
Molecular Mechanisms of Cutaneous Inflammatory Disorder: Atopic Dermatitis


Abstract
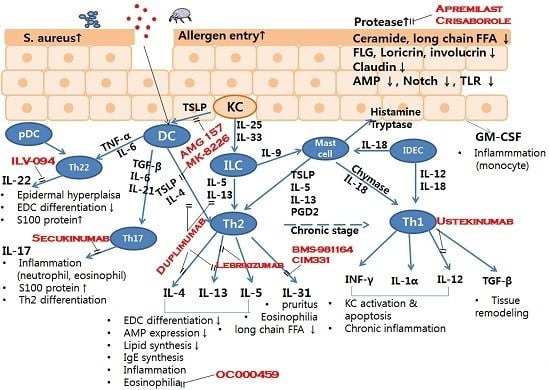
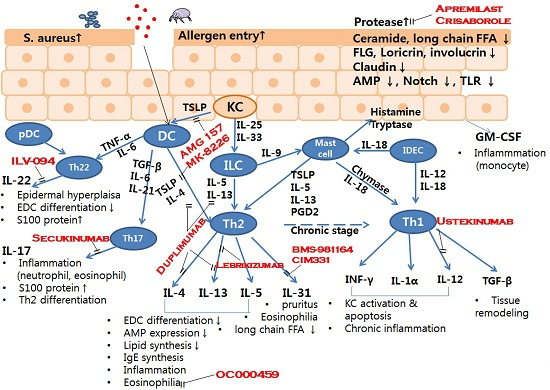
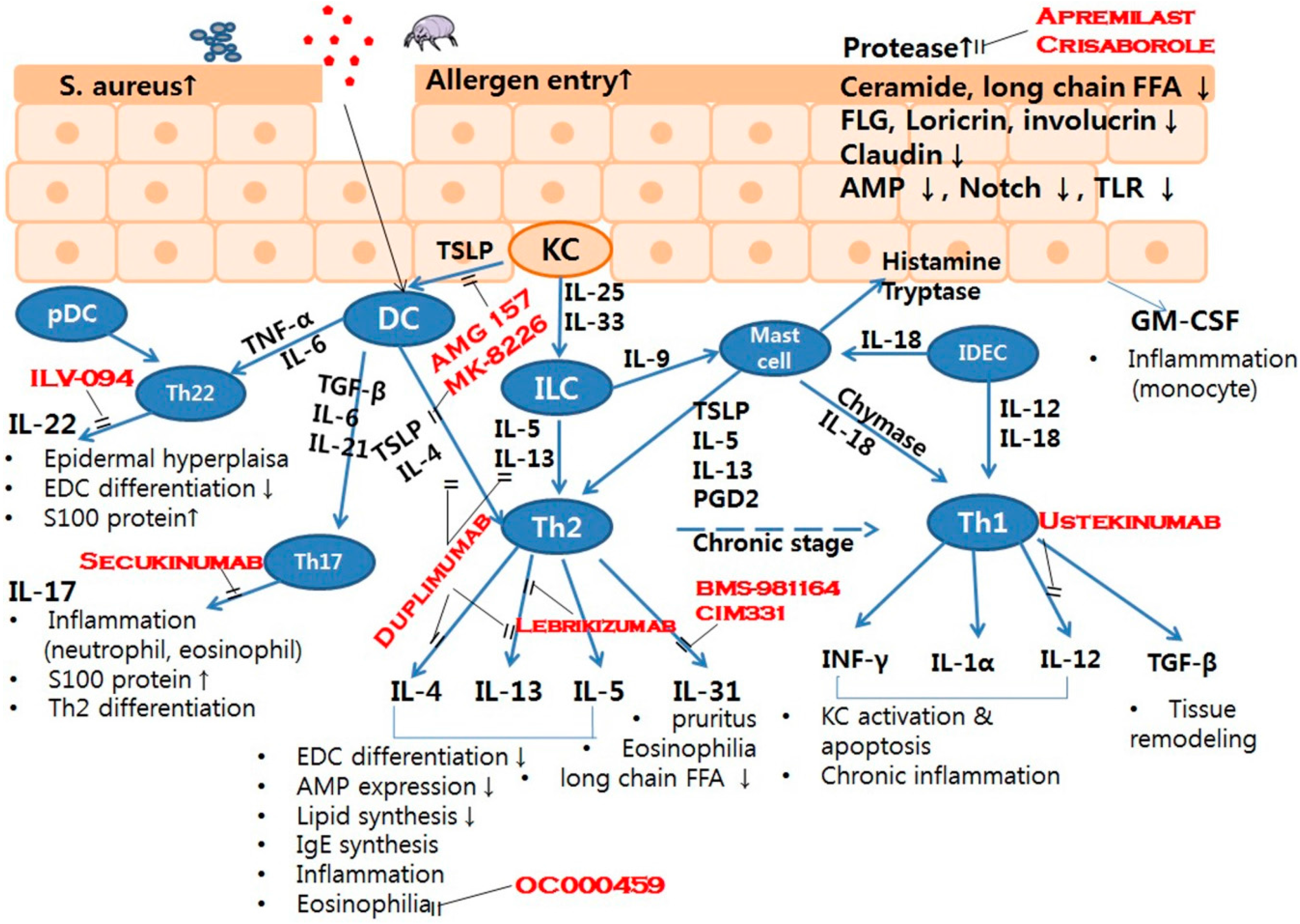
:
1. Introduction
2. Genetics
3. Epigenetics
4. Barrier Dysfunction
5. Immunological Abnormalities
5.1. Innate Immunity
5.2. Adaptive Immunity
5.2.1. Th1/Th2 Imbalance
5.2.2. Th17 Cells
5.2.3. Th22 T Cells
5.2.4. IL-18
5.2.5. IL-31
5.2.6. B cells
5.2.7. Dendritic Cells (DCs)
5.2.8. Chemokines
5.2.9. Mast Cells
5.2.10. Eosinophils and Basophils
5.2.11. Innate Lymphoid Cells
5.3. Signal Pathways
5.3.1. GATA-3
5.3.2. Notch Signaling
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AD | Atopic dermatitis |
| AMPs | Antimicrobial peptides |
| CARD | Caspase recruitment domain |
| CCL | C-C motif chemokine ligand |
| CCR | C-C motif chemokine receptor |
| CLDN1 | Claudin-1 |
| CE | Cornified envelope |
| CXCL | C-X-C motif chemokine ligand |
| DCs | Dendritic cells |
| EDC | Epidermal differentiation complex |
| ERK | Extracellular signal-regulated kinase |
| FcεRI | High affinity IgE receptor |
| FFA | Free fatty acid |
| FLG | Filaggrin gene |
| Foxp3 | Forkhead box P3 |
| GATA-3 | GATA-binding protein 3 |
| GWAS | Genome-wide association study |
| hBD | Human β-defensin |
| HRH4 | Histamine receptor H4 |
| IDECs | Inflammatory dendritic epidermal cells |
| IFNG | IFN-γ gene |
| ILCs | Innate lymphoid cells |
| IL18RAP | Interleukin 18 receptor accessory protein |
| JAK | Janus kinase |
| JNK | c-Jun N-terminal kinase |
| LCs | Langerhans cells |
| LEKTI | Lymphoepithelial Kazal-type trypsin inhibitor |
| mAb | Monoclonal antibodies |
| miR | MicroRNA |
| MMP | Matrix metalloproteinase |
| NALP | NACHT, LRR and PYD domain-containing protein |
| NF-κB | Nuclear factor κ-light-chain-enhancer of activated B cells |
| NOD | Nucleotide-binding oligomerization domain receptors |
| PAR2 | Protease-activated type 2 receptor |
| PDE4 | Phosphodiesterase 4 |
| RANTES | Regulated on activation, normal T cell expressed and secreted |
| SC | Stratum corneum |
| SNP | Single nucleotide polymorphism |
| SP | Serine protease |
| STAT | Signal transducer and activator of transcription |
| Th | T helper |
| TLR | Toll-like receptor |
| TMEM79 | Transmembrane protein 79 |
| Treg | Regulatory T cell |
| TSLP | Thymic stromal lymphopoietin |
| SPINK5 | Serine protease inhibitor Kazal-type 5 |
| SPRR3 | Small proline-rich protein 3 |
References
- Odhiambo, J.A.; Williams, H.C.; Clayton, T.O.; Robertson, C.F.; Asher, M.I. Global variations in prevalence of eczema symptoms in children from isaac phase three. J. Allergy Clin. Immunol. 2009, 124, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Barnes, K.C. An update on the genetics of atopic dermatitis: Scratching the surface in 2009. J. Allergy Clin. Immunol. 2010, 125, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.K.; Kong, K.H.; Khoo, L.; Goh, C.L.; Giam, Y.C. The prevalence and descriptive epidemiology of atopic dermatitis in singapore school children. Br. J. Dermatol. 2002, 146, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Tokura, Y. Extrinsic and intrinsic types of atopic dermatitis. J. Dermatol. Sci. 2010, 58, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Katsarou, A.; Armenaka, M. Atopic dermatitis in older patients: Particular points. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Bantz, S.K.; Zhu, Z.; Zheng, T. The atopic march: Progression from atopic dermatitis to allergic rhinitis and asthma. J. Clin. Cell. Immunol. 2014, 5. [Google Scholar] [CrossRef]
- Andersen, R.M.; Thyssen, J.P.; Maibach, H.I. Qualitative vs. Quantitative atopic dermatitis criteria—In historical and present perspectives. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Oranje, A.P.; Glazenburg, E.J.; Wolkerstorfer, A.; de Waard-van der Spek, F.B. Practical issues on interpretation of scoring atopic dermatitis: The scorad index, objective scorad and the three-item severity score. Br. J. Dermatol. 2007, 157, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Kim, H.J.; Lew, B.L.; Lee, K.H.; Hong, S.P.; Jang, Y.H.; Park, K.Y.; Seo, S.J.; Bae, J.M.; Choi, E.H.; et al. Consensus guidelines for the treatment of atopic dermatitis in korea (Part II): Systemic treatment. Ann. Dermatol. 2015, 27, 578–592. [Google Scholar] [CrossRef] [PubMed]
- Mu, Z.; Zhao, Y.; Liu, X.; Chang, C.; Zhang, J. Molecular biology of atopic dermatitis. Clin. Rev. Allergy Immunol. 2014, 47, 193–218. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Ding, L.; Sivaprasad, U.; Geh, E.; Biagini Myers, J.; Bernstein, J.A.; Khurana Hershey, G.K.; Mersha, T.B. Multiple transcriptome data analysis reveals biologically relevant atopic dermatitis signature genes and pathways. PLoS ONE 2015, 10, e0144316. [Google Scholar] [CrossRef] [PubMed]
- Czarnowicki, T.; Krueger, J.G.; Guttman-Yassky, E. Skin barrier and immune dysregulation in atopic dermatitis: An evolving story with important clinical implications. J. Allergy Clin. Immunol. Pract. 2014, 2, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Boguniewicz, M.; Leung, D.Y. Atopic dermatitis: A disease of altered skin barrier and immune dysregulation. Immunol. Rev. 2011, 242, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.N.; Irvine, A.D.; Terron-Kwiatkowski, A.; Zhao, Y.; Liao, H.; Lee, S.P.; Goudie, D.R.; Sandilands, A.; Campbell, L.E.; Smith, F.J.; et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat. Genet. 2006, 38, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.Y. Our evolving understanding of the functional role of filaggrin in atopic dermatitis. J. Allergy Clin. Immunol. 2009, 124, 494–495. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.; Northstone, K.; Lee, S.P.; Liao, H.; Zhao, Y.; Pembrey, M.; Mukhopadhyay, S.; Smith, G.D.; Palmer, C.N.; McLean, W.H.; et al. The burden of disease associated with filaggrin mutations: A population-based, longitudinal birth cohort study. J. Allergy Clin. Immunol. 2008, 121, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.J.; McLean, W.H. Eczema genetics: Current state of knowledge and future goals. J. Investig. Dermatol. 2009, 129, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, Q.; Liu, J.; Cheng, R.; Zhang, H.; Xue, H.; Bao, Y.; Yao, Z. Mutations analysis in filaggrin gene in Northern China patients with atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Common, J.E.; Haines, R.L.; Balakrishnan, A.; Brown, S.J.; Goh, C.S.; Cordell, H.J.; Sandilands, A.; Campbell, L.E.; Kroboth, K.; et al. Wide spectrum of filaggrin-null mutations in atopic dermatitis highlights differences between Singaporean Chinese and European populations. Br. J. Dermatol. 2011, 165, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Seok, J.; Park, K.Y.; Yoon, Y.; Kim, K.H.; Seo, S.J. Copy-number variation of the filaggrin in Korean patients with atopic dermatitis: What really matters, “number” or “variation”? Br. J. Dermatol. 2016, 174, 1098–1100. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.J.; Kroboth, K.; Sandilands, A.; Campbell, L.E.; Pohler, E.; Kezic, S.; Cordell, H.J.; McLean, W.H.; Irvine, A.D. Intragenic copy number variation within filaggrin contributes to the risk of atopic dermatitis with a dose-dependent effect. J. Investig. Dermatol. 2012, 132, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kezic, S.; O’Regan, G.M.; Yau, N.; Sandilands, A.; Chen, H.; Campbell, L.E.; Kroboth, K.; Watson, R.; Rowland, M.; McLean, W.H.; et al. Levels of filaggrin degradation products are influenced by both filaggrin genotype and atopic dermatitis severity. Allergy 2011, 66, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Margolis, D.J.; Gupta, J.; Apter, A.J.; Ganguly, T.; Hoffstad, O.; Papadopoulos, M.; Rebbeck, T.R.; Mitra, N. Filaggrin-2 variation is associated with more persistent atopic dermatitis in African American subjects. J. Allergy Clin. Immunol. 2014, 133, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, S.; Willis-Owen, S.A.; Kamatani, Y.; Baurecht, H.; Morar, N.; Liang, L.; Edser, P.; Street, T.; Rodriguez, E.; O’Regan, G.M.; et al. A genome-wide association study of atopic dermatitis identifies loci with overlapping effects on asthma and psoriasis. Hum. Mol. Genet. 2013, 22, 4841–4856. [Google Scholar] [CrossRef] [PubMed]
- Marenholz, I.; Rivera, V.A.; Esparza-Gordillo, J.; Bauerfeind, A.; Lee-Kirsch, M.A.; Ciechanowicz, A.; Kurek, M.; Piskackova, T.; Macek, M.; Lee, Y.A. Association screening in the epidermal differentiation complex (EDC) identifies an SPRR3 repeat number variant as a risk factor for eczema. J. Investig. Dermatol. 2011, 131, 1644–1649. [Google Scholar] [CrossRef] [PubMed]
