
Non-Invasive Methods to Monitor Mechanisms of Resistance to Tyrosine Kinase Inhibitors in Non-Small-Cell Lung Cancer: Where Do We Stand?

Abstract
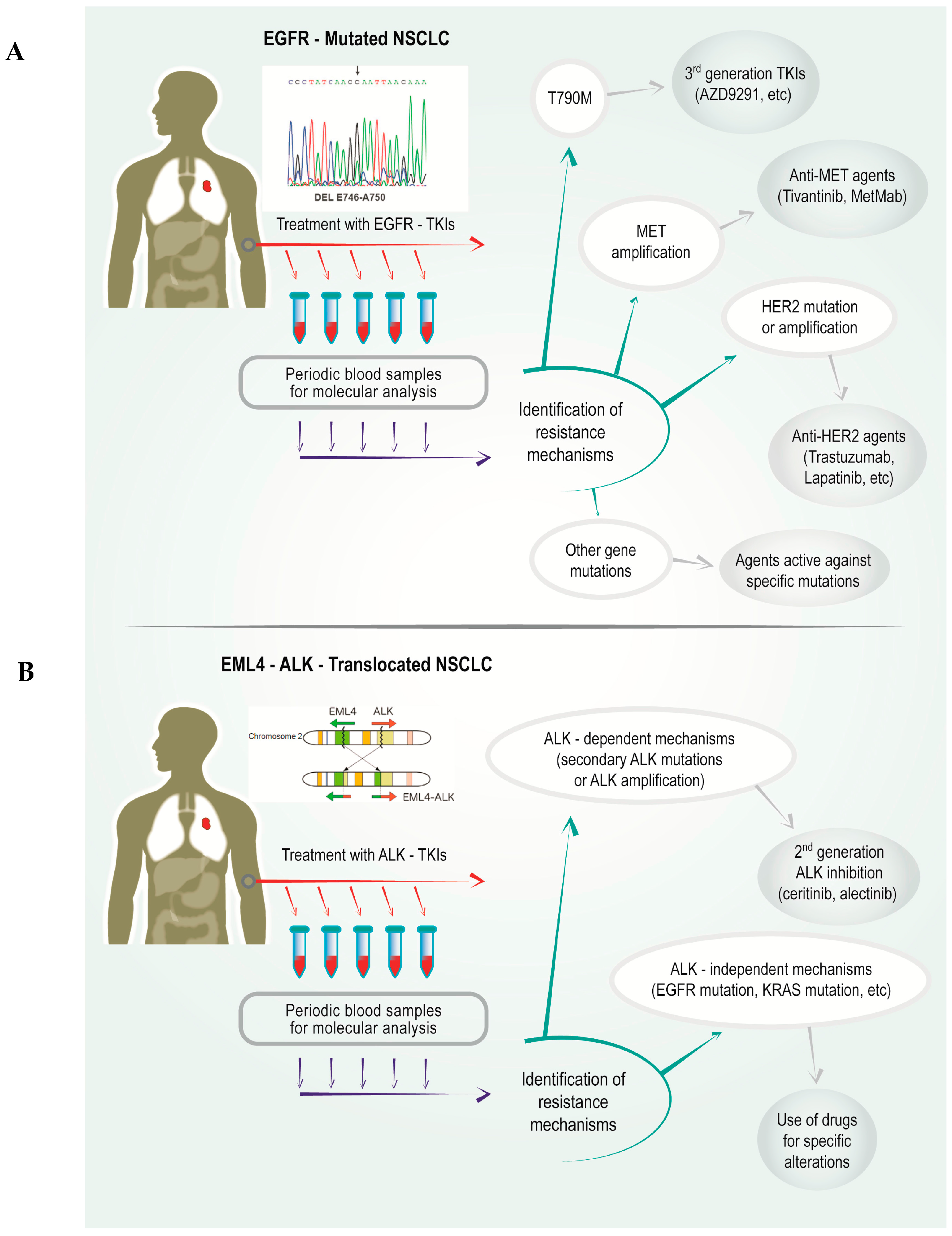
:1. Introduction
2. Acquired Resistance Mechanisms to Epidermal Growth Factor Receptor (EGFR)-Tyrosine Kinase Inhibitors (TKIs) and Possible Strategies for Their Inhibition
2.1. Gatekeeper EGFR T790M Mutation
2.2. Mesenchimal–Epithelial Transition (MET) Amplification
2.3. Insulin-Like Growth Factor-1 Receptor (IGF-1R)
2.4. Human Epidermal Growth Factor Receptor (HER) 2 Mutations
2.5. Activation of Alternative Proliferation Pathways
2.6. Phenotypic Changes
3. Acquired Resistance Mechanisms to Anaplastic Lymphoma Kinase (ALK)-TKIs
3.1. ALK-Dependent Mechanisms
3.1.1. Secondary Mutations at the ALK Gene
3.1.2. ALK Amplification
3.2. ALK-Independent Mechanisms
4. Use of Non-Invasive Methods for the Detection of Resistance Mechanisms to TKIs
4.1. EGFR Mutations
4.2. MET Amplification
4.3. KRAS Mutations
4.4. ALK Alterations
5. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for european patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Yang, J.C.; Wu, Y.L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef]
- Solomon, B.J.; Mok, T.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Janne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Bang, Y.J.; Kwak, E.L.; Iafrate, A.J.; Varella-Garcia, M.; Fox, S.B.; Riely, G.J.; Solomon, B.; Ou, S.H.; Kim, D.W.; et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: Updated results from a phase 1 study. Lancet Oncol. 2012, 13, 1011–1019. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Arcila, M.E.; Sima, C.S.; Riely, G.J.; Chmielecki, J.; Kris, M.G.; Pao, W.; Ladanyi, M.; Miller, V.A. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: Distinct natural history of patients with tumors harboring the T790M mutation. Clin. Cancer Res. 2011, 17, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.; Katakami, N.; Yoshioka, H.; Takeshita, J.; Tanaka, K.; Nanjo, S.; Fujita, S.; Kaji, R.; Imai, Y.; Monden, K.; et al. Rebiopsy of non-small cell lung cancer patients with acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitor: Comparison between T790M mutation-positive and mutation-negative populations. Cancer 2013, 119, 4325–4332. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.A.; Hirsh, V.; Cadranel, J.; Chen, Y.M.; Park, K.; Kim, S.W.; Zhou, C.; Su, W.C.; Wang, M.; Sun, Y.; et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): A phase 2b/3 randomised trial. Lancet Oncol. 2012, 13, 528–538. [Google Scholar] [CrossRef]
