
The Antitumor Effect of Metformin Is Mediated by miR-26a in Breast Cancer

Abstract
:1. Introduction
2. Results
2.1. miR-26a Expression Modulates Cell Viability
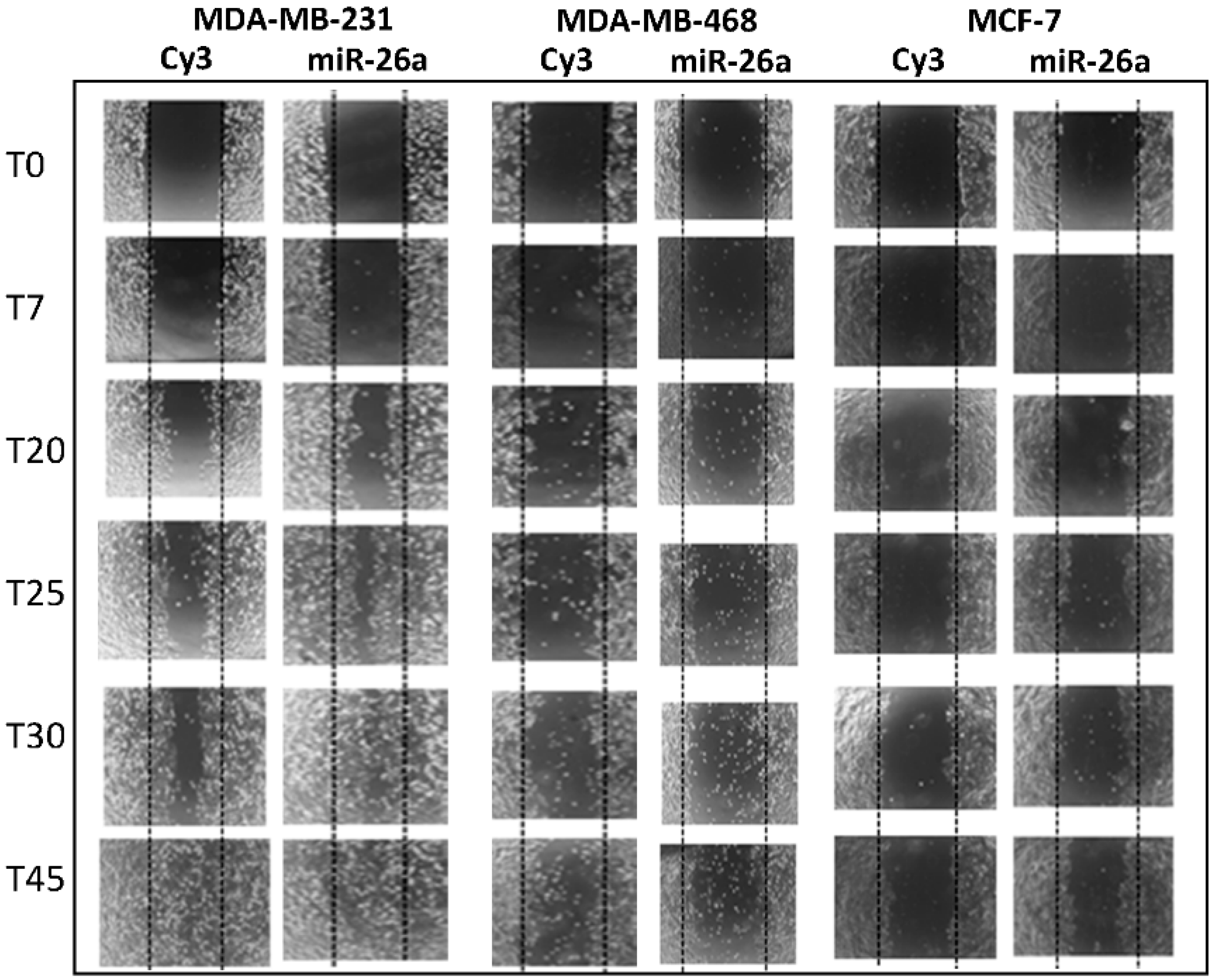
2.2. Effect of miR-26a on Cell Migration
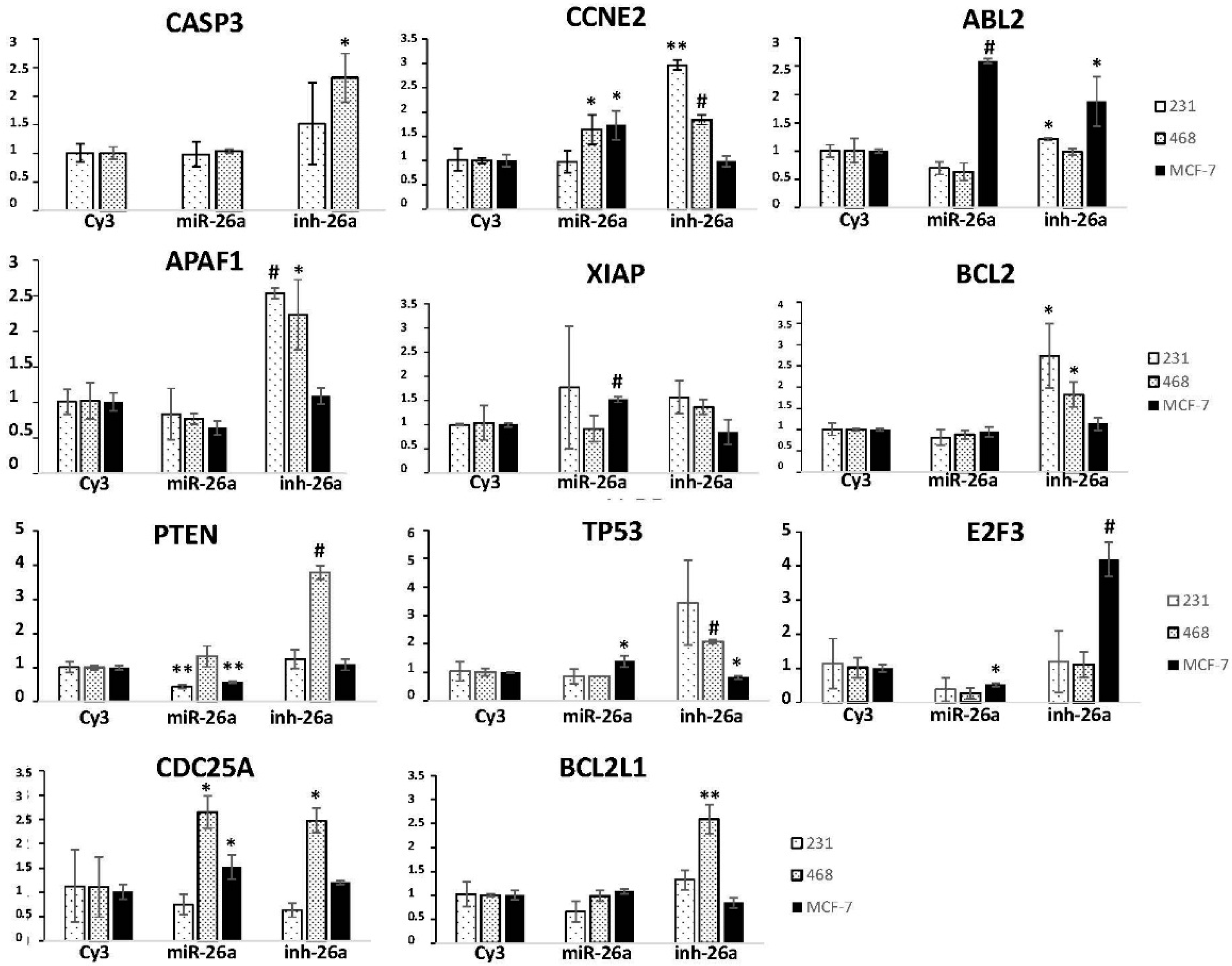
2.3. Evaluation of Potential miR-26a Targets
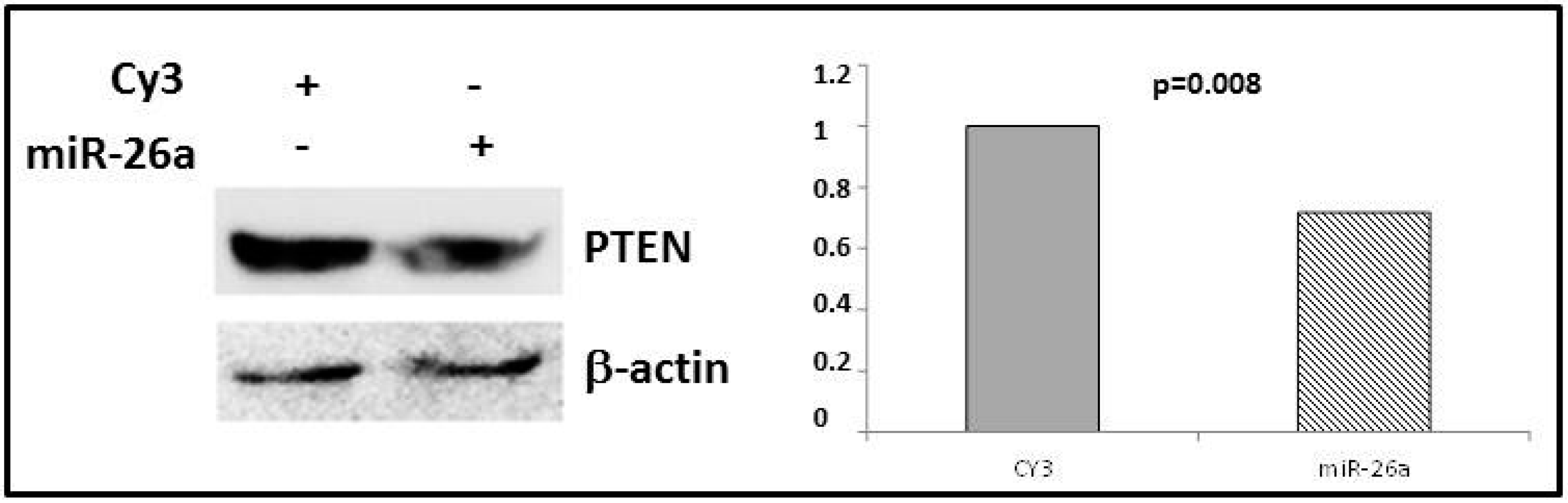
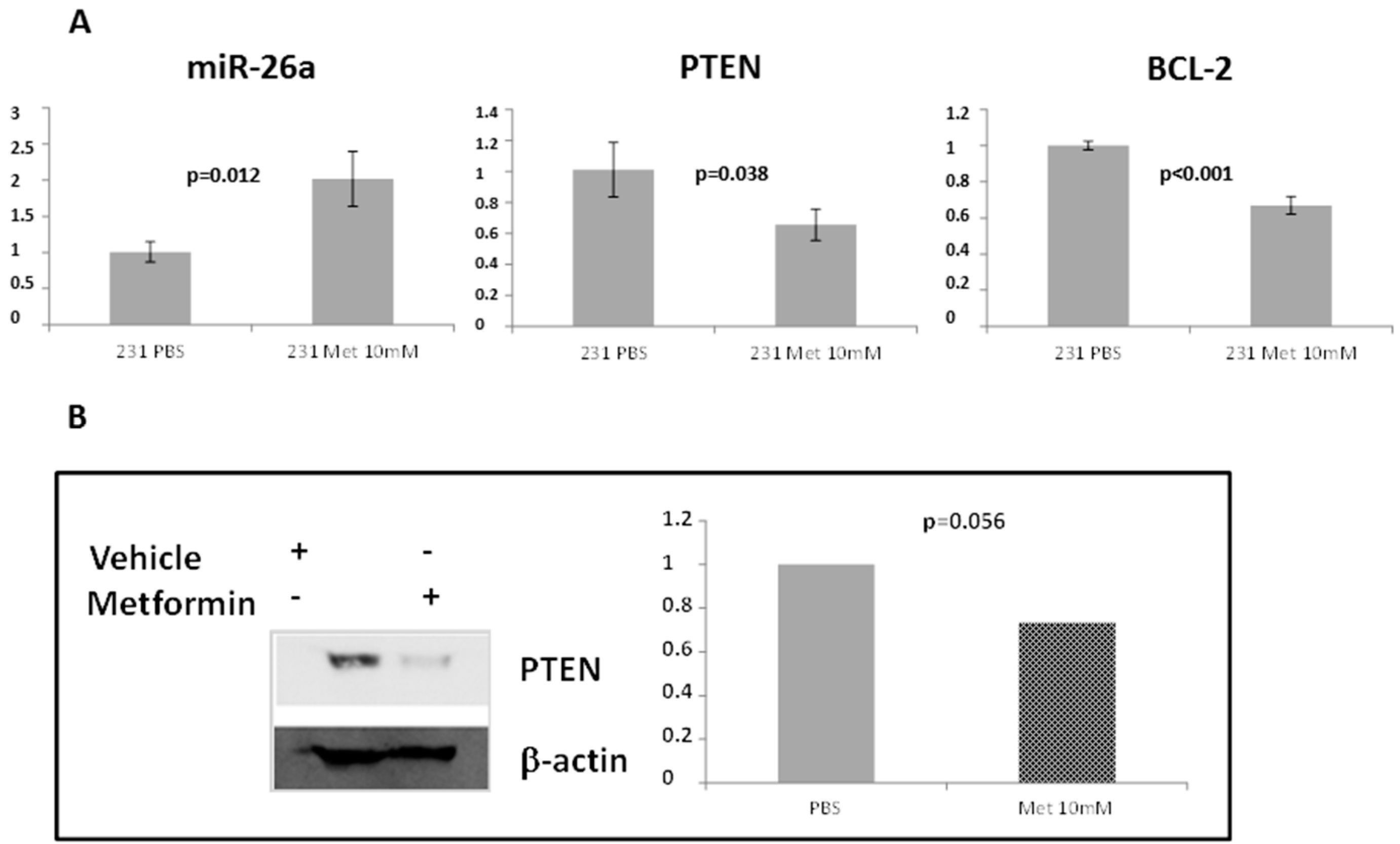
2.4. Phosphatase and Tensin Homolog (PTEN) Regulation by miR-26a
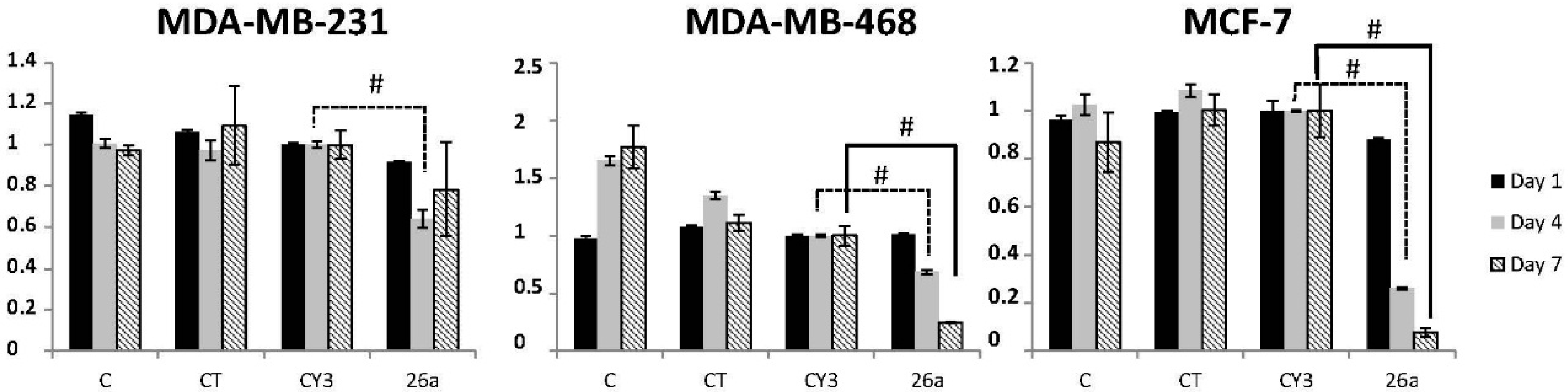
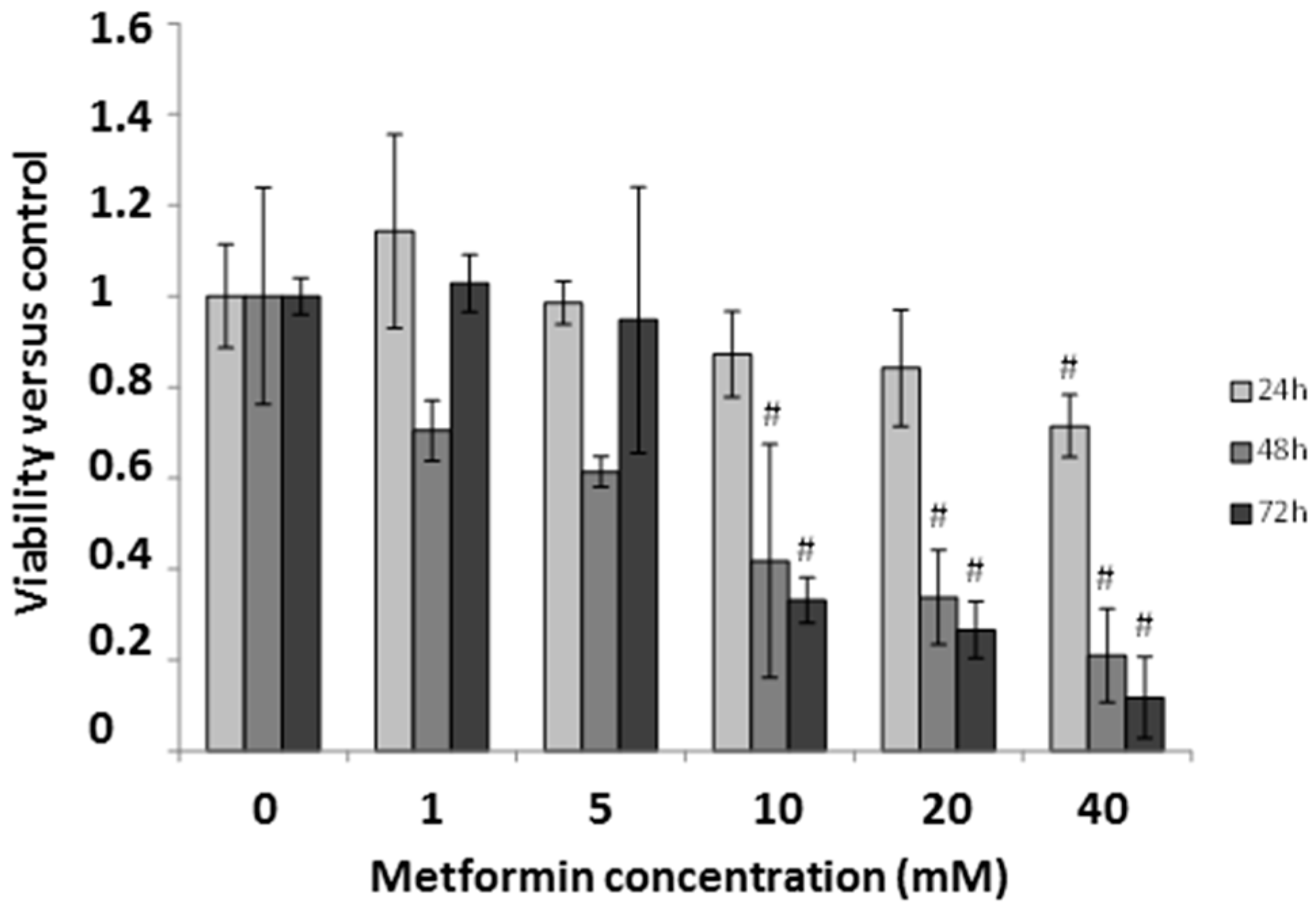
2.5. Effect of Metformin on Breast Cancer Cells
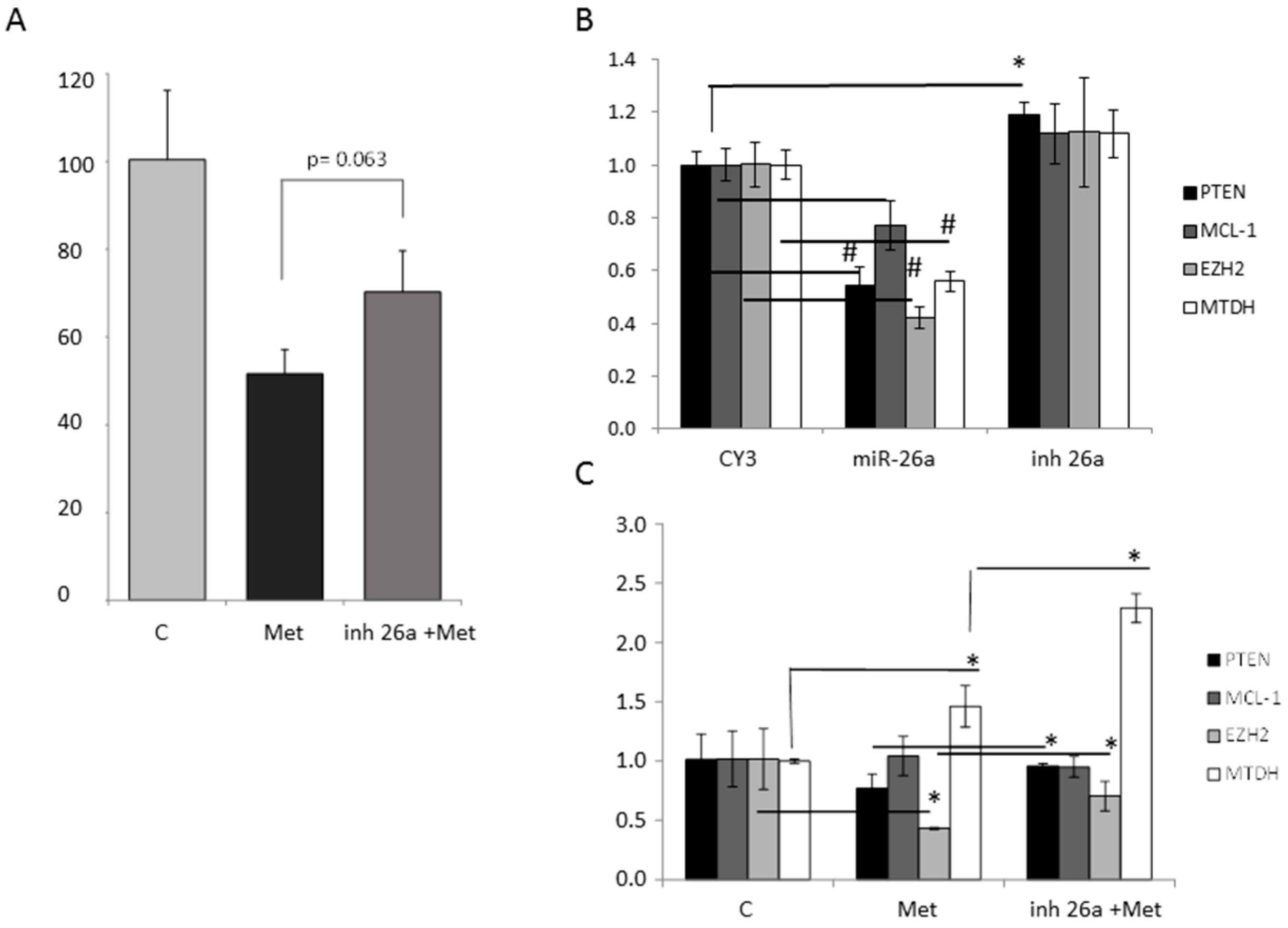
2.6. Effect of Metformin through miR-26a on Breast Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. Cell Viability Assay
4.3. MicroRNA Transfection
4.4. Analysis of miRNA and mRNA Expression by Quantitative Real-Time PCR
4.5. Western Blot Assays
4.6. Wound Healing Assay
4.7. Bioinformatic Databases
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Eroles, P.; Bosch, A.; Perez-Fidalgo, J.A.; Lluch, A. Molecular biology in breast cancer: Intrinsic subtypes and signaling pathways. Cancer Treat. Rev. 2012, 38, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Rivenbark, A.G.; O'Connor, S.M.; Coleman, W.B. Molecular and cellular heterogeneity in breast cancer: Challenges for personalized medicine. Am. J. Pathol. 2013, 183, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.R.; Brown, M.; Cress, R.D.; Parise, C.A.; Caggiano, V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: A population-based study from the California cancer Registry. Cancer 2007, 109, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Dees, E.C.; Sawyer, L.; Gatti, L.; Moore, D.T.; Collichio, F.; Ollila, D.W.; Sartor, C.I.; Graham, M.L.; Perou, C.M. The triple negative paradox: Primary tumor chemosensitivity of breast cancer subtypes. Clin. Cancer Res. 2007, 13, 2329–2334. [Google Scholar] [CrossRef] [PubMed]