- Kelsell, D.P.; Byrne, C. Snping at the epidermal barrier. J. Investig. Dermatol. 2011, 131, 1593–1595. [Google Scholar] [CrossRef] [PubMed]
- Saunders, S.P.; Goh, C.S.; Brown, S.J.; Palmer, C.N.; Porter, R.M.; Cole, C.; Campbell, L.E.; Gierlinski, M.; Barton, G.J.; Schneider, G.; et al. Tmem79/matt is the matted mouse gene and is a predisposing gene for atopic dermatitis in human subjects. J. Allergy Clin. Immunol. 2013, 132, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M.; Wakefield, J.S. Mechanisms of abnormal lamellar body secretion and the dysfunctional skin barrier in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2014, 134, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Shiohama, A.; Kubo, A.; Kawasaki, H.; Ishida-Yamamoto, A.; Yamada, T.; Hachiya, T.; Shimizu, A.; Okano, H.; Kudoh, J.; et al. A homozygous nonsense mutation in the gene for TMEM79, a component for the lamellar granule secretory system, produces spontaneous eczema in an experimental model of atopic dermatitis. J. Allergy Clin. Immunol. 2013, 132, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Lan, C.C.; Tu, H.P.; Wu, C.S.; Ko, Y.C.; Yu, H.S.; Lu, Y.W.; Li, W.C.; Chen, Y.C.; Chen, G.S. Distinct spink5 and IL-31 polymorphisms are associated with atopic eczema and non-atopic hand dermatitis in taiwanese nursing population. Exp. Dermatol. 2011, 20, 975–979. [Google Scholar] [CrossRef] [PubMed]
- Fortugno, P.; Furio, L.; Teson, M.; Berretti, M.; El Hachem, M.; Zambruno, G.; Hovnanian, A.; D’Alessio, M. The 420K LEKTI variant alters LEKTI proteolytic activation and results in protease deregulation: Implications for atopic dermatitis. Hum. Mol. Genet. 2012, 21, 4187–4200. [Google Scholar] [CrossRef] [PubMed]
- De Benedetto, A.; Rafaels, N.M.; McGirt, L.Y.; Ivanov, A.I.; Georas, S.N.; Cheadle, C.; Berger, A.E.; Zhang, K.; Vidyasagar, S.; Yoshida, T.; et al. Tight junction defects in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2011, 127, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.S.; Kang, M.J.; Kwon, J.W.; Lee, S.Y.; Lee, E.; Yang, S.I.; Jung, Y.H.; Hong, K.; Kim, Y.J.; Lee, S.H.; et al. Claudin-1 polymorphism modifies the effect of mold exposure on the development of atopic dermatitis and production of IgE. J. Allergy Clin. Immunol. 2015, 135, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Paternoster, L.; Standl, M.; Chen, C.M.; Ramasamy, A.; Bonnelykke, K.; Duijts, L.; Ferreira, M.A.; Alves, A.C.; Thyssen, J.P.; Albrecht, E.; et al. Meta-analysis of genome-wide association studies identifies three new risk loci for atopic dermatitis. Nat. Genet. 2012, 44, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; Takahashi, A.; Kubo, M.; Tsunoda, T.; Tomita, K.; Sakashita, M.; Yamada, T.; Fujieda, S.; Tanaka, S.; Doi, S.; et al. Genome-wide association study identifies eight new susceptibility loci for atopic dermatitis in the japanese population. Nat. Genet. 2012, 44, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Farinas, M.; Ungar, B.; Correa da Rosa, J.; Ewald, D.A.; Rozenblit, M.; Gonzalez, J.; Xu, H.; Zheng, X.; Peng, X.; Estrada, Y.D.; et al. RNA sequencing atopic dermatitis transcriptome profiling provides insights into novel disease mechanisms with potential therapeutic implications. J. Allergy Clin. Immunol. 2015, 135, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Esaki, H.; Ewald, D.A.; Ungar, B.; Rozenblit, M.; Zheng, X.; Xu, H.; Estrada, Y.D.; Peng, X.; Mitsui, H.; Litman, T.; et al. Identification of novel immune and barrier genes in atopic dermatitis by means of laser capture microdissection. J. Allergy Clin. Immunol. 2015, 135, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Tamari, M.; Hirota, T. Genome-wide association studies of atopic dermatitis. J. Dermatol. 2014, 41, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Paternoster, L.; Standl, M.; Waage, J.; Baurecht, H.; Hotze, M.; Strachan, D.P.; Curtin, J.A.; Bønnelykke, K.; Tian, C.; Takahashi, A.; et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat. Genet. 2015, 47, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Ahmad-Nejad, P.; Mrabet-Dahbi, S.; Breuer, K.; Klotz, M.; Werfel, T.; Herz, U.; Heeg, K.; Neumaier, M.; Renz, H. The toll-like receptor 2 R753Q polymorphism defines a subgroup of patients with atopic dermatitis having severe phenotype. J. Allergy Clin. Immunol. 2004, 113, 565–567. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Schumann, R.R.; Hamann, L.; Neumann, K.; Worm, M.; Heine, G. Association of the toll-like receptor 2 A-16934T promoter polymorphism with severe atopic dermatitis. Allergy 2009, 64, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Potaczek, D.P.; Nastalek, M.; Okumura, K.; Wojas-Pelc, A.; Undas, A.; Nishiyama, C. An association of TLR2–16934A>T polymorphism and severity/phenotype of atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Salpietro, C.; Rigoli, L.; Miraglia Del Giudice, M.; Cuppari, C.; Di Bella, C.; Salpietro, A.; Maiello, N.; La Rosa, M.; Marseglia, G.L.; Leonardi, S.; et al. TLR2 and TLR4 gene polymorphisms and atopic dermatitis in Italian children: A multicenter study. Int. J. Immunopathol. Pharmacol. 2011, 24, 33–40. [Google Scholar] [PubMed]
- Levchenko, L.; Izmailova, O.V.; Shlykova, O.A.; Kaidashev, I.P. Polymorphism 896A/G of TLR4 gene rather than 1196C/T and 2258G/A of TLR2 gene determines severe and complicated course of atopic dermatitis in children. Tsitol. Genet. 2013, 47, 46–53. [Google Scholar] [PubMed]
- Novak, N.; Yu, C.F.; Bussmann, C.; Maintz, L.; Peng, W.M.; Hart, J.; Hagemann, T.; Diaz-Lacava, A.; Baurecht, H.J.; Klopp, N.; et al. Putative association of a TLR9 promoter polymorphism with atopic eczema. Allergy 2007, 62, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Macaluso, F.; Nothnagel, M.; Parwez, Q.; Petrasch-Parwez, E.; Bechara, F.G.; Epplen, J.T.; Hoffjan, S. Polymorphisms in NACHT-LRR (NLR) genes in atopic dermatitis. Exp. Dermatol. 2007, 16, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Lee, J.E.; Namkung, J.H.; Kim, P.S.; Kim, S.; Shin, E.S.; Cho, E.Y.; Yang, J.M. Single nucleotide polymorphisms and the haplotype in the DEFB1 gene are associated with atopic dermatitis in a Korean population. J. Dermatol. Sci. 2009, 54, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.S.; Rafaels, N.M.; Mu, D.; Hand, T.; Murray, T.; Boguniewicz, M.; Hata, T.; Schneider, L.; Hanifin, J.M.; Gallo, R.L.; et al. Genetic variants in thymic stromal lymphopoietin are associated with atopic dermatitis and eczema herpeticum. J. Allergy Clin. Immunol. 2010, 125, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Margolis, D.J.; Kim, B.; Apter, A.J.; Gupta, J.; Hoffstad, O.; Papadopoulos, M.; Mitra, N. Thymic stromal lymphopoietin variation, filaggrin loss of function, and the persistence of atopic dermatitis. JAMA Dermatol. 2014, 150, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Novak, N.; Kruse, S.; Potreck, J.; Maintz, L.; Jenneck, C.; Weidinger, S.; Fimmers, R.; Bieber, T. Single nucleotide polymorphisms of the IL18 gene are associated with atopic eczema. J. Allergy Clin. Immunol. 2005, 115, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Hao, Y.; Zhou, W.; Ma, Y. The relationship between interleukin-18 polymorphisms and allergic disease: A meta-analysis. BioMed Res. Int. 2014, 2014, 290687. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Bin, L.; Rafaels, N.M.; Huang, L.; Potee, J.; Ruczinski, I.; Beaty, T.H.; Paller, A.S.; Schneider, L.C.; Gallo, R.; et al. Targeted deep sequencing identifies rare loss-of-function variants in IFNGR1 for risk of atopic dermatitis complicated by eczema herpeticum. J. Allergy Clin. Immunol. 2015, 136, 1591–1600. [Google Scholar] [CrossRef] [PubMed]
- Namkung, J.H.; Lee, J.E.; Kim, E.; Kim, S.; Kim, S.; Shin, E.S.; Cho, E.Y.; Yang, J.M. Association of single nucleotide polymorphisms in the IL-12 (IL-12a and b) and IL-12 receptor (IL-12rβ1 and β2) genes and gene-gene interactions with atopic dermatitis in koreans. J. Dermatol. Sci. 2010, 57, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Hon, K.L.; Kong, A.P.; Tang, M.F.; Sy, H.Y.; Chan, J.C.; Leung, T.F. Eczema phenotypes are associated with multiple vitamin d pathway genes in chinese children. Allergy 2014, 69, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Hallau, J.; Hamann, L.; Schumann, R.R.; Worm, M.; Heine, G. A promoter polymorphism of the vitamin D metabolism gene CYP24A1 is associated with severe atopic dermatitis in adults. Acta Derm. Venereol. 2016, 96, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Heine, G.; Hoefer, N.; Franke, A.; Nothling, U.; Schumann, R.R.; Hamann, L.; Worm, M. Association of vitamin d receptor gene polymorphisms with severe atopic dermatitis in adults. Br. J. Dermatol. 2013, 168, 855–858. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Arakawa, H.; Suzuki, M.; Kobayashi, Y.; Mochizuki, H.; Kato, M.; Tokuyama, K.; Morikawa, A. Novel dinucleotide repeat polymorphism in the first exon of the STAT-6 gene is associated with allergic diseases. Clin. Exp. Allergy 2001, 31, 1509–1514. [Google Scholar] [CrossRef] [PubMed]
