
Sexually Dimorphic Gene Expression Associated with Growth and Reproduction of Tongue Sole (Cynoglossus semilaevis) Revealed by Brain Transcriptome Analysis

Abstract
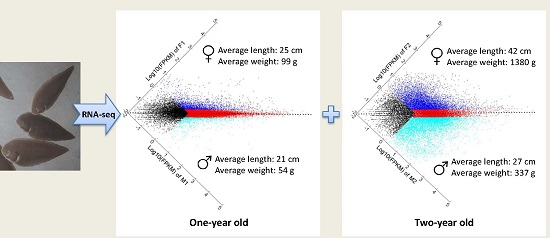
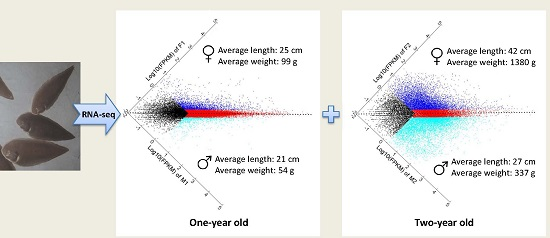
:
1. Introduction
2. Results
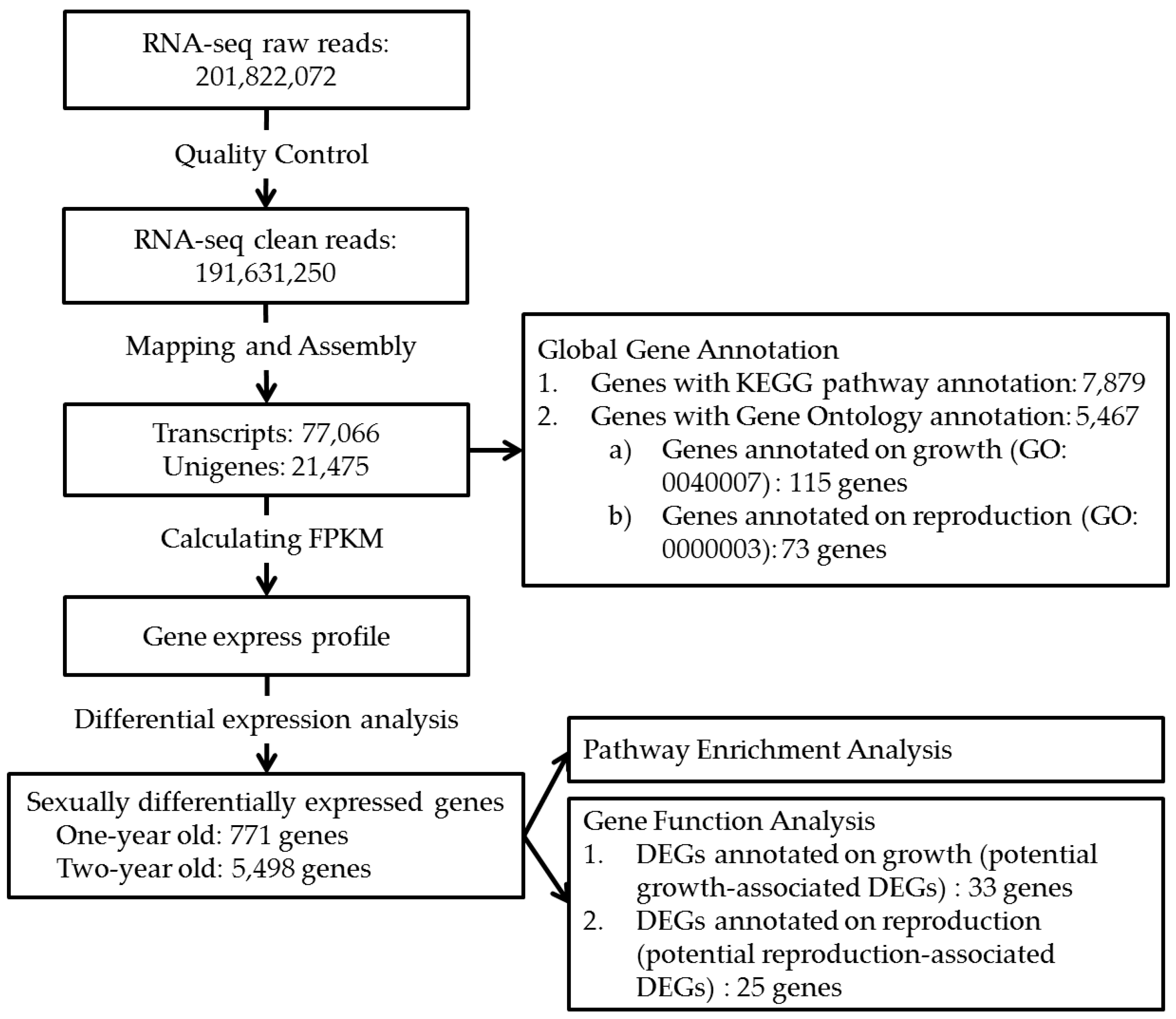
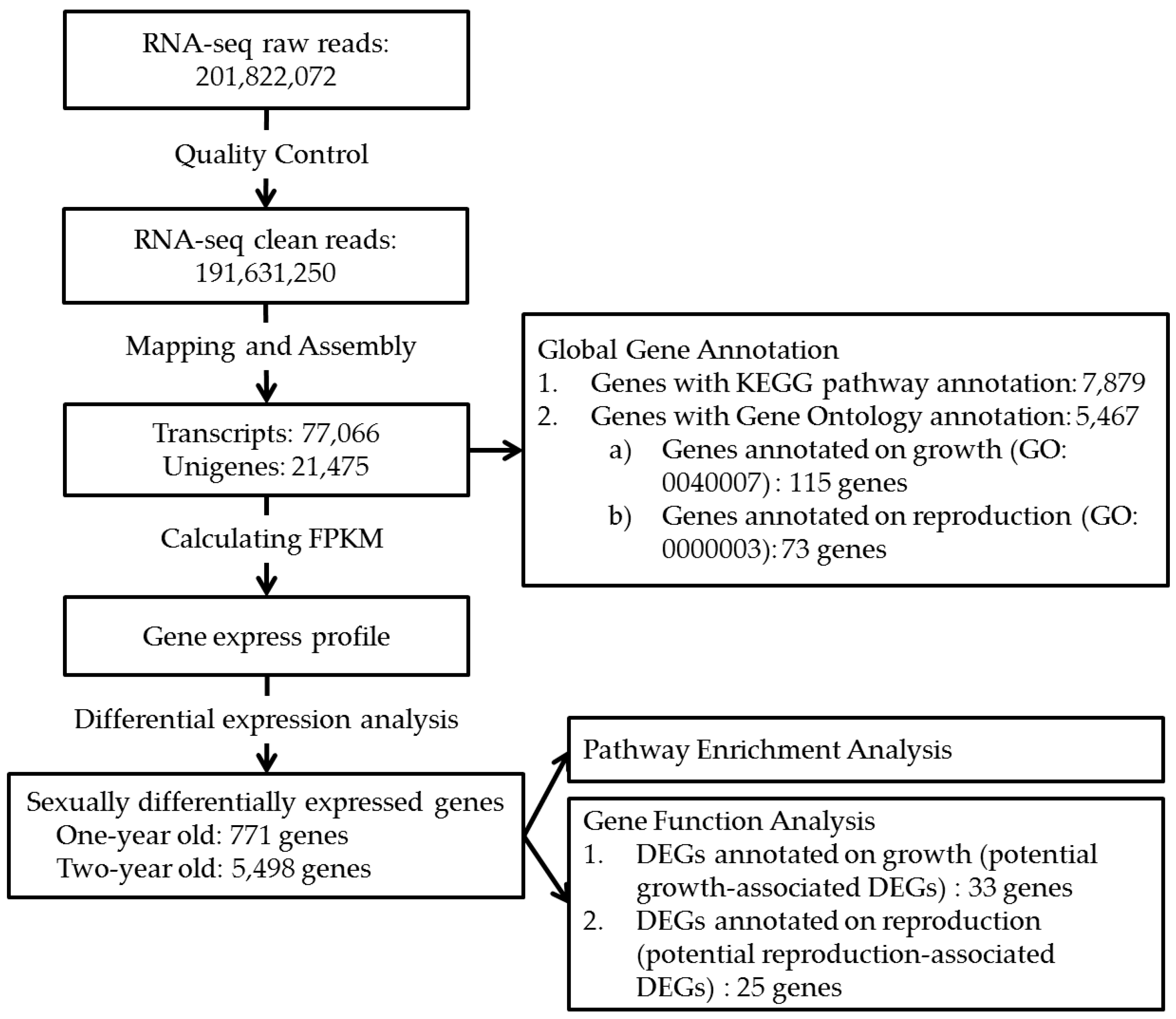
2.1. Transcriptome Sequencing and Assembly
2.2. Gene Identification and Functional Annotation
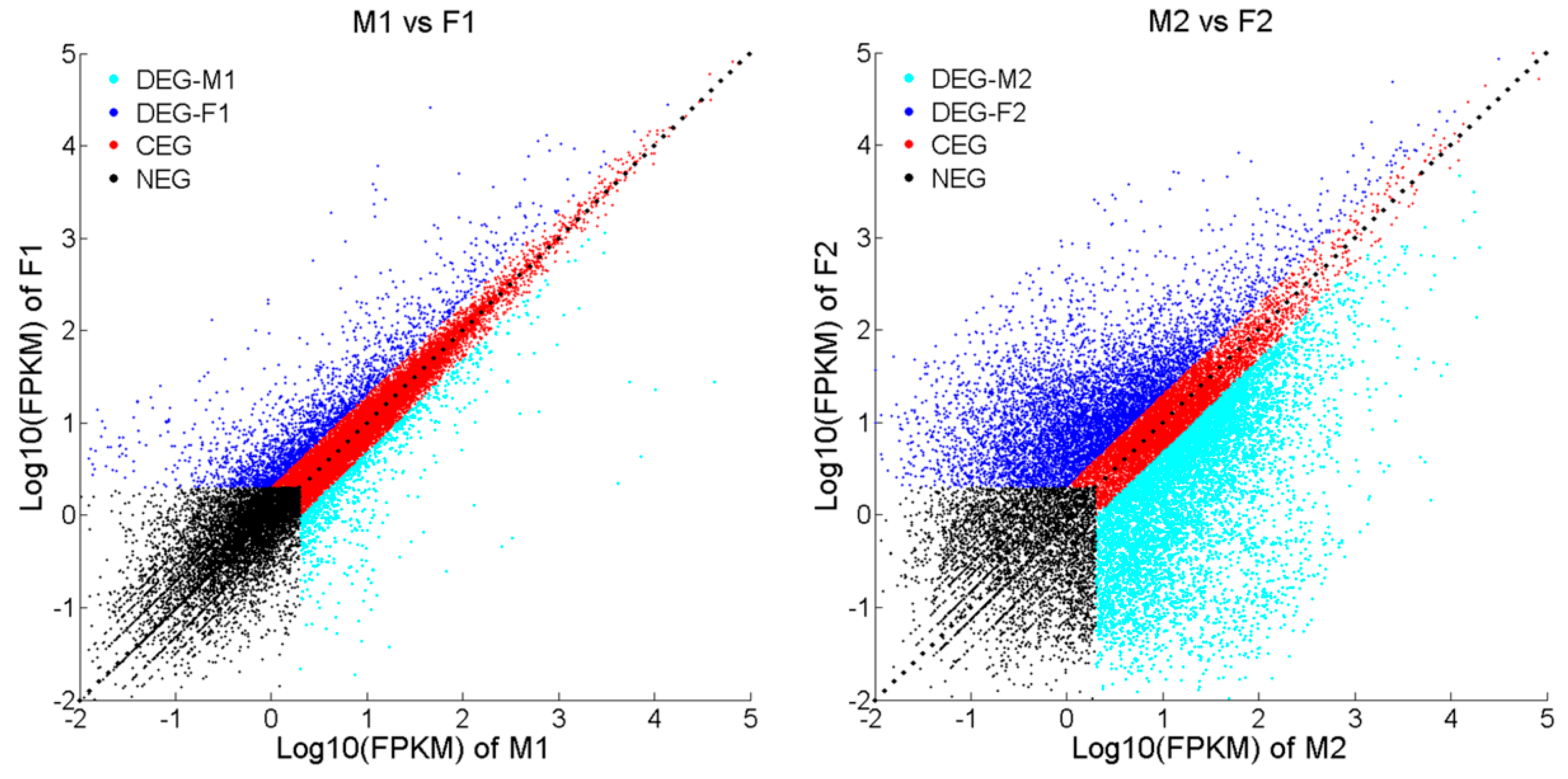
2.3. Differentially Expressed Genes (DEGs) and Gene Enrichment Analysis
2.4. Genes with Sex-Biased Expression Potentially Associated with Growth
2.5. Genes with Sex-Biased Expression Potentially Associated with Reproduction
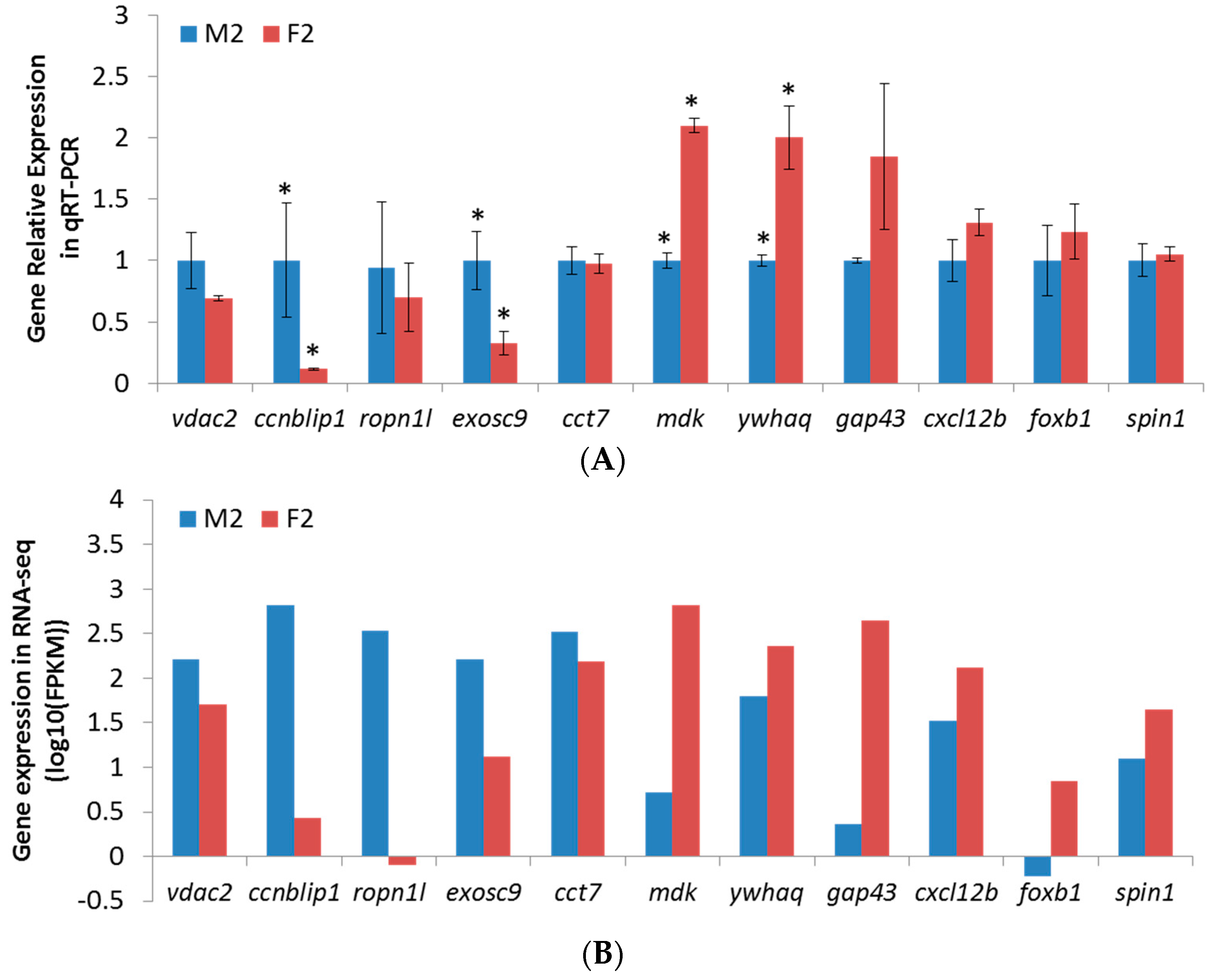
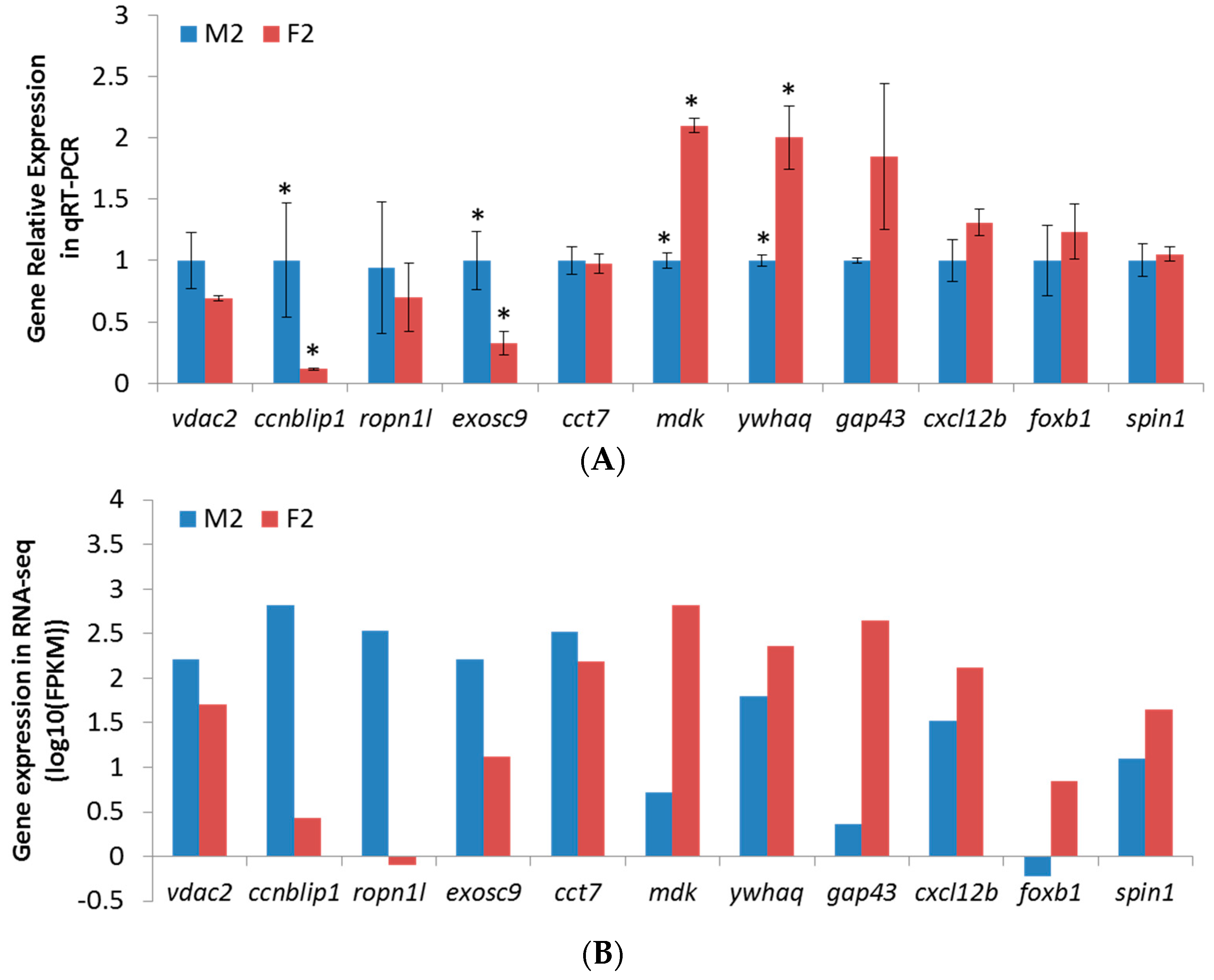
2.6. qPCR Validation of RNA-Seq Results
3. Discussion
3.1. The Brain Transcriptome of Tongue Sole
3.2. Male and Female Brain Expression Pattern
3.3. Sexually Differentially Expressed Genes Potentially Associated with Growth
3.4. Sexually Differentially Expressed Genes Potentially Associated with Reproduction
4. Materials and Methods
4.1. Experimental Fish and Sample Collection
4.2. RNA Isolation
4.3. cDNA Library Construction and Illumina Sequencing
4.4. Sequence Assembly and Analysis
4.5. Functional Annotation and Ontology
4.6. Identification of Differentially Expressed Genes (DEGs) and Pathway Enrichment Analysis
4.7. Experimental Validation by qPCR
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| GO | Gene Ontology |
| DEG | Differentially Expressed Gene |
| NEG | Non-Expressed Gene |
| CEG | Co-Expressed Gene |
| FPKM | Fragments Per Kilobase of exon per Million fragments mapped reads |
| qPCR | quantitative real-time PCR |
References
- Ma, A.J.; Liu, X.Z.; Xu, Y.J.; Liang, Y.; Zhuang, Z.M. Feeding rhythm and growth of the tongue sole, Cynoglossus semilaevis Gunther, during its early life stages. Aquac. Res. 2006, 37, 586–593. [Google Scholar]
- Chen, S.; Zhang, G.; Shao, C.; Huang, Q.; Liu, G.; Zhang, P.; Song, W.; An, N.; Chalopin, D.; Volff, J.N.; et al. Whole-genome sequence of a flatfish provides insights into ZW sex chromosome evolution and adaptation to a benthic lifestyle. Nat. Genet. 2014, 46, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.Y.; Zhang, Q.Q.; Qi, J.; Chen, Y.J.; Zhong, Q.W.; Li, C.M.; Yu, Y.; Li, S.; Wang, Z.G. Identification of differential genes in the ovary relative to the testis and their expression patterns in half-smooth tongue sole (Cynoglossus semilaievis). J. Genet. Genom. 2010, 37, 137–145. [Google Scholar] [CrossRef]
- Baroiller, J.F.; D’Cotta, H.; Saillant, E. Environmental effects on fish sex determination and differentiation. Sex. Dev. 2009, 3, 118–135. [Google Scholar] [CrossRef] [PubMed]
- Ospina-Alvarez, N.; Piferrer, F. Temperature-dependent sex determination in fish revisited: Prevalence, a single sex ratio response pattern, and possible effects of climate change. PLoS ONE 2008, 3, e2837. [Google Scholar] [CrossRef] [PubMed]
- Shang, E.H.H.; Yu, R.M.K.; Wu, R.S.S. Hypoxia affects sex differentiation and development, leading to a male-dominated population in zebrafish (Danio rerio). Environ. Sci. Technol. 2006, 40, 3118–3122. [Google Scholar] [PubMed]
- Francis, R.C. Sexual lability in teleosts: Developmental factors. Q. Rev. Biol. 1992, 67, 18. [Google Scholar] [CrossRef]
- Weltzien, F.A.; Andersson, E.; Andersen, O.; Shalchian-Tabrizi, K.; Norberg, B. The brain-pituitary-gonad axis in male teleosts, with special emphasis on flatfish (Pleuronectiformes). Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2004, 137, 447–477. [Google Scholar] [CrossRef] [PubMed]