- Katakami, N.; Atagi, S.; Goto, K.; Hida, T.; Horai, T.; Inoue, A.; Ichinose, Y.; Koboyashi, K.; Takeda, K.; Kiura, K.; et al. LUX-Lung 4: A phase ii trial of afatinib in patients with advanced non-small-cell lung cancer who progressed during prior treatment with erlotinib, gefitinib, or both. J. Clin. Oncol. 2013, 31, 3335–3341. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Schaefer, G.; Heffron, T.P.; Shao, L.; Ye, X.; Sideris, S.; Malek, S.; Chan, E.; Merchant, M.; La, H.; et al. Noncovalent wild-type-sparing inhibitors of EGFR T790M. Cancer Discov. 2013, 3, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.C.; Salahudeen, A.A.; Wakelee, H.A. Rociletinib, a third generation EGFR tyrosine kinase inhibitor: Current data and future directions. Expert Opin. Pharmacother. 2016, 17, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.A.; Anderton, M.J.; Ashton, S.; Bethel, P.A.; Box, M.; Butterworth, S.; Colclough, N.; Chorley, C.G.; Chuaqui, C.; Cross, D.A.; et al. Structure- and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR). J. Med. Chem. 2013, 56, 7025–7048. [Google Scholar] [CrossRef] [PubMed]
- Janne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Song, A.; Kim, D.W.; Kim, S.; Ahn, Y.O.; Keam, B.; Jeon, Y.K.; Lee, S.H.; Chung, D.H.; Heo, D.S. Mechanisms of acquired resistance to AZD9291: A mutation-selective, irreversible EGFR inhibitor. J. Thorac. Oncol. 2015, 10, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Appleman, L.J. MET signaling pathway: A rational target for cancer therapy. J. Clin. Oncol. 2011, 29, 4837–4838. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ErbB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Scagliotti, G.V.; Novello, S.; Schiller, J.H.; Hirsh, V.; Sequist, L.V.; Soria, J.C.; von Pawel, J.; Schwartz, B.; von Roemeling, R.; Sandler, A.B. Rationale and design of MARQUEE: A phase III, randomized, double-blind study of tivantinib plus erlotinib versus placebo plus erlotinib in previously treated patients with locally advanced or metastatic, nonsquamous, non-small-cell lung cancer. Clin. Lung Cancer 2012, 13, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Aoyama, A.; Yamori, T.; Qi, J.; Oh-hara, T.; Song, Y.; Engelman, J.A.; Fujita, N. Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition. Cancer Res. 2013, 73, 3087–3096. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Yang, R.; Zheng, Z.; Romero, M.; Ross, J.; Bou-Reslan, H.; Carano, R.A.; Kasman, I.; Mai, E.; Young, J.; et al. MetMAb, the one-armed 5D5 anti-c-MET antibody, inhibits orthotopic pancreatic tumor growth and improves survival. Cancer Res. 2008, 68, 4360–4368. [Google Scholar] [CrossRef] [PubMed]
- Spigel, D.; Edelman, M.; O’Byrne, K.; Paz-Ares, L.; Shames, D.; Yu, W.; Paton, V.; Mok, T. Onartuzumab Plus Erlotinib versus Erlotinib in Previously Treated Stage IIIb or IV NSCLC: Results from the Pivotal Phase III Randomized, Multicenter, Placebo-Controlled METLung (OAM49-71g) Global Trial. In Proceedings of the 2014 American Society of Clinical Oncology Annual Meeting, Chicago, IL, USA, 30 May–3 June 2014. No. 15.