- Hussein, Y.M.; Shalaby, S.M.; Nassar, A.; Alzahrani, S.S.; Alharbi, A.S.; Nouh, M. Association between genes encoding components of the IL-4/IL-4 receptor pathway and dermatitis in children. Gene 2014, 545, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Casaca, V.I.; Illi, S.; Klucker, E.; Ballenberger, N.; Schedel, M.; von Mutius, E.; Kabesch, M.; Schaub, B. STAT6 polymorphisms are associated with neonatal regulatory T cells and cytokines and atopic diseases at 3 years. Allergy 2013, 68, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska-Wojdylo, M.; Glen, J.; Zablotna, M.; Rebala, K.; Trzeciak, M.; Sikorska, M.; Ruckemann-Dziurdzinska, K.; Nedoszytko, B.; Florek, A.; Nowicki, R. The frequencies of haplotypes defined by three polymorphisms of the IL-31 gene: −1066, −2057, and IVS2+12 in Polish patients with atopic dermatitis. Int. J. Dermatol. 2015, 54, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Narbutt, J.; Wojtczak, M.; Zalinska, A.; Salinski, A.; Przybylowska-Sygut, K.; Kuna, P.; Majak, P.; Sysa-Jedrzejowska, A.; Lesiak, A. The A/A genotype of an interleukin-17A polymorphism predisposes to increased severity of atopic dermatitis and coexistence with asthma. Clin. Exp. Dermatol. 2015, 40, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.H.; Yu, H.S.; Ko, Y.C.; Chang, W.C.; Chuang, H.Y.; Chen, G.S.; Lee, C.H. Functional regulation of interleukin-31 production by its genetic polymorphism in patients with extrinsic atopic dermatitis. Acta Derm. Venereol. 2012, 92, 430–432. [Google Scholar] [CrossRef] [PubMed]
- Arshad, S.H.; Karmaus, W.; Kurukulaaratchy, R.; Sadeghnejad, A.; Huebner, M.; Ewart, S. Polymorphisms in the interleukin 13 and GATA binding protein 3 genes and the development of eczema during childhood. Br. J. Dermatol. 2008, 158, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Hussein, P.Y.; Zahran, F.; Ashour Wahba, A.; Ahmad, A.S.; Ibrahiem, M.M.; Shalaby, S.M.; El Tarhouny, S.A.; El Sherbiny, H.M.; Bakr, N. Interleukin 10 receptor α subunit (IL-10RA) gene polymorphism and IL-10 serum levels in Egyptian atopic patients. J. Investig. Allergol. Clin. Immunol. 2010, 20, 20–26. [Google Scholar] [PubMed]
- Namkung, J.H.; Lee, J.E.; Kim, E.; Park, G.T.; Yang, H.S.; Jang, H.Y.; Shin, E.S.; Cho, E.Y.; Yang, J.M. An association between IL-9 and IL-9 receptor gene polymorphisms and atopic dermatitis in a Korean population. J. Dermatol. Sci. 2011, 62, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Gharagozlou, M.; Farhadi, E.; Khaledi, M.; Behniafard, N.; Sotoudeh, S.; Salari, R.; Darabi, B.; Fathi, S.M.; Mahmoudi, M.; Aghamohammadi, A.; et al. Association between the interleukin 6 genotype at position −174 and atopic dermatitis. J. Investig. Allergol. Clin. Immunol. 2013, 23, 89–93. [Google Scholar] [PubMed]
- Namkung, J.H.; Lee, J.E.; Kim, E.; Cho, H.J.; Kim, S.; Shin, E.S.; Cho, E.Y.; Yang, J.M. IL-5 and IL-5 receptor α polymorphisms are associated with atopic dermatitis in Koreans. Allergy 2007, 62, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Hoffjan, S.; Beygo, J.; Akkad, D.A.; Parwez, Q.; Petrasch-Parwez, E.; Epplen, J.T. Analysis of variation in the IL7RA and IL2RA genes in atopic dermatitis. J. Dermatol. Sci. 2009, 55, 138–140. [Google Scholar] [CrossRef] [PubMed]
- Nickel, R.G.; Casolaro, V.; Wahn, U.; Beyer, K.; Barnes, K.C.; Plunkett, B.S.; Freidhoff, L.R.; Sengler, C.; Plitt, J.R.; Schleimer, R.P.; et al. Atopic dermatitis is associated with a functional mutation in the promoter of the C-C chemokine rantes. J. Immunol. 2000, 164, 1612–1616. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Roberts, M.H.; Yamamoto, N.; Sugiura, H.; Uehara, M.; Hopkin, J.M. Upregulating promoter polymorphisms of rantes relate to atopic dermatitis. Int. J. Immunogenet. 2006, 33, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Mizutani, H. The role of cytokines/chemokines in the pathogenesis of atopic dermatitis. Curr. Probl. Dermatol. 2011, 41, 80–92. [Google Scholar] [PubMed]
- Yu, B.; Shao, Y.; Zhang, J.; Dong, X.L.; Liu, W.L.; Yang, H.; Liu, L.; Li, M.H.; Yue, C.F.; Fang, Z.Y.; et al. Polymorphisms in human histamine receptor H4 gene are associated with atopic dermatitis. Br. J. Dermatol. 2010, 162, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Ye, T.; Shao, Y.; Zhang, J.; Zhong, Q.; Hu, X.; Zhang, W.; Yu, B. Association between copy-number variations of the human histamine H4 receptor gene and atopic dermatitis in a Chinese population. Clin. Exp. Dermatol. 2013, 38, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Niwa, Y.; Potaczek, D.P.; Kanada, S.; Takagi, A.; Shimokawa, N.; Ito, T.; Mitsuishi, K.; Okubo, Y.; Tajima, M.; Hobo, A.; et al. Fcepsilonrialpha gene (FCER1A) promoter polymorphisms and total serum IgE levels in Japanese atopic dermatitis patients. Int. J. Immunogenet. 2010, 37, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Park, K.Y.; Park, M.K.; Kim, E.J.; Lee, M.K.; Seo, S.J. Fcepsilonri gene promoter polymorphisms and total IgE levels in susceptibility to atopic dermatitis in korea. J. Korean Med. Sci. 2011, 26, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhou, Y.; Lin, L.H.; Wang, J.; Peng, X.; Li, J.; Li, L. Association of polymorphisms in the promoter region of FCER1A gene with atopic dermatitis, chronic uticaria, asthma, and serum immunoglobulin E levels in a Han Chinese population. Hum. Immunol. 2012, 73, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Gao, X.H.; Zhao, L.P.; Di, Z.H.; McHepange, U.O.; Zhang, L.; Chen, H.D.; Wei, H.C. Brain-derived neurotrophic factor gene polymorphisms and serum levels in chinese atopic dermatitis patients. J. Eur. Acad. Dermatol. Venereol. 2009, 23, 1277–1281. [Google Scholar] [CrossRef] [PubMed]
- Blakely, K.; Gooderham, M.; Papp, K. Dupilumab, a monoclonal antibody for atopic dermatitis: A review of current literature. Skin Ther. Lett. 2016, 21, 1–5. [Google Scholar]
- Wang, D.; Beck, L.A. Immunologic targets in atopic dermatitis and emerging therapies: An update. Am. J. Clin. Dermatol. 2016, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Noda, S.; Krueger, J.G.; Guttman-Yassky, E. The translational revolution and use of biologics in patients with inflammatory skin diseases. J. Allergy Clin. Immunol. 2015, 135, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Chang, C.; Lu, Q. The genetics and epigenetics of atopic dermatitis-filaggrin and other polymorphisms. Clin. Rev. Allergy Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Herberth, G.; Bauer, M.; Gasch, M.; Hinz, D.; Roder, S.; Olek, S.; Kohajda, T.; Rolle-Kampczyk, U.; von Bergen, M.; Sack, U.; et al. Maternal and cord blood mir-223 expression associates with prenatal tobacco smoke exposure and low regulatory T-cell numbers. J. Allergy Clin. Immunol. 2014, 133, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Hinz, D.; Bauer, M.; Roder, S.; Olek, S.; Huehn, J.; Sack, U.; Borte, M.; Simon, J.C.; Lehmann, I.; Herberth, G. Cord blood tregs with stable FOXP3 expression are influenced by prenatal environment and associated with atopic dermatitis at the age of one year. Allergy 2012, 67, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K. The role of air pollutants in atopic dermatitis. J. Allergy Clin. Immunol. 2014, 134, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.J.; Guo, Y.L.; Lin, T.J.; Chen, P.C.; Wu, Y.N. GSTM1, GSTP1, prenatal smoke exposure, and atopic dermatitis. Ann. Allergy Asthma Immunol. 2010, 105, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.J.; Chen, S.L.; Lu, T.P.; Chuang, E.Y.; Chen, P.C. Prenatal smoke exposure, DNA methylation, and childhood atopic dermatitis. Clin. Exp. Allergy 2013, 43, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Sekigawa, I.; Ogasawara, H.; Mitsuishi, K.; Hira, K.; Ikeda, S.; Ogawa, H. Expression of DNMT-1 in patients with atopic dermatitis. Arch. Dermatol. Res. 2006, 298, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Wang, P.; Zhao, M.; Liang, G.; Yin, H.; Zhang, G.; Wen, H.; Lu, Q. Demethylation of the FCER1G promoter leads to fcepsilonri overexpression on monocytes of patients with atopic dermatitis. Allergy 2012, 67, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Sonkoly, E.; Janson, P.; Majuri, M.L.; Savinko, T.; Fyhrquist, N.; Eidsmo, L.; Xu, N.; Meisgen, F.; Wei, T.; Bradley, M.; et al. miR-155 is overexpressed in patients with atopic dermatitis and modulates T-cell proliferative responses by targeting cytotoxic T lymphocyte-associated antigen 4. J. Allergy Clin. Immunol. 