- Gahr, M. Male Japanese quails with female brains do not show male sexual behaviors. Proc. Natl. Acad. Sci. USA 2003, 100, 7959–7964. [Google Scholar] [CrossRef] [PubMed]
- Dennis, C. Brain development: The most important sexual organ. Nature 2004, 427, 390–392. [Google Scholar] [CrossRef]
- Davies, W.; Wilkinson, L.S. It is not all hormones: Alternative explanations for sexual differentiation of the brain. Brain Res. 2006, 1126, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Dewing, P.; Shi, T.; Horvath, S.; Vilain, E. Sexually dimorphic gene expression in mouse brain precedes gonadal differentiation. Brain Res. Mol. Brain Res. 2003, 118, 82–90. [Google Scholar] [CrossRef]
- Yang, X.; Schadt, E.E.; Wang, S.; Wang, H.; Arnold, A.P.; Ingram-Drake, L.; Drake, T.A.; Lusis, A.J. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res. 2006, 16, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Parsch, J.; Ellegren, H. The evolutionary causes and consequences of sex-biased gene expression. Nat. Rev. Gene. 2013, 14, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Khatri, P.; Draghici, S.; Ostermeier, G.C.; Krawetz, S.A. Profiling gene expression using onto-express. Genomics 2002, 79, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Parisi, M.; Nuttall, R.; Edwards, P.; Minor, J.; Naiman, D.; Lu, J.; Doctolero, M.; Vainer, M.; Chan, C.; Malley, J.; et al. A survey of ovary-, testis-, and soma-biased gene expression in Drosophila melanogaster adults. Genome Biol. 2004, 5, R40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Small, C.L.; Shima, J.E.; Uzumcu, M.; Skinner, M.K.; Griswold, M.D. Profiling gene expression during the differentiation and development of the murine embryonic gonad. Biol. Reprod. 2005, 72, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Santos, E.M.; Workman, V.L.; Paull, G.C.; Filby, A.L.; van Look, K.J.; Kille, P.; Tyler, C.R. Molecular basis of sex and reproductive status in breeding zebrafish. Physiol. Genom. 2007, 30, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Yuan, J.; Zhou, L.; Sun, L.; Sun, Y.; Yang, S.; Li, M.; Zeng, S.; Huang, B.; Wang, D. Characterization of gonadal transcriptomes from Nile tilapia (Oreochromis niloticus) reveals differentially expressed genes. PLoS ONE 2013, 8, e63604. [Google Scholar] [CrossRef] [PubMed]
- Manousaki, T.; Tsakogiannis, A.; Lagnel, J.; Sarropoulou, E.; Xiang, J.Z.; Papandroulakis, N.; Mylonas, C.C.; Tsigenopoulos, C.S. The sex-specific transcriptome of the hermaphrodite sparid sharpsnout seabream (Diplodus puntazzo). BMC Genom. 2014, 15, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sharma, E.; Kunstner, A.; Fraser, B.A.; Zipprich, G.; Kottler, V.A.; Henz, S.R.; Weigel, D.; Dreyer, C. Transcriptome assemblies for studying sex-biased gene expression in the guppy, Poecilia reticulata. BMC Genom. 2014, 15, 400. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Luan, P.; Zhang, X.; Xue, S.; Peng, L.; Mahbooband, S.; Sun, X. Gonadal transcriptomic analysis of yellow catfish (Pelteobagrus fulvidraco): Identification of sex-related genes and genetic markers. Physiol. Genom. 2014, 46, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.S.; Chen, S.L.; Jiang, Y.L.; Xu, T.J.; Yang, J.F.; Tian, Y.S. Growth differences and differential expression analysis of pituitary adenylate cyclase activating polypeptide (PACAP) and growth hormone-releasing hormone (GHRH) between the sexes in half-smooth tongue sole Cynoglossus semilaevis. Gen. Comp. Endocrinol. 