- Peled, N.; Wynes, M.W.; Ikeda, N.; Ohira, T.; Yoshida, K.; Qian, J.; Ilouze, M.; Brenner, R.; Kato, Y.; Mascaux, C.; et al. Insulin-like growth factor-1 receptor (IGF-1R) as a biomarker for resistance to the tyrosine kinase inhibitor gefitinib in non-small cell lung cancer. Cell. Oncol. 2013, 36, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Woo, J.K.; Kim, E.S.; Hong, W.K.; Lee, H.Y. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res. 2006, 66, 10100–10111. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Rho, J.K.; Jeon, B.S.; Choi, S.J.; Park, S.C.; Lee, S.S.; Kim, H.R.; Kim, C.H.; Lee, J.C. Combined inhibition of IGFR enhances the effects of gefitinib in H1650: A lung cancer cell line with EGFR mutation and primary resistance to EGFR-TK inhibitors. Cancer Chemother. Pharmacol. 2010, 66, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Yao, E.; Shen, R.; Goel, A.; Arcila, M.; Teruya-Feldstein, J.; Zakowski, M.F.; Frankel, S.; Peifer, M.; Thomas, R.K.; et al. High expression levels of total IGF-1R and sensitivity of NSCLC cells in vitro to an anti-IGF-1R antibody (R1507). PLoS ONE 2009, 4, e7273. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, H.; Takahashi, T.; Nomura, M.; Majmudar, K.; Suzuki, M.; Lee, H.; Wistuba, I.I.; Fong, K.M.; Toyooka, S.; Shimizu, N.; et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005, 65, 1642–1646. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.E.; Narasanna, A.; Perez-Torres, M.; Xiang, B.; Wu, F.Y.; Yang, S.; Carpenter, G.; Gazdar, A.F.; Muthuswamy, S.K.; Arteaga, C.L. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 2006, 10, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Takezawa, K.; Pirazzoli, V.; Arcila, M.E.; Nebhan, C.A.; Song, X.; de Stanchina, E.; Ohashi, K.; Janjigian, Y.Y.; Spitzler, P.J.; Melnick, M.A.; et al. HER2 amplification: A potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012, 2, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Cappuzzo, F.; Bemis, L.; Varella-Garcia, M. HER2 mutation and response to trastuzumab therapy in non-small-cell lung cancer. N. Engl. J. Med. 2006, 354, 2619–2621. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Engelman, J.A.; Cantley, L.C. Targeting the PI3K signaling pathway in cancer. Curr. Opin. Genet. Dev. 2010, 20, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Lee, K.K.; Zhang, L.; Gerton, J.L. Stimulation of mTORC1 with l-leucine rescues defects associated with Roberts syndrome. PLoS Genet. 2013, 9, e1003857. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Gogol, M.; Gaudenz, K.; Gerton, J.L. Improved transcription and translation with l-leucine stimulation of mTORC1 in Roberts syndrome. BMC Genom. 2016, 5, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Sowa, N.; Cardenas, M.E.; Gerton, J.L. l-Leucine partially rescues translational and developmental defects associated with zebrafish models of Cornelia de Lange syndrome. Hum. Mol. Genet. 2015, 24, 1540–1555. [Google Scholar] [CrossRef] [PubMed]
- Sos, M.L.; Koker, M.; Weir, B.A.; Heynck, S.; Rabinovsky, R.; Zander, T.; Seeger, J.M.; Weiss, J.; Fischer, F.; Frommolt, P.; et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009, 69, 3256–3261. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Mukohara, T.; Zejnullahu, K.; Lifshits, E.; Borras, A.M.; Gale, C.M.; Naumov, G.N.; Yeap, B.Y.; Jarrell, E.; Sun, J.; et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J. Clin. Investig. 2006, 116, 2695–2706. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [PubMed]