- Libby, G.; Donnelly, L.A.; Donnan, P.T.; Alessi, D.R.; Morris, A.D.; Evans, J.M. New users of metformin are at low risk of incident cancer: A cohort study among people with type 2 diabetes. Diabetes Care 2009, 32, 1620–1625. [Google Scholar] [CrossRef] [PubMed]
- Quinn, B.J.; Kitagawa, H.; Memmott, R.M.; Gills, J.J.; Dennis, P.A. Repositioning metformin for cancer prevention and treatment. Trends Endocrinol. Metab. 2013, 24, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.N.; Nam, J.W. Genomics of microRNA. Trends Genet. 2006, 22, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.L.; Kim, H.; Ying, S.Y. Intron-mediated RNA interference and microRNA (miRNA). Front. Biosci. 2008, 13, 2216–2230. [Google Scholar] [CrossRef] [PubMed]
- Caramuta, S.; Egyhazi, S.; Rodolfo, M.; Witten, D.; Hansson, J.; Larsson, C.; Lui, W.O. MicroRNA expression profiles associated with mutational status and survival in malignant melanoma. J. Investig. Dermatol. 2010, 130, 2062–2070. [Google Scholar] [CrossRef] [PubMed]
- Dillhoff, M.; Liu, J.; Frankel, W.; Croce, C.; Bloomston, M. MicroRNA-21 is overexpressed in pancreatic cancer and a potential predictor of survival. J. Gastrointest. Surg. 2008, 12, 2171–2176. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Ding, J.; Wu, K.; Fan, D. Survival prediction of gastric cancer by a seven-microRNA signature. Gut 2010, 59, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Shimizu, M.; Barbarotto, E.; Nicoloso, M.S.; Dimitri, F.; Sampath, D.; Fabbri, M.; Lerner, S.; Barron, L.L.; Rassenti, L.Z.; et al. microRNA fingerprinting of CLL patients with chromosome 17p deletion identify a miR-21 score that stratifies early survival. Blood 2010, 116, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K.; Yi, M.; Stephens, R.M.; Okamoto, A.; Yokota, J.; Tanaka, T.; et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006, 9, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Zhang, J.; Qian, X.; Han, L.; Zhang, K.; Chen, L.; Liu, J.; Ren, Y.; Yang, M.; Zhang, A.; et al. AC1MMYR2, an inhibitor of dicer-mediated biogenesis of Oncomir miR-21, reverses epithelial-mesenchymal transition and suppresses tumor growth and progression. Cancer Res. 2013, 73, 5519–5531. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.G. miR-34—A microRNA replacement therapy is headed to the clinic. Front. Genet. 2012, 3, 120. [Google Scholar] [CrossRef] [PubMed]
- Trang, P.; Wiggins, J.F.; Daige, C.L.; Cho, C.; Omotola, M.; Brown, D.; Weidhaas, J.B.; Bader, A.G.; Slack, F.J. Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol. Ther. 2011, 19, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Tormo, E.; Pineda, B.; Serna, E.; Guijarro, A.; Ribas, G.; Fores, J.; Chirivella, E.; Climent, J.; Lluch, A.; Eroles, P. MicroRNA Profile in Response to Doxorubicin Treatment in Breast Cancer. J. Cell. Biochem. 2015, 116, 2061–2073. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.Q.; Wang, J.J.; Yan, J.S.; Huang, J.H.; Li, W.; Che, J.P.; Wang, G.C.; Liu, M.; Zheng, J.H. Metformin inhibits cell growth by upregulating microRNA-26a in renal cancer cells. Int. J. Clin. Exp. Med. 2014, 7, 3289–3296. [Google Scholar] [PubMed]
- Bao, B.; Wang, Z.; Ali, S.; Ahmad, A.; Azmi, A.S.; Sarkar, S.H.; Banerjee, S.; Kong, D.; Li, Y.; Thakur, S.; et al. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prev. Res. (Phila) 2012, 5, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Li, L.; Wu, M.; Liu, M.; Xie, X.; Guo, J.; Tang, H. MiR-26a inhibits proliferation and migration of breast cancer through repression of MCL-1. PLoS ONE 2013, 8, e65138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, X.X.; He, J.R.; Zhou, C.X.; Guo, M.; He, M.; Li, M.F.; Chen, G.Q.; Zhao, Q. Pathologically