2011, 170, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Liu, S.F.; Zhuang, Z.M.; Lin, L.; Sun, Z.Z.; Liu, C.L.; Su, Y.Q.; Tang, Q.S. Genomic structure, polymorphism and expression analysis of growth hormone-releasing hormone and pituitary adenylate cyclase activating polypeptide genes in the half-smooth tongue sole (Cynoglossus semilaevis). Genet. Mol. Res. 2011, 10, 3828–3846. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.S.; Liu, H.W.; Chen, S.L.; Jiang, Y.L.; Tian, Y.S. Growth differences and dimorphic expression of growth hormone (GH) in female and male Cynoglossus semilaevis after male sexual maturation. Mar. Genom. 2011, 4, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Liu, S.F.; Zhuang, Z.M.; Lin, L.; Sun, Z.Z.; Liu, C.L.; Ma, H.; Su, Y.Q.; Tang, Q.S. Genomic structure, polymorphism and expression analysis of the growth hormone (GH) gene in female and male Half-smooth tongue sole (Cynoglossus semilaevis). Gene 2012, 493, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Trabzuni, D.; Ramasamy, A.; Imran, S.; Walker, R.; Smith, C.; Weale, M.E.; Hardy, J.; Ryten, M.; North American Brain Expression Consortium. Widespread sex differences in gene expression and splicing in the adult human brain. Nat. Commun. 2013, 4, 2771. [Google Scholar] [CrossRef] [PubMed]
- Sakakima, H.; Yoshida, Y.; Yamazaki, Y.; Matsuda, F.; Ikutomo, M.; Ijiri, K.; Muramatsu, H.; Muramatsu, T.; Kadomatsu, K. Disruption of the Midkine gene (MDK) delays degeneration and regeneration in injured peripheral nerve. J. Neurosci. Res. 2009, 87, 2908–2915. [Google Scholar] [CrossRef] [PubMed]
- Singec, I.; Crain, A.M.; Tobe, B.T.D.; Hou, J.; Talantova, M.; Doctor, K.S.; Choi, J.; Huang, X.; Gutierrez, G.J.; Wolf, D.A.; et al. Midkine (MDK): A newly-recognized endogenously-produced cytokine that, through an autocrine mechanism, is pivotal for neural induction. Neuroreport 2014, 25, 150. [Google Scholar]
- Taylor, T.D.; Noguchi, H.; Totoki, Y.; Toyoda, A.; Kuroki, Y.; Dewar, K.; Lloyd, C.; Itoh, T.; Takeda, T.; Kim, D.W.; et al. Human chromosome 11 DNA sequence and analysis including novel gene identification. Nature 2006, 440, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, T. Midkine: A Promising Molecule for Drug Development to Treat Diseases of the Central Nervous System. Curr. Pharm. Des. 2011, 17, 410–423. [Google Scholar] [CrossRef] [PubMed]
- Weckbach, L.T.; Muramatsu, T.; Walzog, B. Midkine in Inflammation. Sci. World J. 2011, 11, 2491–2505. [Google Scholar] [CrossRef] [PubMed]
- Kaname, T.; Kuwano, A.; Murano, I.; Uehara, K.; Muramatsu, T.; Kajii, T. Midkine gene (MDK), a gene for prenatal differentiation and neuroregulation, maps to band 11p11.2 by fluorescence in situ hybridization. Genomics 1993, 17, 514–515. [Google Scholar] [CrossRef] [PubMed]
- Kadomatsu, K.; Huang, R.P.; Suganuma, T.; Murata, F.; Muramatsu, T. A retinoic acid responsive gene MK found in the teratocarcinoma system is expressed in spatially and temporally controlled manner during mouse embryogenesis. J. Cell Biol. 1990, 110, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.W.; Muramatsu, T.; Kadomatsu, K. Distinct expression of midkine and pleiotrophin in the spinal cord and placental tissues during early mouse development. Dev. Growth Differ. 2000, 42, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, P.J.; Fairhurst, J.L.; Decker, M.M.; Chan, C.P.; Gluzman, Y.; Bohlen, P.; Kovesdi, I. Cloning, characterization and developmental regulation of two members of a novel human gene family of neurite outgrowth-promoting proteins. Growth Factors 1991, 5, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Sortwell, C.E.; Collier, T.J.; Terpstra, B.T.; Thompson, V.B.; O’Malley, J.; Steece-Collier, K.; Wohlgenant, S.M.; Daley, B.F.; Mandel, R.J.; Lipton, J.W. The potential for the trophic factor pleiotrophin (PTN) to protect the Parkinsonian brain. Cell Transplant. 2008, 17, 481. [Google Scholar]