- Gadgeel, S.M.; Wozniak, A. Preclinical rationale for PI3K/Akt/mTOR pathway inhibitors as therapy for epidermal growth factor receptor inhibitor-resistant non-small-cell lung cancer. Clin. Lung Cancer 2013, 14, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Morinaga, R.; Okamoto, I.; Furuta, K.; Kawano, Y.; Sekijima, M.; Dote, K.; Satou, T.; Nishio, K.; Fukuoka, M.; Nakagawa, K. Sequential occurrence of non-small cell and small cell lung cancer with the same EGFR mutation. Lung Cancer 2007, 58, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Witta, S.E.; Jotte, R.M.; Konduri, K.; Neubauer, M.A.; Spira, A.I.; Ruxer, R.L.; Varella-Garcia, M.; Bunn, P.A., Jr.; Hirsch, F.R. Randomized phase II trial of erlotinib with and without entinostat in patients with advanced non-small-cell lung cancer who progressed on prior chemotherapy. J. Clin. Oncol. 2012, 30, 2248–2255. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Januario, T.; Eberhard, D.A.; Cavet, G.; Zhu, W.; Fu, L.; Pham, T.Q.; Soriano, R.; Stinson, J.; Seshagiri, S.; et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin. Cancer Res. 2005, 11, 8686–8698. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.; Buck, E.; Petti, F.; Griffin, G.; Brown, E.; Ramnarine, N.; Iwata, K.K.; Gibson, N.; Haley, J.D. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005, 65, 9455–9462. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.L.; Soda, M.; Yamashita, Y.; Ueno, T.; Takashima, J.; Nakajima, T.; Yatabe, Y.; Takeuchi, K.; Hamada, T.; Haruta, H.; et al. EML4–ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med. 2010, 363, 1734–1739. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Pilling, A.B.; Aisner, D.L.; Kutateladze, T.G.; Le, A.T.; Weickhardt, A.J.; Kondo, K.L.; Linderman, D.J.; Heasley, L.E.; Franklin, W.A.; et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 1472–1482. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Koivunen, J.; Ogino, A.; Yanagita, M.; Nikiforow, S.; Zheng, W.; Lathan, C.; Marcoux, J.P.; Du, J.; Okuda, K.; et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011, 71, 6051–6060. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Shaw, A.T.; Khan, T.M.; Mino-Kenudson, M.; Solomon, B.J.; Halmos, B.; Jessop, N.A.; Wain, J.C.; Yeo, A.T.; Benes, C.; et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci. Transl. Med. 2012, 4, 120ra17. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Katayama, R.; McTigue, M.; Liu, W.; Deng, Y.L.; Brooun, A.; Friboulet, L.; Huang, D.; Falk, M.D.; Timofeevski, S.; et al. Acquired resistance to crizotinib from a mutation in CD74–ROS1. N. Engl. J. Med. 2013, 368, 2395–2401. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Rodig, S.J.; Chirieac, L.R.; Janne, P.A. The biology and treatment of EML4–ALK non-small cell lung cancer. Eur. J. Cancer 2010, 46, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Soda, M.; Sakata, S.; Sugawara, E.; Hatano, S.; Asaka, R.; Nakajima, T.; Mano, H.; Takeuchi, K. KLC1-ALK: A novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue only. PLoS ONE 2012, 7, e31323. [Google Scholar] [CrossRef] [PubMed]
