
Niemann-Pick Type C2 Protein Mediates Hepatic Stellate Cells Activation by Regulating Free Cholesterol Accumulation

 , and
, and 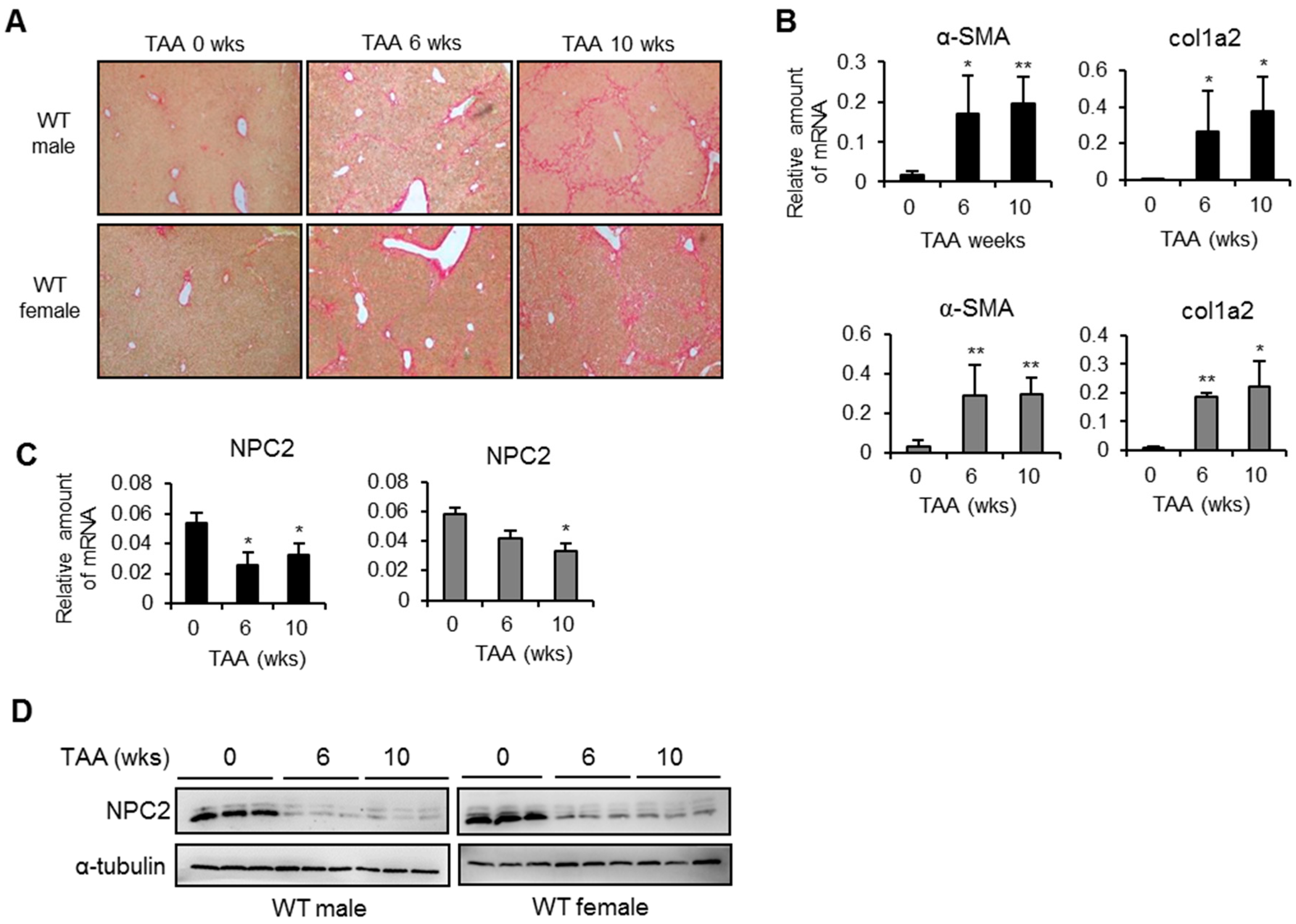{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. NPC2 Is down-Regulated in Liver Fibrosis Tissues
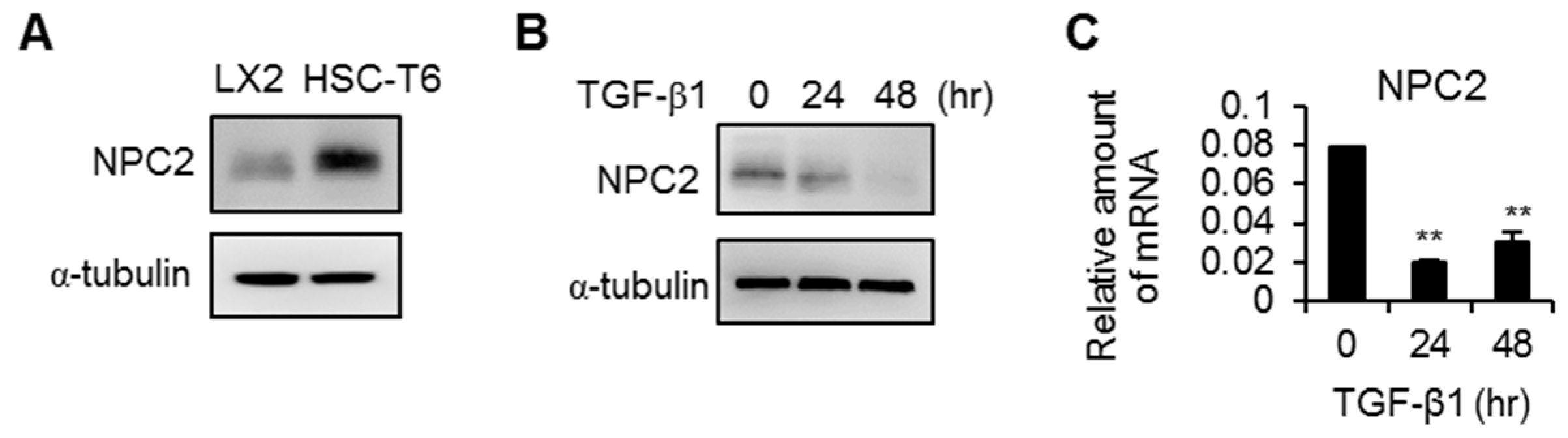
2.2. NPC2 Is Expressed in HSCs but Is down-Regulated in Activated HSCs
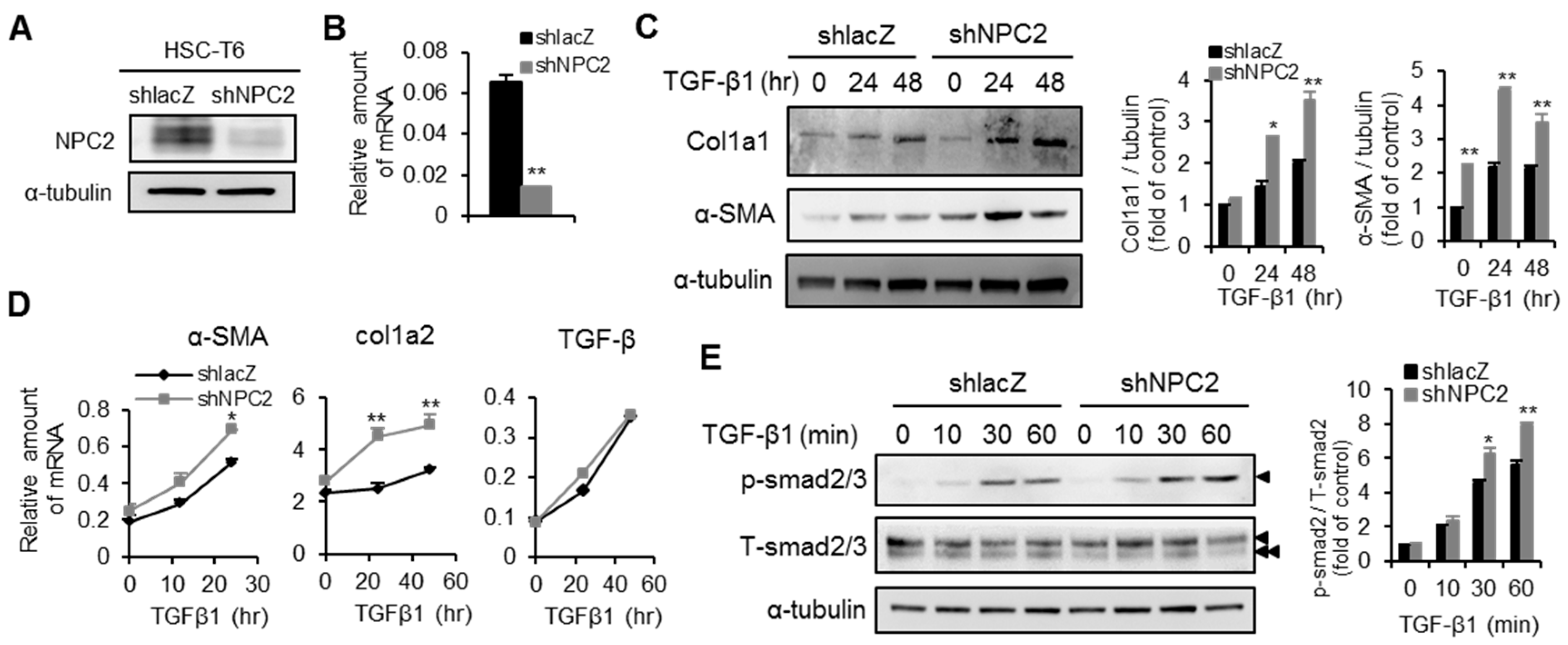
2.3. Knockdown of NPC2 in HSCs Increases TGF-β1-Induced Fibrogenesis
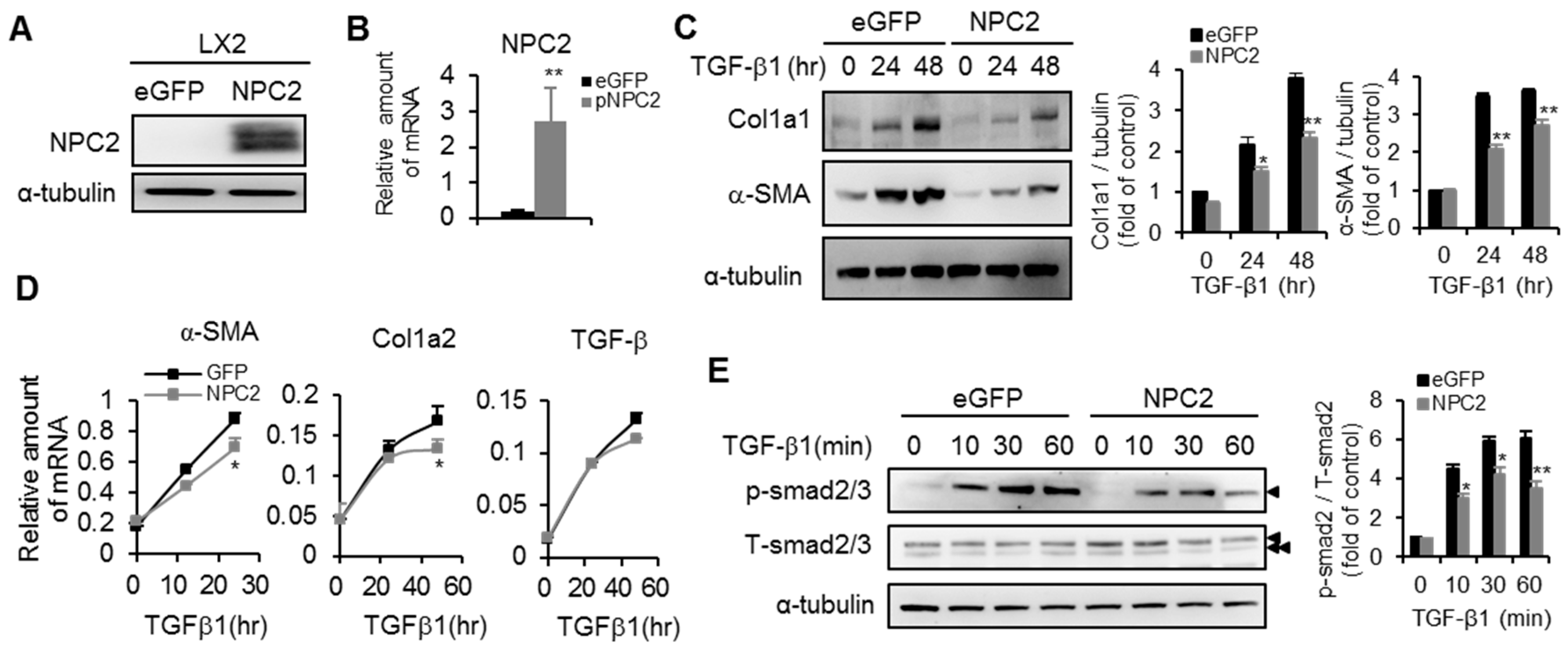
2.4. NPC2 Overexpression Attenuates TGF-β1-Induced Fibrogenesis
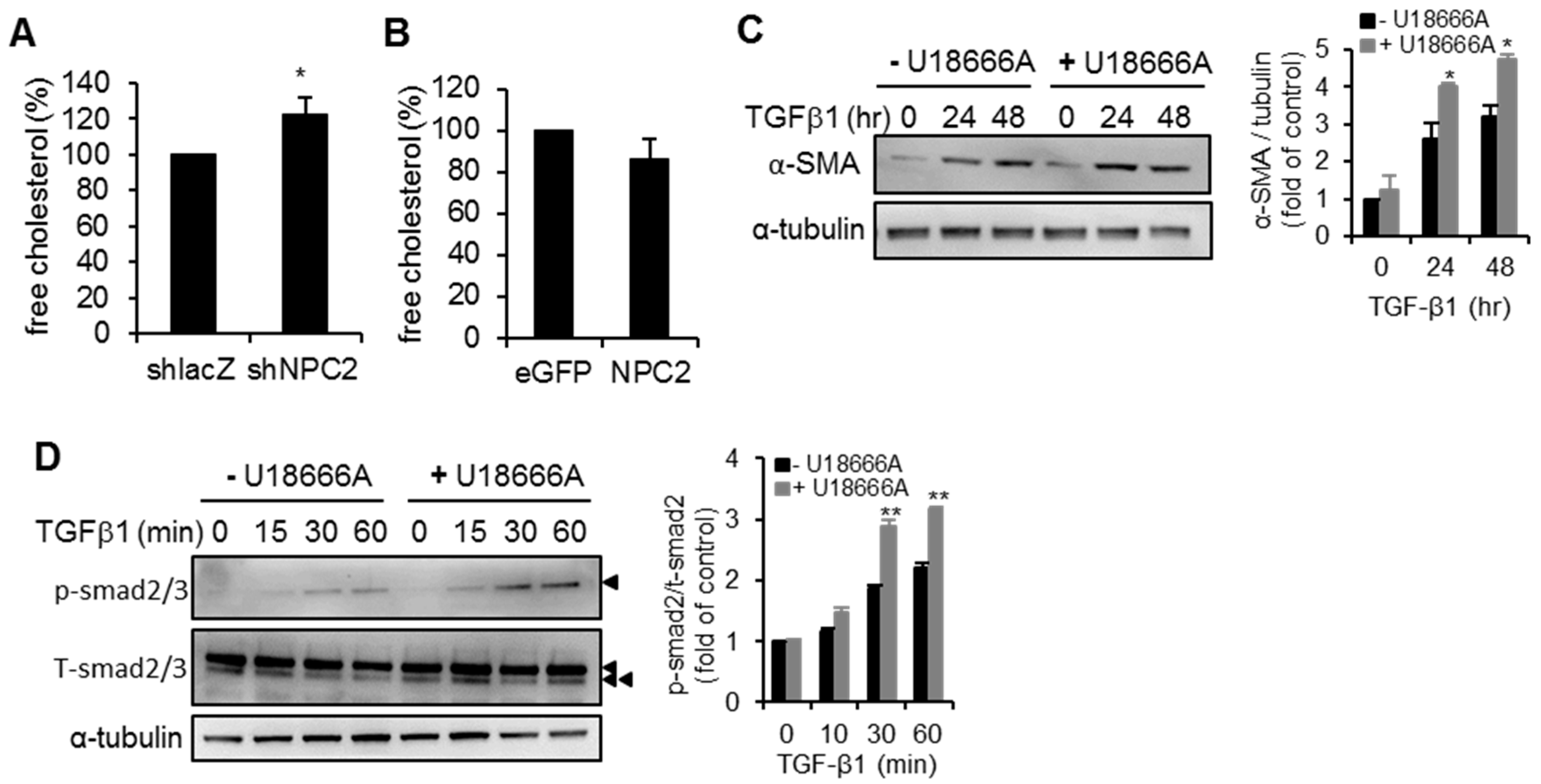
2.5. NPC2 down Regulation Induced Free Cholesterol Accumulation Sensitizes HSCs to TGF-β1 Treatment
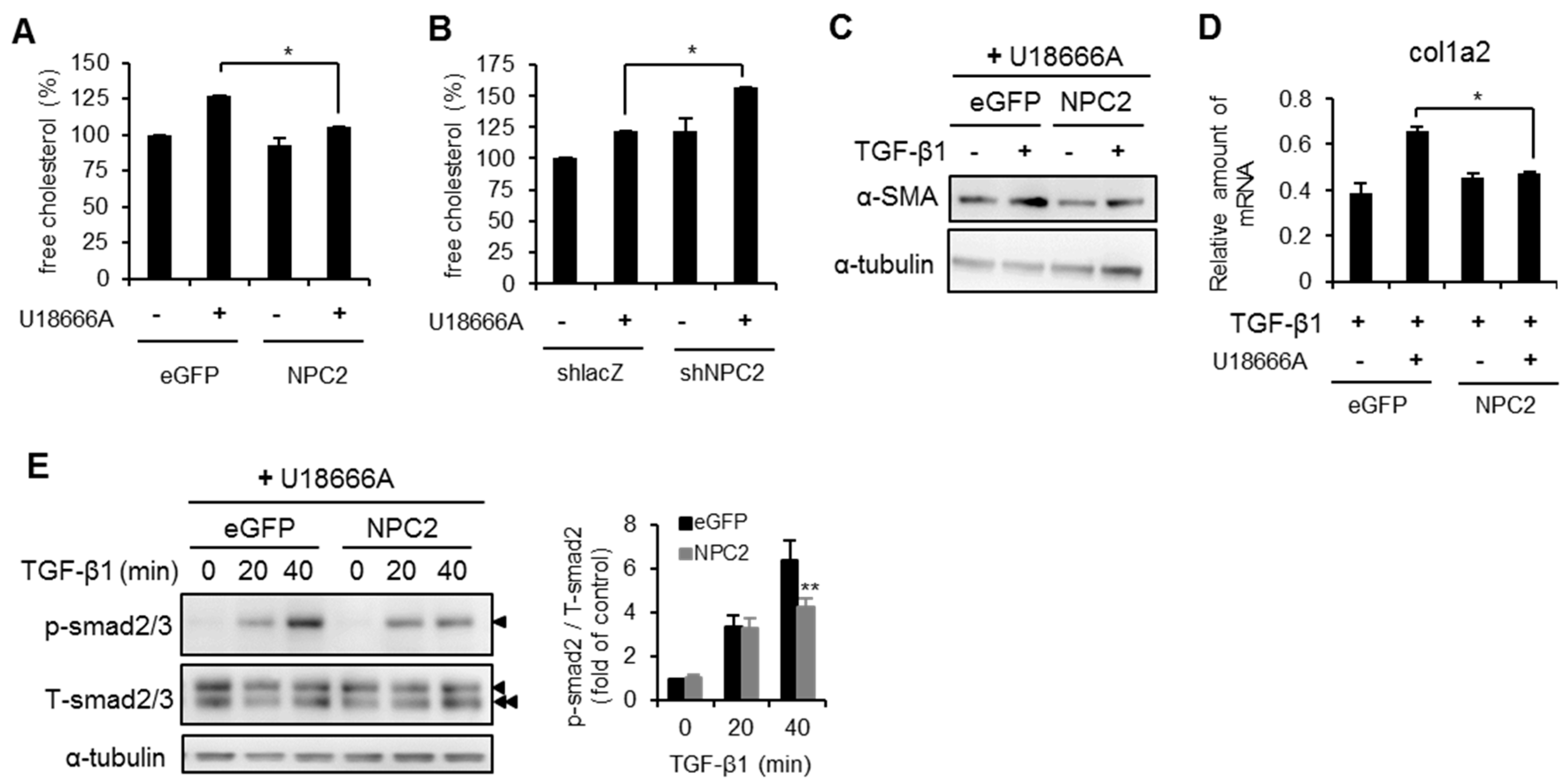
2.6. NPC2 Overexpression Decreases U18666A- and TGF-β1-Induced Free Cholesterol Accumulation and HSCs Activation
3. Discussion
4. Materials and Methods
4.1. Animal Models
4.2. Cell Culture, Plasmids and Lentiviral Infection
4.3. TGF-β1 and U18666A Treatment
4.4. Western Blotting, IHC Staining, and Free Cholesterol Quantification
4.5. Real-Time PCR
4.6. Statistical Analyses
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schuppan, D.; Afdhal, N.H. Liver cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Liver fibrosis—From bench to bedside. J. Hepatol. 2003, 38, S38–S53. [Google Scholar] [CrossRef]