- Xu, C.Y.; Zhu, S.Y.; Wu, M.Y.; Han, W.; Yu, Y. Functional receptors and intracellular signal pathways of midkine (MK) and pleiotrophin (PTN). Biol. Pharm. Bull. 2014, 37, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Human Genome Sequencing, C. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar]
- Muramatsu, H.; Zou, P.; Kurosawa, N.; Ichihara-Tanaka, K.; Maruyama, K.; Inoh, K.; Sakai, T.; Chen, L.; Sato, M.; Muramatsu, T. Female infertility in mice deficient in midkine and pleiotrophin, which form a distinct family of growth factors. Genes Cells 2006, 11, 1405–1417. [Google Scholar] [CrossRef] [PubMed]
- Jee, Y.H.; Lebenthal, Y.; Chaemsaithong, P.; Yan, G.; Peran, I.; Wellstein, A.; Romero, R.; Baron, J. Midkine and pleiotrophin concentrations in amniotic fluid in healthy and complicated pregnancies. PLoS ONE 2016, 11, e0153325. [Google Scholar] [CrossRef] [PubMed]
- De Groen, P.C.; Eggen, B.J.; Gispen, W.H.; Schotman, P.; Schrama, L.H. Cloning and promoter analysis of the human B-50/GAP-43 gene. J. Mol. Neurosci. 1995, 6, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, L.I.; Routtenberg, A. GAP-43: An intrinsic determinant of neuronal development and plasticity. Trends Neurosci. 1997, 20, 84–91. [Google Scholar] [CrossRef]
- Scherer, S.E.; Muzny, D.M.; Buhay, C.J.; Chen, R.; Cree, A.; Ding, Y.; Dugan-Rocha, S.; Gill, R.; Gunaratne, P.; Harris, R.A.; et al. The finished DNA sequence of human chromosome 12. Nature 2006, 440, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Milbrandt, J. Ninjurin2, a novel homophilic adhesion molecule, is expressed in mature sensory and enteric neurons and promotes neurite outgrowth. J. Neurosci. 2000, 20, 187–195. [Google Scholar] [PubMed]
- Bis, J.C.; DeStefano, A.; Liu, X.M.; Brody, J.A.; Choi, S.H.; Verhaaren, B.F.J.; Debette, S.; Ikram, M.A.; Shahar, E.; Butler, K.R.; et al. Associations of NINJ2 sequence variants with incident ischemic stroke in the cohorts for heart and aging in genomic epidemiology (charge) consortium. PLoS ONE 2014, 9, e99798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albig, W.; Bramlage, B.; Gruber, K.; Klobeck, H.G.; Kunz, J.; Doenecke, D. The human replacement histone H3.3B gene (H3F3B). Genomics 1995, 30, 264–272. [Google Scholar] [CrossRef] [PubMed]
- De Luca, P.; Vazquez, E.S.; Moiola, C.P.; Zalazar, F.; Cotignola, J.; Gueron, G.; Gardner, K.; de Siervi, A. BRCA1 loss induces GADD153-mediated doxorubicin resistance in prostate cancer. Mol. Cancer Res. 2011, 9, 1078–1090. [Google Scholar] [CrossRef] [PubMed]
- De Luca, P.; Moiola, C.P.; Zalazar, F.; Gardner, K.; Vazquez, E.S.; de Siervi, A. BRCA1 and p53 regulate critical prostate cancer pathways. Prostate Cancer Prostatic Dis. 2013, 16, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, E.L.; Schilders, G.; Pruijn, G.J. Human cell growth requires a functional cytoplasmic exosome, which is involved in various mRNA decay pathways. RNA 2007, 13, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Mistry, D.S.; Chen, Y.F.; Sen, G.L. Progenitor function in self-renewing human epidermis is maintained by the exosome. Cell Stem Cell 2012, 11, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Strong, E.R.; Schimenti, J.C. Evidence implicating CCNB1IP1, a RING domain-containing protein required for meiotic crossing over in mice, as an E3 SUMO ligase. Genes 2010, 1, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Gronholm, M.; Muranen, T.; Toby, G.G.; Utermark, T.; Hanemann, C.O.; Golemis, E.A.; Carpen, O. A functional association between merlin and HEI10, a cell cycle regulator. Oncogene 2006, 25, 4389–4398. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Lin, M.; Li, K.; Fu, Y.; Liu, X.; Yang, D.; Zhao, Y.; Zheng, J.; Sun, B. Knocking down SMC1A inhibits growth and leads to G2/M arrest in human glioma cells. Int. J. Clin. Exp. Pathol. 2013, 6, 862–869. [Google Scholar] [PubMed]
- Chen, L.; Kass, R.S. A-kinase anchoring proteins: Different partners, different dance. Nat. Cell Biol. 2005, 7, 1050–1051. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cai, Z.M.; Gui, Y.T. Advances in the researches of spermatogenic protein, Ropporin. Natl. J. Androl. 2009, 15, 3. [Google Scholar]
- Tonevitsky, A.G.; Maltseva, D.V.; Abbasi, A.; Samatov, T.R.; Sakharov, D.A.; Shkurnikov, M.U.; Lebedev, A.E.; Galatenko, V.V.; Grigoriev, A.I.; Northoff, H. Dynamically regulated miRNA-mRNA networks revealed by exercise. BMC Physiol. 2013, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, O.A.; Chen, B.; Haase-Pettingell, C.; Ludtke, S.J.; Chiu, W.; King, J.A. Human CCT4 and CCT5 chaperonin subunits expressed in Escherichia coli form biologically active homo-oligomers. J. Biol. Chem. 2013, 288, 17734–17744. [Google Scholar] [CrossRef] [PubMed]
- Damours, O.; Sullivan, R. Evaluating Fertility of Mammalian Spermatozoa e.g., Bovine Spermatozoa Involves Assessing in Spermatozoa Sample such as Bovine Sperm, Amount or Activity of Specific Sperm Fertility Protein, Where Amount/Activity Is Predictive of Fertility. WO2010025548-A1, 11 March 2010. [Google Scholar]
- Grantham, J.; Brackley, K.I.; Willison, K.R. Substantial CCT activity is required for cell cycle progression and cytoskeletal organization in mammalian cells. Exp. Cell Res. 2006, 312, 2309–2324. [Google Scholar] [CrossRef] [PubMed]
- Feldman, D.E.; Thulasiraman, V.; Ferreyra, R.G.; Frydman, J. Formation of the VHL-elongin BC tumor suppressor complex is mediated by the chaperonin TRiC. Mol. Cell 1999, 4, 1051–1061. [Google Scholar] [CrossRef]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Salzberg, S.L. TopHat-Fusion: An algorithm for discovery of novel fusion transcripts. Genome Biol. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Pimentel, H.; Trapnell, C.; Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 2011, 27, 2325–2329. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.S.; Khan, Z.; Kruglyak, L.; Singh, M.; Caudy, A.A. Measuring differential gene expression by short read sequencing: Quantitative comparison to 2-channel gene expression microarrays. BMC Genom. 2009, 10, 221. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, L.; Wang, J.; Shi, W.; Pawar, R.A.; Liu, Y.; Xu, C.; Cong, W.; Hu, Q.; Lu, T.; et al. β-Actin is a useful internal control for tissue-specific gene expression studies using quantitative real-time PCR in the half-smooth tongue sole Cynoglossus semilaevis challenged with LPS or Vibrio anguillarum. Fish Shellfish Immunol. 2010, 29, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO Category | GO Term | Total No. of Genes | M1 vs. F1 | M2 vs. F2 | ||
|---|---|---|---|---|---|---|
| M1 | F1 | M2 | F2 | |||
| Cellular Component | GO:0005576 extracellular region | 310 | 10 | 9 | 40 | 49 |
| GO:0005623 cell | 3238 | 69 | 38 | 425 | 400 | |