2010, 126, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Xue, H.B.; Wang, F.; Shu, C.M.; Zhang, J.H. MicroRNA-155 may be involved in the pathogenesis of atopic dermatitis by modulating the differentiation and function of T Helper type 17 (Th17) cells. Clin. Exp. Immunol. 2015, 181, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Rebane, A.; Runnel, T.; Aab, A.; Maslovskaja, J.; Ruckert, B.; Zimmermann, M.; Plaas, M.; Karner, J.; Treis, A.; Pihlap, M.; et al. MicroRNA-146A alleviates chronic skin inflammation in atopic dermatitis through suppression of innate immune responses in keratinocytes. J. Allergy Clin. Immunol. 2014, 134, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.P.; Nguyen, G.H.; Jin, H.Z. MicroRNA-143 inhibits IL-13-induced dysregulation of the epidermal barrier-related proteins in skin keratinocytes via targeting to IL-13rα1. Mol. Cell. Biochem. 2016, 416, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Hachem, J.P.; Wagberg, F.; Schmuth, M.; Crumrine, D.; Lissens, W.; Jayakumar, A.; Houben, E.; Mauro, T.M.; Leonardsson, G.; Brattsand, M.; et al. Serine protease activity and residual lekti expression determine phenotype in netherton syndrome. J. Investig. Dermatol. 2006, 126, 1609–1621. [Google Scholar] [CrossRef] [PubMed]
- Briot, A.; Deraison, C.; Lacroix, M.; Bonnart, C.; Robin, A.; Besson, C.; Dubus, P.; Hovnanian, A. Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in netherton syndrome. J. Exp. Med. 2009, 206, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Bae, H.C.; Ko, N.Y.; Lee, S.H.; Jeong, S.H.; Lee, H.; Ryu, W.I.; Kye, Y.C.; Son, S.W. Thymic stromal lymphopoietin downregulates filaggrin expression by signal transducer and activator of transcription 3 (STAT3) and extracellular signal-regulated kinase (ERK) phosphorylation in keratinocytes. J. Allergy Clin. Immunol. 2015, 136, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Andoh, T.; Kuraishi, Y. Antipruritic mechanisms of topical e6005, a phosphodiesterase 4 inhibitor: Inhibition of responses to proteinase-activated receptor 2 stimulation mediated by increase in intracellular cyclic amp. J. Dermatol. Sci. 2014, 76, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Volf, E.M.; Au, S.C.; Dumont, N.; Scheinman, P.; Gottlieb, A.B. A phase 2, open-label, investigator-initiated study to evaluate the safety and efficacy of apremilast in subjects with recalcitrant allergic contact or atopic dermatitis. J. Drugs Dermatol. 2012, 11, 341–346. [Google Scholar] [PubMed]
- Samrao, A.; Berry, T.M.; Goreshi, R.; Simpson, E.L. A pilot study of an oral phosphodiesterase inhibitor (apremilast) for atopic dermatitis in adults. Arch. Dermatol. 2012, 148, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Henry, J.; Hsu, C.Y.; Balica, S.; Jean-Decoster, C.; Mechin, M.C.; Hansmann, B.; Rodriguez, E.; Weindinger, S.; Schmitt, A.M.; et al. Defects of filaggrin-like proteins in both lesional and nonlesional atopic skin. J. Allergy Clin. Immunol. 2013, 131, 1094–1102. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.M.; Pfeiffer, S.; Witt, M.; Brautigam, M.; Neumann, C.; Weichenthal, M.; Schwarz, T.; Folster-Holst, R.; Proksch, E. Different effects of pimecrolimus and betamethasone on the skin barrier in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2009, 123, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, A.; Doi, H.; Egawa, G.; Maekawa, A.; Fujita, T.; Nakamizo, S.; Nakashima, C.; Nakajima, S.; Watanabe, T.; Miyachi, Y.; et al. Possible new therapeutic strategy to regulate atopic dermatitis through upregulating filaggrin expression. J. Allergy Clin. Immunol. 2014, 133, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Stout, T.E.; McFarland, T.; Mitchell, J.C.; Appukuttan, B.; Stout, J.T. Recombinant filaggrin is internalized and processed to correct filaggrin deficiency. J. Investig. Dermatol. 2014, 134, 423–429. [Google Scholar] [CrossRef] [PubMed]
- De Benedetto, A.; Slifka, M.K.; Rafaels, N.M.; Kuo, I.H.; Georas, S.N.; Boguniewicz, M.; Hata, T.; Schneider, L.C.; Hanifin, J.M.; Gallo, R.L.; et al. Reductions in claudin-1 may enhance susceptibility to herpes simplex virus 1 infections in atopic dermatitis. J. Allergy Clin. Immunol. 2011, 128, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Gruber, R.; Bornchen, C.; Rose, K.; Daubmann, A.; Volksdorf, T.; Wladykowski, E.; Vidal, Y.S.S.; Peters, E.M.; Danso, M.; Bouwstra, J.A.; et al. Diverse regulation of claudin-1 and claudin-4 in atopic dermatitis. Am. J. Pathol. 2015, 185, 2777–2789. [Google Scholar] [CrossRef] [PubMed]
- Kuo, I.H.; Carpenter-Mendini, A.; Yoshida, T.; McGirt, L.Y.; Ivanov, A.I.; Barnes, K.C.; Gallo, R.L.; Borkowski, A.W.; Yamasaki, K.; Leung, D.Y.; et al. Activation of epidermal Toll-Like receptor 2 enhances tight junction function: Implications for atopic dermatitis and skin barrier repair. J. Investig. Dermatol. 2013, 133, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Farinas, M.; Tintle, S.J.; Shemer, A.; Chiricozzi, A.; Nograles, K.; Cardinale, I.; Duan, S.; Bowcock, A.M.; Krueger, J.G.; Guttman-Yassky, E. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J. Allergy Clin. Immunol. 2011, 127, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Janssens, M.; van Smeden, J.; Gooris, G.S.; Bras, W.; Portale, G.; Caspers, P.J.; Vreeken, R.J.; Hankemeier, T.; Kezic, S.; Wolterbeek, R.; et al. Increase in short-chain ceramides correlates with an altered lipid organization and decreased barrier function in atopic eczema patients. J. Lipid Res. 2012, 53, 2755–2766. [Google Scholar] [CrossRef] [PubMed]
- Van Smeden, J.; Janssens, M.; Kaye, E.C.; Caspers, P.J.; Lavrijsen, A.P.; Vreeken, R.J.; Bouwstra, J.A. The importance of free fatty acid chain length for the skin barrier function in atopic eczema patients. Exp. Dermatol. 2014, 23, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Sawada, E.; Yoshida, N.; Sugiura, A.; Imokawa, G. Th1 cytokines accentuate but Th2 cytokines attenuate ceramide production in the stratum corneum of human epidermal equivalents: An implication for the disrupted barrier mechanism in atopic dermatitis. J. Dermatol. Sci. 2012, 68, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Ong, P.Y.; Ohtake, T.; Brandt, C.; Strickland, I.; Boguniewicz, M.; Ganz, T.; Gallo, R.L.; Leung, D.Y. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N. Engl. J. Med. 2002, 347, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.D.; Novak, N.; Bieber, T.; Pastore, S.; Girolomoni, G.; Boguniewicz, M.; Streib, J.; Wong, C.; Gallo, R.L.; Leung, D.Y. Interleukin-10 downregulates anti-microbial peptide expression in atopic dermatitis. J. Investig. Dermatol. 2005, 125, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Schittek, B. The antimicrobial skin barrier in patients with atopic dermatitis. Curr. Probl. Dermatol. 2011, 41, 54–67. [Google Scholar] [PubMed]
- Son, E.D.; Kim, H.J.; Kim, K.H.; Bin, B.H.; Bae, I.H.; Lim, K.M.; Yu, S.J.; Cho, E.G.; Lee, T.R. S100a7 (psoriasin) inhibits human epidermal differentiation by enhanced IL-6 secretion through IκB/NF-κB signaling. Exp. Dermatol. 2016, 25, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Niebuhr, M.; Langnickel, J.; Sigel, S.; Werfel, T. Dysregulation of CD36 upon TLR-2 stimulation in monocytes from patients with atopic dermatitis and the TLR2 R753Q polymorphism. Exp. Dermatol. 2010, 19, e296–e298. [Google Scholar] [CrossRef] [PubMed]
- Camargo, C.A., Jr.; Ganmaa, D.; Sidbury, R.; Erdenedelger, K.; Radnaakhand, N.; Khandsuren, B. Randomized trial of vitamin d supplementation for winter-related atopic dermatitis in children. J. Allergy Clin. Immunol. 2014, 134, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Hata, T.R.; Audish, D.; Kotol, P.; Coda, A.; Kabigting, F.; Miller, J.; Alexandrescu, D.; Boguniewicz, M.; Taylor, P.; Aertker, L.; et al. A randomized controlled double-blind investigation of the effects of vitamin D dietary supplementation in subjects with atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, U.; Hvid, M.; Johansen, C.; Buchner, M.; Folster-Holst, R.; Deleuran, M.; Vestergaard, C. TSLP, IL-31, IL-33 and SST2 are new biomarkers in endophenotypic profiling of adult and childhood atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Moy, A.P.; Murali, M.; Kroshinsky, D.; Duncan, L.M.; Nazarian, R.M. Immunologic overlap of helper T-cell subtypes 17 and 22 in erythrodermic psoriasis and atopic dermatitis. JAMA Dermatol. 2015, 151, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.D.; Fairchild, H.R.; Kim, B.E.; Bin, L.; Boguniewicz, M.; Redzic, J.S.; Hansen, K.C.; Leung, D.Y. Th2 cytokines act on S100/A11 to downregulate keratinocyte differentiation. J. Investig. Dermatol. 2008, 128, 2248–2258. [Google Scholar] [CrossRef] [PubMed]