- Boland, J.M.; Jang, J.S.; Li, J.; Lee, A.M.; Wampfler, J.A.; Erickson-Johnson, M.R.; Soares, I.; Yang, P.; Jen, J.; Oliveira, A.M.; et al. MET and EGFR mutations identified in ALK-rearranged pulmonary adenocarcinoma: Molecular analysis of 25 ALK-positive cases. J. Thorac. Oncol. 2013, 8, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Esfahani, K.; Agulnik, J.S.; Cohen, V. A systemic review of resistance mechanisms and ongoing clinical trials in ALK-rearranged non-small cell lung cancer. Front. Oncol. 2014, 4, 174. [Google Scholar] [CrossRef] [PubMed]
- Tanizaki, J.; Okamoto, I.; Okabe, T.; Sakai, K.; Tanaka, K.; Hayashi, H.; Kaneda, H.; Takezawa, K.; Kuwata, K.; Yamaguchi, H.; et al. Activation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4–ALK-positive non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 6219–6226. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Zhang, L.; Cheng, Y.; Patel, R.; Wu, H.; Zhang, Y.; Wang, M.; Ji, S.; Belani, C.P.; Yang, J.M.; et al. Induction of autophagy contributes to crizotinib resistance in ALK-positive lung cancer. Cancer Biol. Ther. 2014, 15, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Salgia, R. Mutation testing for directing upfront targeted therapy and post-progression combination therapy strategies in lung adenocarcinoma. Expert Rev. Mol. Diagn. 2016, 16, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Qing, X.; Xiumin, W.; Yali, B.; Chi, S.; Bak, S.H.; Lee, H.Y.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; et al. Longitudinal monitoring of EGFR mutations in plasma predicts outcomes of NSCLC patients treated with EGFR TKIs: Korean lung cancer consortium (KLCC-12-02). Oncotarget 2016, 7, 6984–6993. [Google Scholar] [PubMed]
- Sueoka-Aragane, N.; Katakami, N.; Satouchi, M.; Yokota, S.; Aoe, K.; Iwanaga, K.; Otsuka, K.; Morita, S.; Kimura, S.; Negoro, S.; et al. Monitoring EGFR T790M with Plasma DNA from lung cancer patients in a prospective observational study. Cancer Sci. 2016, 107, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Seki, Y.; Fujiwara, Y.; Kohno, T.; Takai, E.; Sunami, K.; Goto, Y.; Horinouchi, H.; Kanda, S.; Nokihara, H.; Watanabe, S.; et al. Picoliter-droplet digital polymerase chain reaction-based analysis of cell-free plasma DNA to assess EGFR mutations in lung adenocarcinoma that confer resistance to tyrosine-kinase inhibitors. Oncologist 2016, 21, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H.; Azuma, K.; Sakai, K.; Kawahara, A.; Yamada, K.; Tokito, T.; Okamoto, I.; Nishio, K.; Hoshino, T. Digital PCR analysis of plasma cell-free DNA for non-invasive detection of drug resistance mechanisms in EGFR mutant NSCLC: Correlation with paired tumor samples. Oncotarget 2015, 6, 30850–30858. [Google Scholar] [PubMed]