decreased miR-26a antagonizes apoptosis and facilitates carcinogenesis by targeting MTDH and EZH2 in breast cancer. Carcinogenesis 2011, 32, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Eroles, P.; Tormo, E.; Pineda, B.; Espin, E.; Lluch, A. MicroRNAs in breast cancer: One More Turn in Regulation. Curr. Drug Targets 2015, in press. [Google Scholar]
- Kasinski, A.L.; Slack, F.J. Epigenetics and genetics. MicroRNAs en route to the clinic: Progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer 2011, 11, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zheng, J.; Zhang, Y.; Yang, L.; Wang, J.; Ni, J.; Cui, D.; Yu, C.; Cai, Z. Tumor-specific expression of microRNA-26a suppresses human hepatocellular carcinoma growth via cyclin-dependent and -independent pathways. Mol. Ther. 2011, 19, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wu, X.; Wang, C.; Liu, Y.; Zhou, Q.; Xu, K. MiR-26a enhances metastasis potential of lung cancer cells via AKT pathway by targeting PTEN. Biochim. Biophys. Acta 2012, 1822, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Tang, H.; Chen, B.; He, Z.; Deng, M.; Wu, M.; Liu, X.; Yang, L.; Ye, F.; Xie, X. miR-26a suppresses tumour proliferation and metastasis by targeting metadherin in triple negative breast cancer. Cancer Lett. 2015, 357, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.P.; Reijm, E.A.; Sieuwerts, A.M.; Ruigrok-Ritstier, K.; Look, M.P.; Rodriguez-Gonzalez, F.G.; Heine, A.A.; Martens, J.W.; Sleijfer, S.; Foekens, J.A.; et al. High miR-26a and low CDC2 levels associate with decreased EZH2 expression and with favorable outcome on tamoxifen in metastatic breast cancer. Breast Cancer Res. Treat. 2012, 133, 937–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.; Ding, K.; Li, R.; Zhang, W.; Li, G.; Kong, X.; Qian, P.; Lobie, P.E.; Zhu, T. Identification of miR-26 as a key mediator of estrogen stimulated cell proliferation by targeting CHD1, GREB1 and KPNA2. Breast Cancer Res. 2014, 16, R40. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kwon, H.; Lee, J.W.; Lee, S.B.; Park, H.S.; Sohn, G.; Lee, Y.; Koh, B.S.; Yu, J.H.; Son, B.H.; et al. Metformin increases survival in hormone receptor-positive, HER2-positive breast cancer patients with diabetes. Breast Cancer Res. 2015, 17, 64. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, S.; Hernadez-Aya, L.F.; Lei, X.; Meric-Bernstam, F.; Litton, J.K.; Hsu, L.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Effect of metformin on survival outcomes in diabetic patients with triple receptor-negative breast cancer. Cancer 2012, 118, 1202–1211. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Esteva, F.J.; Ensor, J.; Hortobagyi, G.N.; Lee, M.H.; Yeung, S.C. Metformin and thiazolidinediones are associated with improved breast cancer-specific survival of diabetic women with HER2+ breast cancer. Ann. Oncol. 2012, 23, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N.; Berstein, L.M.; Egormin, P.A.; Piskunova, T.S.; Popovich, I.G.; Zabezhinski, M.A.; Kovalenko, I.G.; Poroshina, T.E.; Semenchenko, A.V.; Provinciali, M.; et al. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/Neu transgenic mice. Exp. Gerontol. 2005, 40, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Laurent, K.; Loubat, A.; Giorgetti-Peraldi, S.; Colosetti, P.; Auberger, P.; Tanti, J.F.; Le Marchand-Brustel, Y.; Bost, F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008, 27, 3576–3586. [Google Scholar] [CrossRef] [PubMed]
- Dowling, R.J.; Zakikhani, M.; Fantus, I.G.; Pollak, M.; Sonenberg, N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007, 67, 10804–10812. [Google Scholar] [CrossRef] [PubMed]