- Gittler, J.K.; Shemer, A.; Suarez-Farinas, M.; Fuentes-Duculan, J.; Gulewicz, K.J.; Wang, C.Q.; Mitsui, H.; Cardinale, I.; de Guzman Strong, C.; Krueger, J.G.; et al. Progressive activation of Th2/Th22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 2012, 130, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Sehra, S.; Yao, Y.; Howell, M.D.; Nguyen, E.T.; Kansas, G.S.; Leung, D.Y.; Travers, J.B.; Kaplan, M.H. IL-4 regulates skin homeostasis and the predisposition toward allergic skin inflammation. J. Immunol. 2010, 184, 3186–3190. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.E.; Leung, D.Y.; Boguniewicz, M.; Howell, M.D. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin. Immunol. 2008, 126, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.D.; Kim, B.E.; Gao, P.; Grant, A.V.; Boguniewicz, M.; Debenedetto, A.; Schneider, L.; Beck, L.A.; Barnes, K.C.; Leung, D.Y. Cytokine modulation of atopic dermatitis filaggrin skin expression. J. Allergy Clin. Immunol. 2007, 120, 150–155. [Google Scholar] [CrossRef] [PubMed]
- McAleer, M.A.; Irvine, A.D. The multifunctional role of filaggrin in allergic skin disease. J. Allergy Clin. Immunol. 2013, 131, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Amano, W.; Nakajima, S.; Kunugi, H.; Numata, Y.; Kitoh, A.; Egawa, G.; Dainichi, T.; Honda, T.; Otsuka, A.; Kimoto, Y.; et al. The janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J. Allergy Clin. Immunol. 2015, 136, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Thaci, D.; Simpson, E.L.; Beck, L.A.; Bieber, T.; Blauvelt, A.; Papp, K.; Soong, W.; Worm, M.; Szepietowski, J.C.; Sofen, H.; et al. Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: A randomised, placebo-controlled, dose-ranging phase 2B trial. Lancet 2016, 387, 40–52. [Google Scholar] [CrossRef]
- Toda, M.; Leung, D.Y.; Molet, S.; Boguniewicz, M.; Taha, R.; Christodoulopoulos, P.; Fukuda, T.; Elias, J.A.; Hamid, Q.A. Polarized in vivo expression of IL-11 and IL-17 between acute and chronic skin lesions. J. Allergy Clin. Immunol. 2003, 111, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Nograles, K.E.; Zaba, L.C.; Shemer, A.; Fuentes-Duculan, J.; Cardinale, I.; Kikuchi, T.; Ramon, M.; Bergman, R.; Krueger, J.G.; Guttman-Yassky, E. IL-22-producing “T22” T cells account for upregulated IL-22 in atopic dermatitis despite reduced IL-17-producing Th17 T cells. J. Allergy Clin. Immunol. 2009, 123, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Park, C.O.; Shin, J.U.; Noh, J.Y.; Lee, Y.S.; Lee, N.R.; Kim, H.R.; Noh, S.; Lee, Y.; Lee, J.H.; et al. Damp molecules S100A9 and S100A8 activated by IL-17A and house-dust mites are increased in atopic dermatitis. Exp. Dermatol. 2014, 23, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Farinas, M.; Dhingra, N.; Gittler, J.; Shemer, A.; Cardinale, I.; de Guzman Strong, C.; Krueger, J.G.; Guttman-Yassky, E. Intrinsic atopic dermatitis shows similar Th2 and higher Th17 immune activation compared with extrinsic atopic dermatitis. J. Allergy Clin. Immunol. 2013, 132, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Noda, S.; Suarez-Farinas, M.; Ungar, B.; Kim, S.J.; de Guzman Strong, C.; Xu, H.; Peng, X.; Estrada, Y.D.; Nakajima, S.; Honda, T.; et al. The asian atopic dermatitis phenotype combines features of atopic dermatitis and psoriasis with increased Th17 polarization. J. Allergy Clin. Immunol. 2015, 136, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Czarnowicki, T.; Gonzalez, J.; Shemer, A.; Malajian, D.; Xu, H.; Zheng, X.; Khattri, S.; Gilleaudeau, P.; Sullivan-Whalen, M.; Suarez-Farinas, M.; et al. Severe atopic dermatitis is characterized by selective expansion of circulating TH2/TC2 and TH22/TC22, but not TH17/TC17, cells within the skin-homing T-cell population. J. Allergy Clin. Immunol. 2015, 136, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Gutowska-Owsiak, D.; Schaupp, A.L.; Salimi, M.; Taylor, S.; Ogg, G.S. Interleukin-22 downregulates filaggrin expression and affects expression of profilaggrin processing enzymes. Br. J. Dermatol. 2011, 165, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Witte, E.; Wallace, E.; Docke, W.D.; Kunz, S.; Asadullah, K.; Volk, H.D.; Sterry, W.; Sabat, R. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: A potential role in psoriasis. Eur. J. Immunol. 2006, 36, 1309–1323. [Google Scholar] [CrossRef] [PubMed]
- Niebuhr, M.; Mainardy, J.; Heratizadeh, A.; Satzger, I.; Werfel, T. Staphylococcal exotoxins induce interleukin 22 in human Th22 cells. Int. Arch. Allergy Immunol. 2014, 165, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Kim, H.; Kim, Y.; Choi, J.; Jeon, J.; Hwang, Y.; Kang, J.S.; Lee, W.J. The crucial role of IL-22 and its receptor on tarc production and T cell migration by HDM extract. Exp. Dermatol. 2016, 25, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Boniface, K.; Bernard, F.X.; Garcia, M.; Gurney, A.L.; Lecron, J.C.; Morel, F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J. Immunol. 2005, 174, 3695–3702. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.W.; Li, J.; Kodangattil, S.R.; Luxenberg, D.P.; Bennett, F.; Martino, M.; Collins, M.; Dunussi-Joannopoulos, K.; Gill, D.S.; Wolfman, N.M.; et al. IL-22R, IL-10R2, and IL-22BP binding sites are topologically juxtaposed on adjacent and overlapping surfaces of IL-22. J. Mol. Biol. 2008, 382, 1168–1183. [Google Scholar] [CrossRef] [PubMed]
- Witte, E.; Witte, K.; Warszawska, K.; Sabat, R.; Wolk, K. Interleukin-22: A cytokine produced by T, NK and NKT cell subsets, with importance in the innate immune defense and tissue protection. Cytokine Growth Factor Rev. 2010, 21, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.S.; Maekawa, Y.; Kitamura, A.; Tanigaki, K.; Yoshimoto, T.; Kishihara, K.; Yasutomo, K. Notch signaling drives IL-22 secretion in CD4+ T cells by stimulating the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2010, 107, 5943–5948. [Google Scholar] [CrossRef] [PubMed]
- Czarnowicki, T.; Esaki, H.; Gonzalez, J.; Malajian, D.; Shemer, A.; Noda, S.; Talasila, S.; Berry, A.; Gray, J.; Becker, L.; et al. Early pediatric atopic dermatitis shows only a cutaneous lymphocyte antigen (CLA)(+) TH2/TH1 cell imbalance, whereas adults acquire CLA+ TH22/TC22 cell subsets. J. Allergy Clin. Immunol. 2015, 136, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Aihara, M.; Kirino, M.; Harada, I.; Komori-Yamaguchi, J.; Yamaguchi, Y.; Nagashima, Y.; Ikezawa, Z. Interleukin-18 is elevated in the horny layer in patients with atopic dermatitis and is associated with staphylococcus aureus colonization. Br. J. Dermatol. 2011, 164, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Choi, J.K.; Jung, H.J.; Park, K.H.; Jang, Y.H.; Lee, W.J.; Lee, S.J.; Kim, S.H.; Kang, H.Y.; Kim, J.M.; et al. Effects of topical application of a recombinant staphylococcal enterotoxin a on dncb and dust mite extract-induced atopic dermatitis-like lesions in a murine model. Eur. J. Dermatol. 2014, 24, 186–193. [Google Scholar] [PubMed]
- Lee, J.H.; Cho, D.H.; Park, H.J. IL-18 and cutaneous inflammatory diseases. Int. J. Mol. Sci. 2015, 16, 29357–29369. [Google Scholar] [CrossRef] [PubMed]
- Novak, N.; Valenta, R.; Bohle, B.; Laffer, S.; Haberstok, J.; Kraft, S.; Bieber, T. Fcepsilonri engagement of langerhans cell-like dendritic cells and inflammatory dendritic epidermal cell-like dendritic cells induces chemotactic signals and different T-cell phenotypes in vitro. J. Allergy Clin. Immunol. 2004, 113, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, T.; Tsutsui, H.; Tominaga, K.; Hoshino, K.; Okamura, H.; Akira, S.; Paul, W.E.; Nakanishi, K. IL-18, although antiallergic when administered with IL-12, stimulates IL-4 and histamine release by basophils. Proc. Natl. Acad. Sci. USA 1999, 96, 13962–13966. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Yagita, H.; Ortaldo, J.R.; Wiltrout, R.H.; Young, H.A. In vivo administration of IL-18 can induce IgE production through Th2 cytokine induction and up-regulation of CD40 ligand (CD154) expression on CD4+ T cells. Eur. J. Immunol. 2000, 30, 1998–2006. [Google Scholar] [CrossRef]