- Iredale, J.P.; Thompson, A.; Henderson, N.C. Extracellular matrix degradation in liver fibrosis: Biochemistry and regulation. Biochim. Biophys. Acta 2013, 1832, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P. Models of liver fibrosis: Exploring the dynamic nature of inflammation and repair in a solid organ. J. Clin. Investig. 2007, 117, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 2011, 6, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.J.; Parola, M. Liver fibrogenic cells. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic fibrosis—Overview. Toxicology 2008, 254, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, A.; Curley, S.A.; Wu, X.; Brown, P.; Hwang, J.P.; Shetty, K.; Yao, Z.X.; He, A.R.; Li, S.; Katz, L.; et al. Hepatic stem cells and transforming growth factor β in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Lee, U.E.; Friedman, S.L. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, Y.; Okazaki, I. Emerging insights into transforming growth factor β Smad signal in hepatic fibrogenesis. Gut 2007, 56, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Fallowfield, J.A. Therapeutic targets in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G709–G715. [Google Scholar] [CrossRef] [PubMed]
- Yata, Y.; Gotwals, P.; Koteliansky, V.; Rockey, D.C. Dose-dependent inhibition of hepatic fibrosis in mice by a TGF-β soluble receptor: Implications for antifibrotic therapy. Hepatology 2002, 35, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T.; Millat, G. Structure and function of the NPC2 protein. Biochim. Biophys. Acta 2004, 1685, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Naureckiene, S.; Sleat, D.E.; Lackland, H.; Fensom, A.; Vanier, M.T.; Wattiaux, R.; Jadot, M.; Lobel, P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science 2000, 290, 2298–2301. [Google Scholar] [CrossRef] [PubMed]
- Storch, J.; Xu, Z. Niemann-Pick C2 (NPC2) and intracellular cholesterol trafficking. Biochim. Biophys. Acta 2009, 1791, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Ko, D.C.; Binkley, J.; Sidow, A.; Scott, M.P. The integrity of a cholesterol-binding pocket in Niemann-Pick C2 protein is necessary to control lysosome cholesterol levels. Proc. Natl. Acad. Sci. USA 2003, 100, 2518–2525. [Google Scholar] [CrossRef] [PubMed]
- Sleat, D.E.; Wiseman, J.A.; El-Banna, M.; Price, S.M.; Verot, L.; Shen, M.M.; Tint, G.S.; Vanier, M.T.; Walkley, S.U.; Lobel, P. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc. Natl. Acad. Sci. USA 2004, 101, 5886–5891. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Maxfield, F.R. Lipid and cholesterol trafficking in NPC. Biochim. Biophys. Acta 2004, 1685, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.J.; Fang, C.C.; Yen, C.H.; Hsu, S.M.; Wang, C.K.; Huang, S.F.; Liang, Y.C.; Lin, Y.Y.; Chu, Y.T.; Arthur Chen, Y.M. Niemann-Pick type C2 protein regulates liver cancer progression via modulating ERK1/2 pathway: Clinicopathological correlations and therapeutical implications. Int. J. Cancer 2015, 137, 1341–1351. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.J.; Lin, M.W.; Yen, C.H.; Lin, Y.T.; Wang, C.K.; Huang, S.F.; Chen, K.H.; Yang, C.P.; Chen, T.L.; Hou, M.F.; et al. Characterization of Niemann-Pick type C2 protein expression in multiple cancers using a novel NPC2 monoclonal antibody. PLoS ONE 2013, 8, e77586. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N.; Morrow, O.B.; Connole, M.L.; Lee, S.P. Association between dietary nutrient composition and the incidence of cirrhosis or liver cancer in the united states population. Hepatology 2009, 50, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Franzen, L.E.; Mathiesen, U.L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Statins in non-alcoholic fatty liver disease and chronically elevated liver enzymes: A histopathological follow-up study. J. Hepatol. 