| GO:0019012 virion | 11 | 3 | 0 | 5 | 2 | |
| GO:0031974 membrane-enclosed lumen | 408 | 8 | 2 | 83 | 31 | |
| GO:0031975 envelope | 105 | 7 | 0 | 22 | 18 | |
| GO:0032991 macromolecular complex | 1014 | 34 | 26 | 175 | 151 | |
| GO:0043226 organelle | 1669 | 35 | 28 | 282 | 212 | |
| GO:0044421 extracellular region part | 214 | 3 | 4 | 34 | 33 | |
| GO:0044422 organelle part | 1008 | 29 | 15 | 193 | 127 | |
| GO:0044423 virion part | 11 | 3 | 0 | 5 | 2 | |
| GO:0044456 synapse part | 75 | 3 | 1 | 3 | 23 | |
| GO:0044464 cell part | 3238 | 69 | 38 | 425 | 400 | |
| GO:0045202 synapse | 102 | 6 | 1 | 5 | 28 | |
| Molecular Function | GO:0003824 catalytic activity | 2105 | 34 | 13 | 298 | 210 |
| GO:0005198 structural molecule activity | 112 | 9 | 18 | 20 | 42 | |
| GO:0005215 transporter activity | 393 | 19 | 5 | 30 | 68 | |
| GO:0005488 binding | 2780 | 53 | 31 | 380 | 318 | |
| GO:0009055 electron carrier activity | 6 | 0 | 0 | 1 | 1 | |
| GO:0015457 auxiliary transport protein activity | 7 | 0 | 1 | 1 | 2 | |
| GO:0016209 antioxidant activity | 14 | 0 | 0 | 1 | 2 | |
| GO:0030234 enzyme regulator activity | 200 | 0 | 4 | 22 | 35 | |
| GO:0030528 transcription regulator activity | 188 | 2 | 1 | 14 | 14 | |
| GO:0042056 chemoattractant activity | 2 | 0 | 0 | 0 | 1 | |
| GO:0045182 translation regulator activity | 26 | 1 | 0 | 8 | 2 | |
| GO:0045499 chemorepellent activity | 1 | 0 | 0 | 0 | 0 | |
| GO:0060089 molecular transducer activity | 430 | 3 | 2 | 19 | 35 | |
| Biological Process | GO:0000003 reproduction | 73 | 0 | 0 | 20 | 5 |
| GO:0001906 cell killing | 3 | 0 | 0 | 0 | 1 | |
| GO:0002376 immune system process | 189 | 2 | 1 | 28 | 26 | |
| GO:0008152 metabolic process | 2756 | 46 | 37 | 406 | 297 | |
| GO:0008371 obsolete biological process | 58 | 4 | 0 | 4 | 8 | |
| GO:0009987 cellular process | 3553 | 71 | 48 | 502 | 403 | |
| GO:0010926 anatomical structure formation | 378 | 8 | 8 | 78 | 43 | |
| GO:0016032 viral reproduction | 30 | 1 | 1 | 8 | 8 | |
| GO:0016043 cellular component organization | 652 | 15 | 8 | 134 | 77 | |
| GO:0016265 death | 169 | 2 | 2 | 32 | 14 | |
| GO:0022414 reproductive process | 72 | 1 | 1 | 19 | 10 | |
| GO:0022610 biological adhesion | 113 | 1 | 0 | 13 | 15 | |
| GO:0032501 multicellular organismal process | 1315 | 34 | 17 | 151 | 174 | |
| GO:0032502 developmental process | 1124 | 26 | 14 | 147 | 129 | |
| GO:0040007 growth | 115 | 5 | 3 | 12 | 18 | |
| GO:0040011 locomotion | 152 | 1 | 1 | 18 | 22 | |
| GO:0043473 pigmentation | 1784 | 33 | 26 | 198 | 216 | |
| GO:0044085 cellular component biogenesis | 327 | 11 | 22 | 75 | 58 | |
| GO:0048511 rhythmic process | 11 | 0 | 0 | 0 | 1 | |
| GO:0050896 response to stimulus | 606 | 19 | 9 | 81 | 83 | |
| GO:0051179 localization | 955 | 30 | 11 | 120 | 147 | |
| GO:0051234 establishment of localization | 790 | 30 | 10 | 100 | 130 | |
| GO:0051704 multi-organism process | 55 | 2 | 1 | 11 | 11 | |
| GO:0065007 biological regulation | 1927 | 38 | 29 | 219 | 250 | |
| All genes annotated by GO | 5467 | 104 | 63 | 711 | 619 | |
| Gene | FPKM | p-Value | Annotation | GO Function | |
|---|---|---|---|---|---|
| M2 | F2 | M2 vs. F2 | |||
| DEGs Up Regulated in M2 | |||||
| h3f3b | 670.6 | 193.2 | 1.9E-56 | Histone H3.3 | Positive regulation of cell growth (GO:0030307) |
| hsc70 | 247.9 | 0.1 | 5.5E-34 | Heat shock cognate 70 kDa protein | Fin regeneration (GO:0031101) |
| exosc9 | 164.6 | 13.2 | 1.7E-32 | Exosome complex component RRP45 | Positive regulation of cell growth (GO:0030307) |
| vdac2 | 164.4 | 51.1 | 7.9E-14 | Voltage-dependent anion-selective channel protein 2 | Fin regeneration (GO:0031101) |