- Sorensen, B.S.; Wu, L.; Wei, W.; Tsai, J.; Weber, B.; Nexo, E.; Meldgaard, P. Monitoring of epidermal growth factor receptor tyrosine kinase inhibitor-sensitizing and resistance mutations in the plasma DNA of patients with advanced non-small cell lung cancer during treatment with erlotinib. Cancer 2014, 120, 3896–3901. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, R.; Wang, S.; Zhong, J.; Wu, M.; Zhao, J.; Duan, J.; Zhuo, M.; An, T.; Wang, Y.; et al. Quantification and dynamic monitoring of EGFR T790M in plasma cell-free DNA by digital PCR for prognosis of EGFR-TKI treatment in advanced NSCLC. PLoS ONE 2014, 9, e110780. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Ye, X.; Zhang, M.Z.; Sun, Y.; Wang, J.Y.; Ni, J.; Zhang, H.P.; Zhang, L.; Luo, J.; Zhang, J.; et al. Plasma EGFR T790M ctDNA status is associated with clinical outcome in advanced NSCLC patients with acquired EGFR-TKI resistance. Sci. Rep. 2016, 6, 20913. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Horiike, A.; Irwin, D.L.; Kudo, K.; Fujita, Y.; Tanimoto, A.; Sakatani, T.; Saito, R.; Kaburaki, K.; Yanagitani, N.; et al. Detection of epidermal growth factor receptor T790M mutation in plasma DNA from patients refractory to epidermal growth factor receptor tyrosine kinase inhibitor. Cancer Sci. 2013, 104, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, T.K.; Sequist, L.V.; Heymach, J.V.; Riely, G.J.; Janne, P.A.; Koch, W.H.; Sullivan, J.P.; Fox, D.B.; Maher, R.; Muzikansky, A.; et al. Detection of T790M, the acquired resistance EGFR mutation, by tumor biopsy versus noninvasive blood-based analyses. Clin. Cancer Res. 2016, 22, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Marcq, M.; Vallee, A.; Bizieux, A.; Denis, M.G. Detection of EGFR mutations in the plasma of patients with lung adenocarcinoma for real-time monitoring of therapeutic response to tyrosine kinase inhibitors? J. Thorac. Oncol. 2014, 9, e49–e50. [Google Scholar] [CrossRef] [PubMed]
- Del Re, M.; Tiseo, M.; Bordi, P.; D’Incecco, A.; Camerini, A.; Petrini, I.; Lucchesi, M.; Inno, A.; Spada, D.; Vasile, E.; et al. Contribution of KRAS mutations and c.2369C > T (p.T790M) EGFR to acquired resistance to EGFR-TKIs in EGFR mutant NSCLC: A study on circulating tumor DNA. Oncotarget 2016. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Arcila, M.E.; Chmielecki, J.; Ladanyi, M.; Miller, V.A.; Pao, W. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin. Cancer Res. 2011, 17, 5530–5537. [Google Scholar] [CrossRef] [PubMed]
- Karlovich, C.; Goldman, J.W.; Sun, J.M.; Mann, E.; Sequist, L.V.; Konopa, K.; Wen, W.; Angenendt, P.; Horn, L.; Spigel, D.; et al. Assessment of EGFR mutation status in matched plasma and tumor tissue of NSCLC patients from a phase I study of rociletinib (CO-1686). Clin. Cancer Res. 2016, 22, 2386–2395. [Google Scholar] [CrossRef] [PubMed]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, A.; Palma, J.F.; Felicioni, L.; de Pas, T.M.; Chiari, R.; Del Grammastro, M.; Filice, G.; Ludovini, V.; Brandes, A.A.; Chella, A.; et al. Early prediction of response to tyrosine kinase inhibitors by quantification of EGFR mutations in plasma of NSCLC patients. J. Thorac. Oncol. 