- Lee, H.J.; Kwon, Y.S.; Park, C.O.; Oh, S.H.; Lee, J.H.; Wu, W.H.; Chang, N.S.; Lee, M.G.; Lee, K.H. Corticotropin-releasing factor decreases IL-18 in the monocyte-derived dendritic cell. Exp. Dermatol. 2009, 18, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Tsutsui, H.; Murakami, T.; Yumikura-Futatsugi, S.; Yamanaka, K.; Tanaka, M.; Iwakura, Y.; Suzuki, N.; Takeda, K.; Akira, S.; et al. IL-18 contributes to the spontaneous development of atopic dermatitis-like inflammatory skin lesion independently of IgE/STAT6 under specific pathogen-free conditions. Proc. Natl. Acad. Sci. USA 2002, 99, 11340–11345. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.J.; Zou, Y.X.; Zeng, S.W. Risk factors for and expression of immune and inflammatory factors in atopic dermatitis in chinese population: A birth cohort study. Mol. Cell. Probes 2016, 30, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Tsutsui, H.; Yoshimoto, T.; Kotani, M.; Matsumoto, M.; Fujita, A.; Wang, W.; Higa, S.; Koshimoto, T.; Nakanishi, K.; et al. Interleukin-18 is elevated in the sera from patients with atopic dermatitis and from atopic dermatitis model mice, NC/NGA. Int. Arch. Allergy Immunol. 2001, 125, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Thijs, J.; Krastev, T.; Weidinger, S.; Buckens, C.F.; de Bruin-Weller, M.; Bruijnzeel-Koomen, C.; Flohr, C.; Hijnen, D. Biomarkers for atopic dermatitis: A systematic review and meta-analysis. Curr. Opin. Allergy Clin. Immunol. 2015, 15, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Zedan, K.; Rasheed, Z.; Farouk, Y.; Alzolibani, A.A.; Bin Saif, G.; Ismail, H.A.; Al Robaee, A.A. Immunoglobulin E, interleukin-18 and interleukin-12 in patients with atopic dermatitis: Correlation with disease activity. J. Clin. Diagn. Res. 2015, 9, Wc01–Wc05. [Google Scholar] [CrossRef] [PubMed]
- Sonkoly, E.; Muller, A.; Lauerma, A.I.; Pivarcsi, A.; Soto, H.; Kemeny, L.; Alenius, H.; Dieu-Nosjean, M.C.; Meller, S.; Rieker, J.; et al. IL-31: A new link between t cells and pruritus in atopic skin inflammation. J. Allergy Clin. Immunol. 2006, 117, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, M.H.; Hasan, Z.E.; Shaheen, K.Y. Serum measurement of interleukin-31 (IL-31) in paediatric atopic dermatitis: Elevated levels correlate with severity scoring. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, C.; Marquardt, Y.; Czaja, K.; Wenzel, J.; Frank, J.; Luscher-Firzlaff, J.; Luscher, B.; Baron, J.M. IL-31 regulates differentiation and filaggrin expression in human organotypic skin models. J. Allergy Clin. Immunol. 2012, 129, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Danso, M.O.; van Drongelen, V.; Mulder, A.; van Esch, J.; Scott, H.; van Smeden, J.; El Ghalbzouri, A.; Bouwstra, J.A. TNF-α and Th2 cytokines induce atopic dermatitis-like features on epidermal differentiation proteins and stratum corneum lipids in human skin equivalents. J. Investig. Dermatol. 2014, 134, 1941–1950. [Google Scholar] [PubMed]
- Zhang, Q.; Putheti, P.; Zhou, Q.; Liu, Q.; Gao, W. Structures and biological functions of IL-31 and IL-31 receptors. Cytokine Growth Factor Rev. 2008, 19, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Fujii, E.; Watanabe, T.; Takashima, Y.; Matsushita, H.; Furuhashi, T.; Morita, A. Distribution of IL-31 and its receptor expressing cells in skin of atopic dermatitis. J. Dermatol. Sci. 2014, 74, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Feld, M.; Garcia, R.; Buddenkotte, J.; Katayama, S.; Lewis, K.; Muirhead, G.; Hevezi, P.; Plesser, K.; Schrumpf, H.; Krjutskov, K.; et al. The pruritus- and TH2-associated cytokine IL-31 promotes growth of sensory nerves. J. Allergy Clin. Immunol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, O.; Furue, M.; Nakagawa, H.; Shiramoto, M.; Hanada, R.; Matsuki, S.; Imayama, S.; Kato, M.; Hasebe, I.; Taira, K.; et al. The first trial of CIM331, a humanized antihuman interleukin-31 receptor a antibody, in healthy volunteers and patients with atopic dermatitis to evaluate safety, tolerability and pharmacokinetics of a single dose in a randomized, double-blind, placebo-controlled study. Br. J. Dermatol. 2016, 174, 296–304. [Google Scholar] [PubMed]
- Lee, C.H.; Hong, C.H.; Yu, W.T.; Chuang, H.Y.; Huang, S.K.; Chen, G.S.; Yoshioka, T.; Sakata, M.; Liao, W.T.; Ko, Y.C.; et al. Mechanistic correlations between two itch biomarkers, cytokine interleukin-31 and neuropeptide beta-endorphin, via STAT3/calcium axis in atopic dermatitis. Br. J. Dermatol. 2012, 167, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Hawro, T.; Saluja, R.; Weller, K.; Altrichter, S.; Metz, M.; Maurer, M. Interleukin-31 does not induce immediate itch in atopic dermatitis patients and healthy controls after skin challenge. Allergy 2014, 69, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Hanel, K.H.; Pfaff, C.M.; Cornelissen, C. Control of the physical and antimicrobial skin barrier by an IL-31-IL-1 signaling network. J. Immunol. 2016, 196, 3233–3244. [Google Scholar] [CrossRef] [PubMed]
- Jirapongsananuruk, O.; Hofer, M.F.; Trumble, A.E.; Norris, D.A.; Leung, D.Y. Enhanced expression of B7.2 (CD86) in patients with atopic dermatitis: A potential role in the modulation of IgE synthesis. J. Immunol. 1998, 160, 4622–4627. [Google Scholar] [PubMed]
- Navarrete-Dechent, C.; Perez-Mateluna, G.; Silva-Valenzuela, S.; Vera-Kellet, C.; Borzutzky, A. Humoral and cellular autoreactivity to epidermal proteins in atopic dermatitis. Arch. Immunol. Ther. Exp. 2016. [Google Scholar] [CrossRef] [PubMed]
- Heil, P.M.; Maurer, D.; Klein, B.; Hultsch, T.; Stingl, G. Omalizumab therapy in atopic dermatitis: Depletion of IgE does not improve the clinical course—A randomized, placebo-controlled and double blind pilot study. J. Dtsch. Dermatol. Ges. 2010, 8, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Hosli, S.; Kostylina, G.; Yawalkar, N.; Simon, H.U. Anti-CD20 (rituximab) treatment improves atopic eczema. J. Allergy Clin. Immunol. 2008, 121, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Wollenberg, A.; Wen, S.; Bieber, T. Phenotyping of epidermal dendritic cells: Clinical applications of a flow cytometric micromethod. Cytometry 1999, 37, 147–155. [Google Scholar] [CrossRef]
- Wollenberg, A.; Sharma, S.; von Bubnoff, D.; Geiger, E.; Haberstok, J.; Bieber, T. Topical tacrolimus (FK506) leads to profound phenotypic and functional alterations of epidermal antigen-presenting dendritic cells in atopic dermatitis. J. Allergy Clin. Immunol. 2001, 107, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Wollenberg, A.; Wagner, M.; Gunther, S.; Towarowski, A.; Tuma, E.; Moderer, M.; Rothenfusser, S.; Wetzel, S.; Endres, S.; Hartmann, G. Plasmacytoid dendritic cells: A new cutaneous dendritic cell subset with distinct role in inflammatory skin diseases. J. Investig. Dermatol. 2002, 119, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Wang, Y.H.; Duramad, O.; Hori, T.; Delespesse, G.J.; Watanabe, N.; Qin, F.X.; Yao, Z.; Cao, W.; Liu, Y.J. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J. Exp. Med. 2005, 202, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Gros, E.; Petzold, S.; Maintz, L.; Bieber, T.; Novak, N. Reduced ifn-gamma receptor expression and attenuated IFN-γ response by dendritic cells in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2011, 128, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.F. Thymic stromal lymphopoietin and allergic disease. J. Allergy Clin. Immunol. 2012, 130, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Nograles, K.E.; Kikuchi, T.; Gonzalez, J.; Carucci, J.A.; Krueger, J.G. Human langerhans cells induce distinct IL-22-producing CD4+ T cells lacking IL-17 production. Proc. Natl. Acad. Sci. USA 2009, 106, 21795–21800. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, D.; Stark, H.; Chazot, P.L.; Shenton, F.C.; Leurs, R.; Werfel, T.; Gutzmer, R. Human inflammatory dendritic epidermal cells express a functional histamine H4 receptor. J. Investig. Dermatol. 2008, 128, 1696–1703. [Google Scholar] [CrossRef] [PubMed]