| ppp2r1a | 237.7 | 108.1 | 9.4E-11 | Serine/threonine-protein phosphatase 2A 65 kDa regulatory subunit A β isoform | Growth (GO:0040007) |
| timm50 | 44.6 | 12.1 | 2.5E-05 | Mitochondrial import inner membrane translocase subunit TIM50 | Growth (GO:0040007) |
| bmp2 | 30.6 | 6.1 | 5.3E-05 | Bone morphogenetic protein 2 | Growth (GO:0040007) |
| ascl1 | 17.0 | 1.1 | 9.1E-05 | Achaete-scute homolog 1 | Sensory epithelium regeneration (GO:0070654) |
| suv420h1 | 31.8 | 7.3 | 1.1E-04 | Histone-lysine N-methyltransferase SUV420H1 | Regulation of multicellular organism growth (GO:0040014) |
| orc1 | 15.7 | 1.1 | 1.8E-04 | Origin recognition complex subunit 1 | Regulation of multicellular organism growth (GO:0040014) |
| nenf | 26.8 | 5.7 | 2.2E-04 | Neudesin neurotrophic factor | Growth (GO:0040007) |
| dnajc2 | 42.6 | 16.0 | 9.6E-04 | dnaJ homolog subfamily C member 2 | Negative regulation of cell growth (GO:0030308) |
| DEGs Down Regulated in M2 | |||||
| mdk | 5.3 | 667.8 | 1.7E-157 | Midkine | Growth (GO:0040007) |
| gap43 | 2.3 | 442.4 | 2.3E-100 | Growth associated protein 43 | Tissue regeneration (GO:0042246) |
| ptn | 30.3 | 287.1 | 3.1E-56 | Pleiotrophin | Growth (GO:0040007) |
| gja1 | 1.9 | 222.0 | 6.1E-54 | Gap junction α-1 protein | Fin regeneration (GO:0031101) |
| ninj2 | 55.5 | 255.5 | 1.1E-34 | Ninjurin-2 | Tissue regeneration (GO:0042246) |
| cxcl12b | 33.3 | 131.8 | 1.1E-16 | Stromal cell-derived factor 1 precursor | Fin regeneration (GO:0031101) |
| fgf12 | 2.0 | 56.1 | 5.0E-15 | Fibroblast growth factor 12 | Growth (GO:0040007) |
| gcfc2 | 23.4 | 102.5 | 2.3E-14 | GC-rich sequence DNA-binding factor 2 | Fin regeneration (GO:0031101) |
| sox2 | 0.7 | 38.5 | 5.3E-11 | Transcription factor SOX-2 | Fin regeneration (GO:0031101) |
| fyna | 2.8 | 38.2 | 1.0E-09 | Tyrosine-protein kinase fyna | Fin regeneration (GO:0031101) |
| epha4 | 1.7 | 34.5 | 1.9E-09 | Ephrin type-A receptor 4 | Negative regulation of collateral sprouting of intact axon in response to injury (GO:0048685) |
| bdnf | 1.9 | 29.3 | 6.6E-08 | Brain-derived neurotrophic factor | Growth (GO:0040007) |
| cdh2 | 0.6 | 24.8 | 1.5E-07 | cadherin-2-like | Tissue regeneration (GO:0042246) |
| rerg | 3.0 | 29.9 | 2.6E-07 | Ras-related and estrogen-regulated growth inhibitor | Negative regulation of cell growth (GO:0030308) |
| ctgf | 2.5 | 22.9 | 9.4E-06 | Connective tissue growth factor | Regulation of cell growth (GO:0001558) |
| igfbp1 | 0.2 | 17.9 | 9.8E-06 | Insulin-like growth factor-binding protein 1 | Regulation of cell growth (GO:0001558) |
| klf6 | 2.6 | 22.5 | 1.3E-05 | Krueppel-like factor 6 | Organ growth (GO:0035265) |
| sptbn1 | 7.0 | 27.5 | 1.7E-04 | Spectrin β chain, non-erythrocytic 1 | Positive regulation of multicellular organism growth (GO:0040018) |
| Gene | FPKM | p-Value | Annotation | GO Function | |
|---|---|---|---|---|---|
| M2 | F2 | M2 vs. F2 | |||
| pleaDEGs Up Regulated in M2 | |||||
| ccnb1ip1 | 661.4 | 2.7 | 3.6E-138 | E3 ubiquitin-protein ligase CCNB1IP1 | Reproductive cellular process (GO:0048610) |
| rsph1 | 516.7 | 5.7 | 1.4E-118 | Radial spoke head 1 homolog | Reproduction (GO:0000003) |
| ropn1l | 343.4 | 0.8 | 1.5E-66 | Ropporin-1-like protein | Spermatid development (GO:0007286) |
| alkbh5 | 180.5 | 17.9 | 4.5E-33 | RNA demethylase ALKBH5 | Spermatogenesis (GO:0007283) |
| henmt1 | 150.3 | 0.0 | 2.4E-29 | Small RNA 2’-O-methyltransferase | Oocyte development (GO:0048599) |
| cct5 | 358.3 | 136.8 | 3.4E-21 | Chaperonin containing T-complex polypeptide 5 | Binding of sperm to zona pellucida (GO:0007339) |