2015, 10, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.S.; Yang, T.Y.; Tsai, C.R.; Chen, K.C.; Hsu, K.H.; Tsai, M.H.; Yu, S.L.; Su, K.Y.; Chen, J.J.; Chang, G.C. Dynamic plasma EGFR mutation status as a predictor of EGFR-TKI efficacy in patients with EGFR-mutant lung adenocarcinoma. J. Thorac. Oncol. 2015, 10, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Imamura, F.; Uchida, J.; Kukita, Y.; Kumagai, T.; Nishino, K.; Inoue, T.; Kimura, M.; Oba, S.; Kato, K. Monitoring of treatment responses and clonal evolution of tumor cells by circulating tumor DNA of heterogeneous mutant EGFR genes in lung cancer. Lung Cancer 2016, 94, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Brugger, W.; Triller, N.; Blasinska-Morawiec, M.; Curescu, S.; Sakalauskas, R.; Manikhas, G.M.; Mazieres, J.; Whittom, R.; Ward, C.; Mayne, K.; et al. Prospective molecular marker analyses of EGFR and KRAS from a randomized, placebo-controlled study of erlotinib maintenance therapy in advanced non-small-cell lung cancer. J. Clin. Oncol. 2011, 29, 4113–4120. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Shepherd, F.A.; Hirsh, V.; Mok, T.; Socinski, M.A.; Gervais, R.; Liao, M.L.; Bischoff, H.; Reck, M.; Sellers, M.V.; et al. Molecular predictors of outcome with gefitinib and docetaxel in previously treated non-small-cell lung cancer: Data from the randomized phase III INTEREST trial. J. Clin. Oncol. 2010, 28, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Ulivi, P.; Delmonte, A.; Chiadini, E.; Calistri, D.; Papi, M.; Mariotti, M.; Verlicchi, A.; Ragazzini, A.; Capelli, L.; Gamboni, A.; et al. Gene mutation analysis in EGFR wild type NSCLC responsive to erlotinib: Are there features to guide patient selection? Int. J. Mol. Sci. 2014, 16, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.Q.; da Cunha Santos, G.; Ding, K.; Sakurada, A.; Cutz, J.C.; Liu, N.; Zhang, T.; Marrano, P.; Whitehead, M.; Squire, J.A.; et al. Role of KRAS and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada Clinical Trials Group Study BR.21. J. Clin. Oncol. 2008, 26, 4268–4275. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.P.; Heigener, D.; Schott-von-Romer, K.; Gutz, S.; Laack, E.; Digel, W.; Guschall, W.R.; Franke, A.; Bodenstein, H.; Schmidtgen, C.; et al. Epidermal growth factor receptor-related tumor markers and clinical outcomes with erlotinib in non-small cell lung cancer: An analysis of patients from German centers in the TRUST study. J. Thorac. Oncol. 2008, 3, 1446–1453. [Google Scholar] [CrossRef] [PubMed]
- Ilie, M.; Long, E.; Butori, C.; Hofman, V.; Coelle, C.; Mauro, V.; Zahaf, K.; Marquette, C.H.; Mouroux, J.; Paterlini-Brechot, P.; et al. ALK-gene rearrangement: A comparative analysis on circulating tumour cells and tumour tissue from patients with lung adenocarcinoma. Ann. Oncol. 2012, 23, 2907–2913. [Google Scholar] [CrossRef] [PubMed]