2007, 47, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Teratani, T.; Tomita, K.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Tominaga, S.; Hiroi, S.; Irie, R.; Okada, Y.; et al. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 2012, 142, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Usui, S.; Furuhashi, H.; Kimura, A.; Nishiyama, K.; et al. Acyl-CoA:Cholesterol acyltransferase 1 mediates liver fibrosis by regulating free cholesterol accumulation in hepatic stellate cells. J. Hepatol. 2014, 61, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Okada, Y.; Kurihara, C.; Irie, R.; Yokoyama, H.; et al. Free cholesterol accumulation in hepatic stellate cells: Mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014, 59, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Kirchhoff, C.; Osterhoff, C.; Young, L. Molecular cloning and characterization of HE1, a major secretory protein of the human epididymis. Biol. Reprod. 1996, 54, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Maxfield, F.R.; Tabas, I. Role of cholesterol and lipid organization in disease. Nature 2005, 438, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J.; Mangelsdorf, D.J. The liver X receptor gene team: Potential new players in atherosclerosis. Nat. Med. 2002, 8, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, Y.A. Multidrug permeases and subcellular cholesterol transport. Nat. Rev. Mol. Cell Biol. 2001, 2, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Harrison, K.D.; Miao, R.Q.; Fernandez-Hernando, C.; Suarez, Y.; Davalos, A.; Sessa, W.C. Nogo-B receptor stabilizes Niemann-Pick Type C2 protein and regulates intracellular cholesterol trafficking. Cell Metab. 2009, 10, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Mari, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; Garcia-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and FAS-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Simeonova, P.P.; Gallucci, R.M.; Hulderman, T.; Wilson, R.; Kommineni, C.; Rao, M.; Luster, M.I. The role of tumor necrosis factor-α in liver toxicity, inflammation, and fibrosis induced by carbon tetrachloride. Toxicol. Appl. Pharmacol. 2001, 177, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Bohan, A.; Chen, W.S.; Denson, L.A.; Held, M.A.; Boyer, J.L. Tumor necrosis factor α-dependent up-regulation of LRH-1 and MRP3(ABCC3) reduces liver injury in obstructive cholestasis. J. Biol. Chem. 2003, 278, 36688–36698. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.; Gressner, A.M.; Weiskirchen, R. Immortal hepatic stellate cell lines: Useful tools to study hepatic stellate cell biology and function? J. Cell. Mol. Med. 2007, 11, 704–722. [Google Scholar] [CrossRef] [PubMed]







© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Twu, Y.-C.; Lee, T.-S.; Lin, Y.-L.; Hsu, S.-M.; Wang, Y.-H.; Liao, C.-Y.; Wang, C.-K.; Liang, Y.-C.; Liao, Y.-J. Niemann-Pick Type C2 Protein Mediates Hepatic Stellate Cells Activation by Regulating Free Cholesterol Accumulation. Int. J. Mol. Sci. 2016, 17, 1122. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17071122
Twu Y-C, Lee T-S, Lin Y-L, Hsu S-M, Wang Y-H, Liao C-Y, Wang C-K, Liang Y-C, Liao Y-J. Niemann-Pick Type C2 Protein Mediates Hepatic Stellate Cells Activation by Regulating Free Cholesterol Accumulation. International Journal of Molecular Sciences. 2016; 17(7):1122. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17071122
Chicago/Turabian StyleTwu, Yuh-Ching, Tzong-Shyuan Lee, Yun-Lian Lin, Shih-Ming Hsu, Yuan-Hsi Wang, Chia-Yu Liao, Chung-Kwe Wang, Yu-Chih Liang, and Yi-Jen Liao. 2016. "Niemann-Pick Type C2 Protein Mediates Hepatic Stellate Cells Activation by Regulating Free Cholesterol Accumulation" International Journal of Molecular Sciences 17, no. 7: 1122. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17071122