaporins: Important but elusive drug targets. Nat. Rev. Drug Discov. 2014, 13, 259–277. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.; Rees, S.; Kalindjian, S.; Philpott, K. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, P.; Barril, X. Understanding and predicting druggability. A high-throughput method for detection of drug binding sites. J. Med. Chem. 2010, 53, 5858–5867. [Google Scholar] [CrossRef] [PubMed]
- Hussein, H.A.; Borrel, A.; Geneix, C.; Petitjean, M.; Regad, L.; Camproux, A.-C. PockDrug-Server: A new web server for predicting pocket druggability on holo and apo proteins. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Edfeldt, F.N.B.; Folmer, R.H.A.; Breeze, A.L. Fragment screening to predict druggability (ligandability) and lead discovery success. Drug Discov. Today 2011, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Kandoi, G.; Acencio, M.L.; Lemke, N. Prediction of druggable proteins using machine learning and systems biology: A mini-review. Syst. Biol. 2015, 6, 366. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.R.; Napoleon, R.L.; Yueh, C.; Whitty, A.; Vajda, S. New frontiers in druggability. J. Med. Chem. 2015, 58, 9063–9088. [Google Scholar] [CrossRef] [PubMed]
- Cruciani, G.; Pastor, M.; Guba, W. VolSurf: A new tool for the pharmacokinetic optimization of lead compounds. Eur. J. Pharm. Sci. 2000, 11 (Suppl. S2), S29–S39. [Google Scholar] [CrossRef]
- Henrich, S.; Salo-Ahen, O.M.H.; Huang, B.; Rippmann, F.F.; Cruciani, G.; Wade, R.C. Computational approaches to identifying and characterizing protein binding sites for ligand design. J. Mol. Recognit. 2010, 23, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Sirci, F.; Goracci, L.; Rodríguez, D.; van Muijlwijk-Koezen, J.; Gutiérrez-de-Terán, H.; Mannhold, R. Ligand-, structure- and pharmacophore-based molecular fingerprints: A case study on adenosine A1, A2A, A2B, and A3 receptor antagonists. J. Comput. Aided Mol. Des. 2012, 26, 1247–1266. [Google Scholar] [CrossRef] [PubMed]
- Goodford, P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.C.; Coleman, R.G.; Smyth, K.T.; Cao, Q.; Soulard, P.; Caffrey, D.R.; Salzberg, A.C.; Huang, E.S. Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 2007, 25, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Wolburg, H.; Wolburg-Buchholz, K.; Fallier-Becker, P.; Noell, S.; Mack, A.F. Structure and functions of aquaporin-4-based orthogonal arrays of particles. Int. Rev. Cell. Mol. Biol. 2011, 287, 1–41. [Google Scholar] [PubMed]
- Crane, J.M.; Verkman, A.S. Reversible, temperature-dependent supramolecular assembly of aquaporin-4 orthogonal arrays in live cell membranes. Biophys. J. 2009, 97, 3010–3018. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Kim, B.; Friesner, R.A.; Berne, B.J. Replica exchange with solute tempering: A method for sampling biological systems in explicit water. Proc. Natl. Acad. Sci. USA 2005, 102, 13749–13754. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Ripphausen, P.; Nisius, B.; Bajorath, J. State-of-the-art in ligand-based virtual screening. Drug Discov. Today 2011, 16, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Nie, A.; An, J.; Huang, Z. Structure-based virtual screening of chemical libraries for drug discovery. Curr. Opin. Chem. Biol. 2006, 10, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Loving, K.A.; Lin, A.; Cheng, A.C. Structure-based druggability assessment of the mammalian structural proteome with inclusion of light protein flexibility. PLoS Comput. Biol. 2014, 10, e1003741. [Google Scholar] [CrossRef] [PubMed]
- Koska, J.; Spassov, V.Z.; Maynard, A.J.; Yan, L.; Austin, N.; Flook, P.K.; Venkatachalam, C.M. Fully automated molecular mechanics based induced fit protein-ligand docking method. J. Chem. Inf. Model. 2008, 48, 1965–1973. [Google Scholar] [CrossRef] [PubMed]
- Bolia, A.; Gerek, Z.N.; Ozkan, S.B. BP-Dock: A flexible docking scheme for exploring protein-ligand interactions based on unbound structures. J. Chem. Inf. Model. 2014, 54, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.I.; Bauman, J.E.; Bologa, C.G.; Buranda, T.; Chigaev, A.; Edwards, B.S.; Jarvik, J.W.; Gresham, H.D.; Haynes, M.K.; Hjelle, B.; et al. Drug repurposing from an academic perspective. Drug Discov. Today Ther. Strateg. 2011, 8, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.L.; Waters, M.F.R.; Lockwood, D.N.J. The role of thalidomide in the management of erythema nodosum leprosum. Lepr. Rev. 2007, 78, 197–215. [Google Scholar] [PubMed]
- Boolell, M.; Allen, M.J.; Ballard, S.A.; Gepi-Attee, S.; Muirhead, G.J.; Naylor, A.M.; Osterloh, I.H.; Gingell, C. Sildenafil: An orally active type 5 cyclic GMP-specific phosphodiesterase inhibitor for the treatment of penile erectile dysfunction. Int. J. Impot. Res. 1996, 8, 47–52. [Google Scholar]
- Tobinick, E.L. The value of drug repositioning in the current pharmaceutical market. Drug News Perspect. 2009, 22, 119–125. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Effect on AQP4 | Reference |
|---|---|---|
| 2-(Nicotinamido)-1,3,4-thiadiazole | Inhibition of water permeability | [36,37] |
| Sumatriptan | Inhibition of water permeability | [37] |
| Rizatriptan | Inhibition of water permeability | [37] |
| Acetazolamide | Inhibition of water permeability | [34] |
| Arbidol | Blockage of NMO-IgG binding | [38] |
| Berbamine | Blockage of NMO-IgG binding | [38] |
| Tamarixetin | Blockage of NMO-IgG binding | [38] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mangiatordi, G.F.; Alberga, D.; Trisciuzzi, D.; Lattanzi, G.; Nicolotti, O. Human Aquaporin-4 and Molecular Modeling: Historical Perspective and View to the Future. Int. J. Mol. Sci. 2016, 17, 1119. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17071119
Mangiatordi GF, Alberga D, Trisciuzzi D, Lattanzi G, Nicolotti O. Human Aquaporin-4 and Molecular Modeling: Historical Perspective and View to the Future. International Journal of Molecular Sciences. 2016; 17(7):1119. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17071119
Chicago/Turabian StyleMangiatordi, Giuseppe Felice, Domenico Alberga, Daniela Trisciuzzi, Gianluca Lattanzi, and Orazio Nicolotti. 2016. "Human Aquaporin-4 and Molecular Modeling: Historical Perspective and View to the Future" International Journal of Molecular Sciences 17, no. 7: 1119. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17071119