- Pailler, E.; Adam, J.; Barthelemy, A.; Oulhen, M.; Auger, N.; Valent, A.; Borget, I.; Planchard, D.; Taylor, M.; Andre, F.; et al. Detection of circulating tumor cells harboring a unique ALK rearrangement in ALK-positive non-small-cell lung cancer. J. Clin. Oncol. 2013, 31, 2273–2281. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Xu, D.; Wang, Z.; Xiang, X.; Tang, B.; Li, S.; Hou, M.; Zhang, Y.; Chen, J.F.; Lin, M.; et al. Detecting ALK-rearrangement of CTC enriched by nanovelcro chip in advanced NSCLC patients. Oncotarget 2016. [Google Scholar] [CrossRef]
- Tan, C.L.; Lim, T.H.; Lim, T.K.; Tan, D.S.; Chua, Y.W.; Ang, M.K.; Pang, B.; Lim, C.T.; Takano, A.; Lim, A.S.; et al. Concordance of anaplastic lymphoma kinase (ALK) gene rearrangements between circulating tumor cells and tumor in non-small cell lung cancer. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.J.; Karachaliou, N.; Berenguer, J.; Gimenez-Capitan, A.; Schellen, P.; Teixido, C.; Tannous, J.; Kuiper, J.L.; Drees, E.; Grabowska, M.; et al. Rearranged EML4–ALK fusion transcripts sequester in circulating blood platelets and enable blood-based crizotinib response monitoring in non-small-cell lung cancer. Oncotarget 2016, 7, 1066–1075. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Author (Reference) | Year | Methodology | No. of Patients * | T790M Detected in Plasma (%) | Concordance between T790M in Plasma/CTCs and Tumor Re-Biopsy (%) | T790M Identified Prior to Clinical Disease Progression |
|---|---|---|---|---|---|---|
| Lee et al. [59] | 2016 | ddPCR | 79 | 29 | NE | Yes |
| Sueoka-Aragane et al. [60] | 2016 | MBP-QP | 87 | 40 | 50 | Yes |
| Seki et al. [61] | 2016 | Picoliter ddPCR | 35 | 44 | 80 | NE |
| Ishii et al. [62] | 2015 | ddPCR | 18 | 56 | 83 | NE |
| Sorensen et al. [63] | 2014 | Cobas EGFR blood test | 23 | 39 | NE | Yes |
| Wang et al. [64] | 2014 | DHPLC | 135 | 43 | NE | NE |
| Zheng et al. [65] | 2016 | ddPCR | 117 | 47 | NE | Yes |
| Sakai et al. [66] | 2013 | SABER-Sequenom MassARRAY | 75 | 28 | NE | NE |
| Sunderasan et al. [67] | 2016 | CTC-enriched PCR method | 28 | 50 | 57/74 † | NE |
| fctDNA-Cobas EGFR mut test | 32 | 50 | 60/61 † | NE | ||
| CTCs and fctDNA together | 23 | 100 | 65/69 † | NE | ||
| Marcq et al. [68] | 2014 | ARMS | 2 | 50 | NE | NE |
| Del Re et al. [69] | 2016 | ddPCR | 33 | 33 | 62.5 | NE |
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ulivi, P. Non-Invasive Methods to Monitor Mechanisms of Resistance to Tyrosine Kinase Inhibitors in Non-Small-Cell Lung Cancer: Where Do We Stand? Int. J. Mol. Sci. 2016, 17, 1186. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17071186
Ulivi P. Non-Invasive Methods to Monitor Mechanisms of Resistance to Tyrosine Kinase Inhibitors in Non-Small-Cell Lung Cancer: Where Do We Stand? International Journal of Molecular Sciences. 2016; 17(7):1186. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17071186
Chicago/Turabian StyleUlivi, Paola. 2016. "Non-Invasive Methods to Monitor Mechanisms of Resistance to Tyrosine Kinase Inhibitors in Non-Small-Cell Lung Cancer: Where Do We Stand?" International Journal of Molecular Sciences 17, no. 7: 1186. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17071186