- Martin-Castillo, B.; Dorca, J.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Lopez-Bonet, E.; Garcia, M.; del Barco, S.; Menendez, J.A. Incorporating the antidiabetic drug metformin in HER2-positive breast cancer treated with neo-adjuvant chemotherapy and trastuzumab: An ongoing clinical-translational research experience at the Catalan Institute of Oncology. Ann. Oncol. 2010, 21, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yuan, Y.; Huang, L.; Qiao, M.; Zhang, Y. Metformin alters the expression profiles of microRNAs in human pancreatic cancer cells. Diabetes Res. Clin. Pract. 2012, 96, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Huse, J.T.; Brennan, C.; Hambardzumyan, D.; Wee, B.; Pena, J.; Rouhanifard, S.H.; Sohn-Lee, C.; le Sage, C.; Agami, R.; Tuschl, T.; et al. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 2009, 23, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Huang, W.; Jiang, X.; Pennicooke, B.; Park, P.J.; Johnson, M.D. Integrative genome analysis reveals an oncomir/oncogene cluster regulating glioblastoma survivorship. Proc. Natl. Acad. Sci. USA 2010, 107, 2183–2188. [Google Scholar] [CrossRef] [PubMed]
- Hou, G.; Zhang, S.; Zhang, X.; Wang, P.; Hao, X.; Zhang, J. Clinical pathological characteristics and prognostic analysis of 1,013 breast cancer patients with diabetes. Breast Cancer Res. Treat. 2013, 137, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Lega, I.C.; Austin, P.C.; Gruneir, A.; Goodwin, P.J.; Rochon, P.A.; Lipscombe, L.L. Association between metformin therapy and mortality after breast cancer: A population-based study. Diabetes Care 2013, 36, 3018–3026. [Google Scholar] [CrossRef] [PubMed]
- Peeters, P.J.; Bazelier, M.T.; Vestergaard, P.; Leufkens, H.G.; Schmidt, M.K.; de Vries, F.; de Bruin, M.L. Use of metformin and survival of diabetic women with breast cancer. Curr. Drug Saf. 2003, 8, 357–363. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhang, S.; Hou, G.; Zhang, X.; Hao, X.; Zhang, J. Clinical pathological characteristics and prognostic analysis of diabetic women with luminal subtype breast cancer. Tumour Biol. 2014, 35, 2035–2045. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Liang, J.; Yang, X.; Wang, Y.; Zhao, Y.; Wu, H.; Sun, L.; Zhang, Y.; Chen, Y.; Li, R.; et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol. Cell. Biol. 2007, 27, 5105–5119. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Kleine-Kohlbrecher, D.; Dietrich, N.; Pasini, D.; Gargiulo, G.; Beekman, C.; Theilgaard-Monch, K.; Minucci, S.; Porse, B.T.; Marine, J.C.; et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007, 21, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Chourrout, D.; Vervoort, M.; Leblanc, B.; Cavalli, G. Genome regulation by polycomb and trithorax proteins. Cell 2007, 128, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [PubMed]
- Heering, J.; Erlmann, P.; Olayioye, M.A. Simultaneous loss of the DLC1 and PTEN tumor suppressors enhances breast cancer cell migration. Exp. Cell Res. 2009, 315, 2505–2514. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Gu, J.; Matsumoto, K.; Aota, S.; Parsons, R.; Yamada, K.M. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 1998, 280, 1614–1617. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Gu, J.; Takino, T.; Yamada, K.M. Tumor suppressor PTEN inhibition of cell invasion, migration, and growth: Differential involvement of focal adhesion kinase and p130Cas. Cancer Res. 1999, 59, 442–449. [Google Scholar] [PubMed]
- Wu, P.P.; Zhu, H.Y.; Sun, X.F.; Chen, L.X.; Zhou, Q.; Chen, J. MicroRNA-141 regulates the tumour suppressor DLC1 in colorectal cancer. Neoplasma 2015, 62, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.J.; Li, J.S.; Zhou, H.; Xiao, H.X.; Li, Y.; Zhou, T. MicroRNA-106b promotes colorectal cancer cell migration and invasion by directly targeting DLC1. J. Exp. Clin. Cancer Res. 2015, 34, 73. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Shopsowitz, K.E.; Wu, C.; Liu, G.; Dreaden, E.C.; Hammond, P.T. Periodic-shRNA molecules are capable of gene silencing, cytotoxicity and innate immune activation in cancer cells. Nucleic Acids Res. 2016, 44, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Gyongyosi, A.; Docs, O.; Czimmerer, Z.; Orosz, L.; Horvath, A.; Torok, O.; Mehes, G.; Nagy, L.; Balint, B.L. Measuring expression levels of small regulatory RNA molecules from body fluids and formalin-fixed, paraffin-embedded samples. Methods Mol. Biol. 2014, 1182, 105–119. [Google Scholar] [PubMed]
- Rodriguez, L.G.; Wu, X.; Guan, J.L. Wound-healing assay. Methods Mol. Biol. 2005, 294, 23–29. [Google Scholar] [PubMed]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miR-26a Target Genes | |
|---|---|
| CASP3 | Theoretical |
| CCNE2 | Demonstrated |
| ABL2 | Theoretical |
| APAF1 | Theoretical |
| XIAP | Theoretical |
| BCL2 | Theoretical |
| PTEN | Demonstrated |
| TP53 | Theoretical |
| E2F3 | Theoretical |
| CDC25A | Theoretical |
| BCL2L1 | Theoretical |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabello, P.; Pineda, B.; Tormo, E.; Lluch, A.; Eroles, P. The Antitumor Effect of Metformin Is Mediated by miR-26a in Breast Cancer. Int. J. Mol. Sci. 2016, 17, 1298. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17081298
Cabello P, Pineda B, Tormo E, Lluch A, Eroles P. The Antitumor Effect of Metformin Is Mediated by miR-26a in Breast Cancer. International Journal of Molecular Sciences. 2016; 17(8):1298. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17081298
Chicago/Turabian StyleCabello, Paula, Begoña Pineda, Eduardo Tormo, Ana Lluch, and Pilar Eroles. 2016. "The Antitumor Effect of Metformin Is Mediated by miR-26a in Breast Cancer" International Journal of Molecular Sciences 17, no. 8: 1298. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17081298