| ovol1 | 112.1 | 0.1 | 3.2E-19 | Putative transcription factor Ovo-like 1 | Spermatogenesis (GO:0007283) |
| klhl10 | 54.7 | 0.0 | 7.0E-14 | Kelch-like protein 10 | Spermatid development (GO:0007286) |
| cct7 | 329.3 | 153.7 | 1.4E-13 | Cct7 protein | Binding of sperm to zona pellucida (GO:0007339) |
| cct4 | 305.8 | 147.9 | 8.8E-12 | Cct4 protein | Binding of sperm to zona pellucida (GO:0007339) |
| fem1b | 44.6 | 3.0 | 2.5E-10 | Protein fem-1 homolog B-like | Epithelial cell maturation involved in prostate gland development (GO:0060743) |
| sf1 | 103.4 | 33.7 | 9.2E-09 | Splicing factor 1 | Leydig cell differentiation (GO:0033327) |
| styx | 69.8 | 20.2 | 3.6E-07 | Serine/threonine/tyrosine-interacting protein | Spermatogenesis (GO:0007283) |
| ptbp1 | 45.3 | 9.4 | 1.2E-06 | Polypyrimidine tract-binding protein 1-like | Positive regulation of cortical granule exocytosis by elevation of cytosolic calcium ion concentration (GO:0060472) |
| tbp | 41.8 | 9.0 | 4.4E-06 | TATA box binding protein | Spermatogenesis (GO:0007283) |
| pld6 | 24.9 | 2.6 | 1.1E-05 | Phospholipase D family member 6 | P granule organization (GO:0030719) |
| polr2h | 50.9 | 15.8 | 3.2E-05 | DNA-directed RNA polymerases I%2C II%2C and III subunit RPABC3 | Positive regulation of viral transcription (GO:0050434) |
| pacrg | 38.8 | 10.5 | 7.8E-05 | Parkin coregulated gene protein | Spermatid development (GO:0007286) |
| tceb1 | 137.6 | 75.3 | 1.2E-04 | Transcription elongation factor B polypeptide 1 | Positive regulation of viral transcription (GO:0050434) |
| tdrd9 | 13.3 | 0.6 | 2.9E-04 | Putative ATP-dependent RNA helicase TDRD9 | Fertilization (GO:0009566); DNA methylation during gametogenesis (GO:0043046) |
| DEGs Down Regulated in M2 | |||||
| ywhaq | 63.5 | 229.4 | 1.1E-25 | YWHAQ protein | Germarium-derived oocyte fate determination (GO:0007294) |
| cxcl12b | 33.3 | 131.8 | 1.1E-16 | Stromal cell-derived factor 1-like | Germ cell migration (GO:0008354) |
| spin1 | 12.4 | 44.6 | 4.0E-06 | Spindlin-1-like | Gamete generation (GO:0007276) |
| stau2 | 41.7 | 80.2 | 1.1E-04 | Double-stranded RNA-binding protein Staufen homolog 2 | Germ cell migration (GO:0008354) |
| sptbn1 | 7.0 | 27.5 | 1.7E-04 | Spectrin β chain, non-erythrocytic 1 | Fertilization (GO:0009566) |
| Gene Annotation | DEGs | Non-DEGs | All Transcribed Genes |
|---|---|---|---|
| Category A | a | b | a + b |
| Other categories | c | d | c + d |
| All categories | a + c | b + d | a + b + c + d |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Zheng, M.; Liu, J.; Liu, Y.; Lu, J.; Sun, X. Sexually Dimorphic Gene Expression Associated with Growth and Reproduction of Tongue Sole (Cynoglossus semilaevis) Revealed by Brain Transcriptome Analysis. Int. J. Mol. Sci. 2016, 17, 1402. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091402
Wang P, Zheng M, Liu J, Liu Y, Lu J, Sun X. Sexually Dimorphic Gene Expression Associated with Growth and Reproduction of Tongue Sole (Cynoglossus semilaevis) Revealed by Brain Transcriptome Analysis. International Journal of Molecular Sciences. 2016; 17(9):1402. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091402
Chicago/Turabian StyleWang, Pingping, Min Zheng, Jian Liu, Yongzhuang Liu, Jianguo Lu, and Xiaowen Sun. 2016. "Sexually Dimorphic Gene Expression Associated with Growth and Reproduction of Tongue Sole (Cynoglossus semilaevis) Revealed by Brain Transcriptome Analysis" International Journal of Molecular Sciences 17, no. 9: 1402. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091402