- Gschwandtner, M.; Schakel, K.; Werfel, T.; Gutzmer, R. Histamine H4 receptor activation on human slan-dendritic cells down-regulates their pro-inflammatory capacity. Immunology 2011, 132, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.; Ahrens, K.; Devalaraja, M.; Dymond, M.; Fagura, M.; Hargreaves, A.; Holt, A.; Peers, I.; Price, S.; Reens, J.; et al. Use of a canine model of atopic dermatitis to investigate the efficacy of a CCR4 antagonist in allergen-induced skin inflammation in a randomized study. J. Investig. Dermatol. 2016, 136, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Morita, E.; Takahashi, H.; Niihara, H.; Dekio, I.; Sumikawa, Y.; Murakami, Y.; Matsunaka, H. Stratum corneum tarc level is a new indicator of lesional skin inflammation in atopic dermatitis. Allergy 2010, 65, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Miyahara, H.; Okazaki, N.; Nagakura, T.; Korematsu, S.; Izumi, T. Elevated umbilical cord serum TARC/CCL17 levels predict the development of atopic dermatitis in infancy. Clin. Exp. Allergy 2011, 41, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Onoue, A.; Kabashima, K.; Kobayashi, M.; Mori, T.; Tokura, Y. Induction of eosinophIL- and Th2-attracting epidermal chemokines and cutaneous late-phase reaction in tape-stripped skin. Exp. Dermatol. 2009, 18, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Miyano, K.; Matsushita, S.; Tsuchida, T.; Nakamura, K. Inhibitory effect of a histamine 4 receptor antagonist on CCL17 and CCL22 production by monocyte-derived langerhans cells in patients with atopic dermatitis. J. Dermatol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kleinewietfeld, M.; Puentes, F.; Borsellino, G.; Battistini, L.; Rotzschke, O.; Falk, K. CCR6 expression defines regulatory effector/memory-like cells within the CD25+CD4+ T-cell subset. Blood 2005, 105, 2877–2886. [Google Scholar] [CrossRef] [PubMed]
- Esche, C.; de Benedetto, A.; Beck, L.A. Keratinocytes in atopic dermatitis: Inflammatory signals. Curr. Allergy Asthma Rep. 2004, 4, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Gahr, N.; Folster-Holst, R.; Weichenthal, M.; Christophers, E.; Schroder, J.M.; Bartels, J. Dermal fibroblasts from acute inflamed atopic dermatitis lesions display increased eotaxin/CCL11 responsiveness to interleukin-4 stimulation. Br. J. Dermatol. 2011, 164, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Yawalkar, N.; Uguccioni, M.; Scharer, J.; Braunwalder, J.; Karlen, S.; Dewald, B.; Braathen, L.R.; Baggiolini, M. Enhanced expression of eotaxin and CCR3 in atopic dermatitis. J. Investig. Dermatol. 1999, 113, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Park, C.W.; Lee, B.H.; Han, H.J.; Lee, C.H.; Ahn, H.K. Tacrolimus decreases the expression of eotaxin, CCR3, rantes and interleukin-5 in atopic dermatitis. Br. J. Dermatol. 2005, 152, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.; Lan, H.; Zeng, K.; Zhao, X. Serum fractalkine (CX3CL1) concentration correlates with clinical severity in pediatric atopic dermatitis patients. Ann. Clin. Lab. Sci. 2016, 46, 168–173. [Google Scholar] [PubMed]
- Echigo, T.; Hasegawa, M.; Shimada, Y.; Takehara, K.; Sato, S. Expression of fractalkine and its receptor, CX3CR1, in atopic dermatitis: Possible contribution to skin inflammation. J. Allergy Clin. Immunol. 2004, 113, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Siegel, A.M.; Stone, K.D.; Cruse, G.; Lawrence, M.G.; Olivera, A.; Jung, M.Y.; Barber, J.S.; Freeman, A.F.; Holland, S.M.; O’Brien, M.; et al. Diminished allergic disease in patients with STAT3 mutations reveals a role for STAT3 signaling in mast cell degranulation. J. Allergy Clin. Immunol. 2013, 132, 1388–1396. [Google Scholar] [CrossRef] [PubMed]
- Mekori, Y.A.; Metcalfe, D.D. Mast cell-T cell interactions. J. Allergy Clin. Immunol. 1999, 104, 517–523. [Google Scholar] [CrossRef]
- Theiner, G.; Gessner, A.; Lutz, M.B. The mast cell mediator PGD2 suppresses IL-12 release by dendritic cells leading to Th2 polarized immune responses in vivo. Immunobiology 2006, 211, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Horsmanheimo, L.; Harvima, I.T.; Jarvikallio, A.; Harvima, R.J.; Naukkarinen, A.; Horsmanheimo, M. Mast cells are one major source of interleukin-4 in atopic dermatitis. Br. J. Dermatol. 1994, 131, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Nagarkar, D.R.; Poposki, J.A.; Comeau, M.R.; Biyasheva, A.; Avila, P.C.; Schleimer, R.P.; Kato, A. Airway epithelial cells activate Th2 cytokine production in mast cells through IL-1 and thymic stromal lymphopoietin. J. Allergy Clin. Immunol. 2012, 130, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Gauchat, J.F.; Henchoz, S.; Mazzei, G.; Aubry, J.P.; Brunner, T.; Blasey, H.; Life, P.; Talabot, D.; Flores-Romo, L.; Thompson, J.; et al. Induction of human IgE synthesis in B cells by mast cells and basophils. Nature 1993, 365, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Jawdat, D.M.; Albert, E.J.; Rowden, G.; Haidl, I.D.; Marshall, J.S. IgE-mediated mast cell activation induces langerhans cell migration in vivo. J. Immunol. 2004, 173, 5275–5282. [Google Scholar] [CrossRef] [PubMed]
- Suto, H.; Nakae, S.; Kakurai, M.; Sedgwick, J.D.; Tsai, M.; Galli, S.J. Mast cell-associated tnf promotes dendritic cell migration. J. Immunol. 2006, 176, 4102–4112. [Google Scholar] [CrossRef] [PubMed]
- Modena, B.D.; Dazy, K.; White, A.A. Emerging concepts: Mast cell involvement in allergic diseases. Transl. Res. 2016, 174, 98–121. [Google Scholar] [CrossRef] [PubMed]
- Saluja, R.; Ketelaar, M.E.; Hawro, T.; Church, M.K.; Maurer, M.; Nawijn, M.C. The role of the IL-33/IL-1RL1 axis in mast cell and basophil activation in allergic disorders. Mol. Immunol. 2015, 63, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Sehra, S.; Serezani, A.P.; Ocana, J.A.; Travers, J.B.; Kaplan, M.H. Mast cells regulate epidermal barrier function and the development of allergic skin inflammation. J. Investig. Dermatol. 2016, 136, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Braathen, L.R.; Simon, H.U. Eosinophils and atopic dermatitis. Allergy 2004, 59, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chen, Q.; Su, S.B.; Zhang, P.; Kurosaka, K.; Caspi, R.R.; Michalek, S.M.; Rosenberg, H.F.; Zhang, N.; Oppenheim, J.J. EosinophIL-derived neurotoxin acts as an alarmin to activate the TLR2-MyD88 signal pathway in dendritic cells and enhances Th2 immune responses. J. Exp. Med. 2008, 205, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Abu-Ghazaleh, R.I.; Gleich, G.J.; Prendergast, F.G. Interaction of eosinophil granule major basic protein with synthetic lipid bilayers: A mechanism for toxicity. J. Membr. Biol. 1992, 128, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Kagi, M.K.; Joller-Jemelka, H.; Wuthrich, B. Correlation of eosinophils, eosinophil cationic protein and soluble interleukin-2 receptor with the clinical activity of atopic dermatitis. Dermatology 1992, 185, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.F.; Wong, C.K.; Ho, A.W.; Hu, S.; Chen, D.P.; Lam, C.W. Activation of human eosinophils and epidermal keratinocytes by Th2 cytokine IL-31: Implication for the immunopathogenesis of atopic dermatitis. Int. Immunol. 2010, 22, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Kunsleben, N.; Rudrich, U.; Gehring, M.; Novak, N.; Kapp, A.; Raap, U. IL-31 induces chemotaxis, calcium mobilization, release of reactive oxygen species, and CCL26 in eosinophils, which are capable to release IL-31. J. Investig. Dermatol. 2015, 135, 1908–1911. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Wang, K.; Siracusa, M.C.; Saenz, S.A.; Brestoff, J.R.; Monticelli, L.A.; Noti, M.; Tait Wojno, E.D.; Fung, T.C.; Kubo, M.; et al. Basophils promote innate lymphoid cell responses in inflamed skin. J. Immunol. 2014, 193, 3717–3725. [Google Scholar] [CrossRef] [PubMed]
- Jiao, D.; Wong, C.K.; Qiu, H.N.; Dong, J.; Cai, Z.; Chu, M.; Hon, K.L.; Tsang, M.S.; Lam, C.W. NOD2 and TLR2 ligands trigger the activation of basophils and eosinophils by interacting with dermal fibroblasts in atopic dermatitis-like skin inflammation. Cell. Mol. Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Chu, I.M.; Hon, K.L.; Tsang, M.S.; Lam, C.W. Aberrant expression of bacterial pattern recognition receptor NOD2 of basophils and microbicidal peptides in atopic dermatitis. Molecules 2016, 21, 417. [Google Scholar] [CrossRef] [PubMed]
- Spits, H.; Cupedo, T. Innate lymphoid cells: Emerging insights in development, lineage relationships, and function. Annu. Rev. Immunol. 2012, 30, 647–675. [Google Scholar] [CrossRef] [PubMed]
- Salimi, M.; Barlow, J.L.; Saunders, S.P.; Xue, L.; Gutowska-Owsiak, D.; Wang, X.; Huang, L.C.; Johnson, D.; Scanlon, S.T.; McKenzie, A.N.; et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J. Exp. Med. 2013, 210, 2939–2950. [Google Scholar] [CrossRef] [PubMed]
- Roediger, B.; Kyle, R.; Le Gros, G.; Weninger, W. Dermal group 2 innate lymphoid cells in atopic dermatitis and allergy. Curr. Opin. Immunol. 2014, 31, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Doherty, T.A.; Broide, D.H. Group 2 innate lymphoid cells: New players in human allergic diseases. J. Investig. Allergol. Clin. Immunol. 2015, 25, 1–11. [Google Scholar] [PubMed]
- Saunders, S.P.; Moran, T.; Floudas, A.; Wurlod, F.; Kaszlikowska, A.; Salimi, M.; Quinn, E.M.; Oliphant, C.J.; Nunez, G.; McManus, R.; et al. Spontaneous atopic dermatitis is mediated by innate immunity, with the secondary lung inflammation of the atopic march requiring adaptive immunity. J. Allergy Clin. Immunol. 2016, 137, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S. Innate lymphoid cells in the skin. J. Investig. Dermatol. 2015, 135, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Savinko, T.; Matikainen, S.; Saarialho-Kere, U.; Lehto, M.; Wang, G.; Lehtimaki, S.; Karisola, P.; Reunala, T.; Wolff, H.; Lauerma, A.; et al. IL-33 and ST2 in atopic dermatitis: Expression profiles and modulation by triggering factors. J. Investig. Dermatol. 2012, 132, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Salimi, M.; Panse, I.; Mjosberg, J.M.; McKenzie, A.N.; Spits, H.; Klenerman, P.; Ogg, G. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on Th2 cells. J. Allergy Clin. Immunol. 2014, 133, 1184–1194. [Google Scholar] [CrossRef] [PubMed]
- Halim, T.Y.; Hwang, Y.Y.; Scanlon, S.T.; Zaghouani, H.; Garbi, N.; Fallon, P.G.; McKenzie, A.N. Group 2 innate lymphoid cells license dendritic cells to potentiate memory Th2 cell responses. Nat. Immunol. 2016, 17, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Roediger, B.; Kyle, R.; Yip, K.H.; Sumaria, N.; Guy, T.V.; Kim, B.S.; Mitchell, A.J.; Tay, S.S.; Jain, R.; Forbes-Blom, E.; et al. Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat. Immunol. 2013, 14, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Siracusa, M.C.; Saenz, S.A.; Noti, M.; Monticelli, L.A.; Sonnenberg, G.F.; Hepworth, M.R.; van Voorhees, A.S.; Comeau, M.R.; Artis, D. Tslp elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.Y. Gata3: A master of many trades in immune regulation. Trends Immunol. 2014, 35, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Ho, I.C.; Tai, T.S.; Pai, S.Y. Gata3 and the T-cell lineage: Essential functions before and after T-helper-2-cell differentiation. Nat. Rev. Immunol. 2009, 9, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Milner, J.D. Altered T-cell receptor signaling in the pathogenesis of allergic disease. J. Allergy Clin. Immunol. 2011, 127, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Choi, E.J.; Choi, H.; Lee, K.S.; Kim, H.R.; Na, B.R.; Kwon, M.S.; Jeong, G.S.; Choi, H.G.; Choi, E.Y.; et al. Oral administration of 4-hydroxy-3-methoxycinnamaldehyde attenuates atopic dermatitis by inhibiting T cell and keratinocyte activation. PLoS ONE 2015, 10, e0144521. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Myers, R.A.; Lee, J.H.; Igartua, C.; Lee, K.E.; Kim, Y.H.; Kim, E.J.; Yoon, D.; Lee, J.S.; Hirota, T.; et al. Genome-wide association study of recalcitrant atopic dermatitis in Korean children. J. Allergy Clin. Immunol. 2015, 136, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Hussein, Y.M.; Alzahrani, S.S.; Alharthi, A.A.; Alhazmi, A.S.; Ghonaim, M.M.; Alghamdy, A.A.; El Askary, A. Gene polymorphism of interleukin-4, interleukin-4 receptor and STAT6 in children with atopic dermatitis in Taif, Saudi Arabia. Immunol. Investig. 2016, 45, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Q.; Shi, H.Z. CD28/CTLA-4–CD80/CD86 and ICOS–B7RP-1 costimulatory pathway in bronchial asthma. Allergy 2006, 61, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C. Does therapeutic intervention in atopic dermatitis normalize epidermal notch deficiency? Exp. Dermatol. 2014, 23, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C. The potential role of impaired notch signalling in atopic dermatitis. Acta Derm. Venereol. 2015, 95, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, C.; Liu, Z.; Liu, X.; Han, C.; Cao, X.; Li, N. Notch signal suppresses toll-like receptor-triggered inflammatory responses in macrophages by inhibiting extracellular signal-regulated kinase 1/2-mediated nuclear factor κB activation. J. Biol. Chem. 2012, 287, 6208–6217. [Google Scholar] [CrossRef] [PubMed]
- Bailis, W.; Yashiro-Ohtani, Y.; Fang, T.C.; Hatton, R.D.; Weaver, C.T.; Artis, D.; Pear, W.S. Notch simultaneously orchestrates multiple helper t cell programs independently of cytokine signals. Immunity 2013, 39, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.R.; The, L.; Batia, L.M.; Beattie, K.; Katibah, G.E.; McClain, S.P.; Pellegrino, M.; Estandian, D.M.; Bautista, D.M. The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell 2013, 155, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M.; Hatano, Y.; Williams, M.L. Basis for the barrier abnormality in atopic dermatitis: Outside-inside-outside pathogenic mechanisms. J. Allergy Clin. Immunol. 2008, 121, 1337–1343. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Gene | Locus | Alleles or Mutation | SNP | Population | Disease Severity | Reference |
|---|---|---|---|---|---|---|
| Epidermal Differentiation Complex | ||||||
| FLG | 1q21.3 | Loss-of-function Copy number variation | R501X, 2282del4 | European, Chinese, Singaporean | Increases the risk of AD correlated with severity | [14,16,18,19,20,21,22] |
| FLG2 | 1q21.3 | Premature stop codon | rs12568784, Q2053del224 rs16833974 | African American | More persistent AD | [23] |
| SPINK5 | 5q31 | Loss-of-function | rs2303070 T | Taiwanese | Increases the risk of AD | [30] |
| E420K | Italian | Increases the risk of AD | [31] | |||
| SPRR3 | 1q21.3 | Copy number variation | rs28989168 | German | Increases the risk of AD | [25] |
| TMEM79 | Missense mutation | rs6684514 | Ireland | Increases the risk of AD | [27] | |
| claudin-1 | 3q28 | Haplotype-tagging | rs893051 | African American | Increases the risk of AD | [32] |
| AG or GG genotype | rs9290929 | Korean | Mold infection | [33] | ||
| Innate Immunity | ||||||
| TLR2 | 4 | Missense mutation | R753Q | German, Italian | Severe AD | [40] |
| A | 16934T | German, Japanese | Severe AD | [41,42] | ||
| TLR4 | 9 | N/A | D299G | Italian | Increased in AD | [43] |
| 896A/G | N/A | Ukrainian | Increased viral respiratory infections | [44] | ||
| TLR9 | 3 | TT | C-1237T | German | Intrinsic AD | [45] |
| NOD1 | N/A | rs2907748 rs2907749 | German | Allergen sensitization | [46] | |
| hBD 1 | 8 | haplotype CT | rs5743399 | Korean | allergen sensitization | [47] |
| N/A | rs5743409 | Korean | AD | [47] | ||
| TSLP | 5q22 | C/T | rs1898671 | European American | Eczema herpeticum | [48] |
| Adaptive Immunity | ||||||
| IL-18 | 11q22 | G-allele | rs1946518 rs187238 | Chinese | Low risk of AD | [51] |
| IL18RAP | 2q12 | N/A | rs6419573 | Japanese | Increase the risk of AD | [35] |
| IL-12 | chr3 | IVS-798A/T, haplotype TA | rs582504, rs582054, rs2243151 | Korean | Increase the risk of AD | [53] |
| IL-12RB | chr5 chr1 | TT AA | rs438421, rs2066446 | Korean | Allergen sensitization | [53] |
| IFNG/IFNGR1 | 12/6q23-24 | Loss-of-function | V14M and Y397C | African American | Eczema herpeticum | [52] |
| IL-4 | 5q31-33 | T allele | 590 C/T of IL-4 promoter | Egyptian | Increase the risk of AD | [58] |
| IL-4Rα | 16 | Gain of function | I50V, Q576R | Egyptian | Increase the risk of AD | [58] |
| IL-13 | 5q31.1 | N/A | rs12188917 | Association with asthma | [34] | |
| STAT6 | 12 | Minor allele homozygotes | rs324011 | German | Low risk of AD | [59] |
| IL-31 | 12 | Haplotype AAA and GAA | 1066, −2057, and ivs2 + 12 | polish | High IL-31 serum level severe pruritus | [60] |
| IL-17A | AA genotype | 152 G/A | polish | Severe AD in coexistence of asthma | [61] | |
| FCER1A | 1 | N/A | promoter | Japanese | Allergen sensitization | [74] |
| Chemokines | N/A | |||||
| RANTES | 17.35 | 28G | N/A | German | Allergen sensitization | [69] |
| 403A overexpression | N/A | Japanese, German | Allergen sensitization | [70] | ||
| Vitamin D Pathway | ||||||
| Cyp24a1 | 20q54 | Major C allele | rs2248359 | German | Severe AD | [55] |
| VDR | 20q13 | AT | rs7975232 | Chinese | Severe, eosinophilia and high IgE levels | [54] |
| Nerve Growth Factor Pathway | ||||||
| BDNF | 11 | T | C270T | Chinese | Intrinsic AD and male sex | [77] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.E.; Kim, J.S.; Cho, D.H.; Park, H.J. Molecular Mechanisms of Cutaneous Inflammatory Disorder: Atopic Dermatitis. Int. J. Mol. Sci. 2016, 17, 1234. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17081234
Kim JE, Kim JS, Cho DH, Park HJ. Molecular Mechanisms of Cutaneous Inflammatory Disorder: Atopic Dermatitis. International Journal of Molecular Sciences. 2016; 17(8):1234. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17081234
Chicago/Turabian StyleKim, Jung Eun, Jong Sic Kim, Dae Ho Cho, and Hyun Jeong Park. 2016. "Molecular Mechanisms of Cutaneous Inflammatory Disorder: Atopic Dermatitis" International Journal of Molecular Sciences 17, no. 8: 1234. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17081234