
Biomass Smoke Exposure Enhances Rhinovirus-Induced Inflammation in Primary Lung Fibroblasts


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
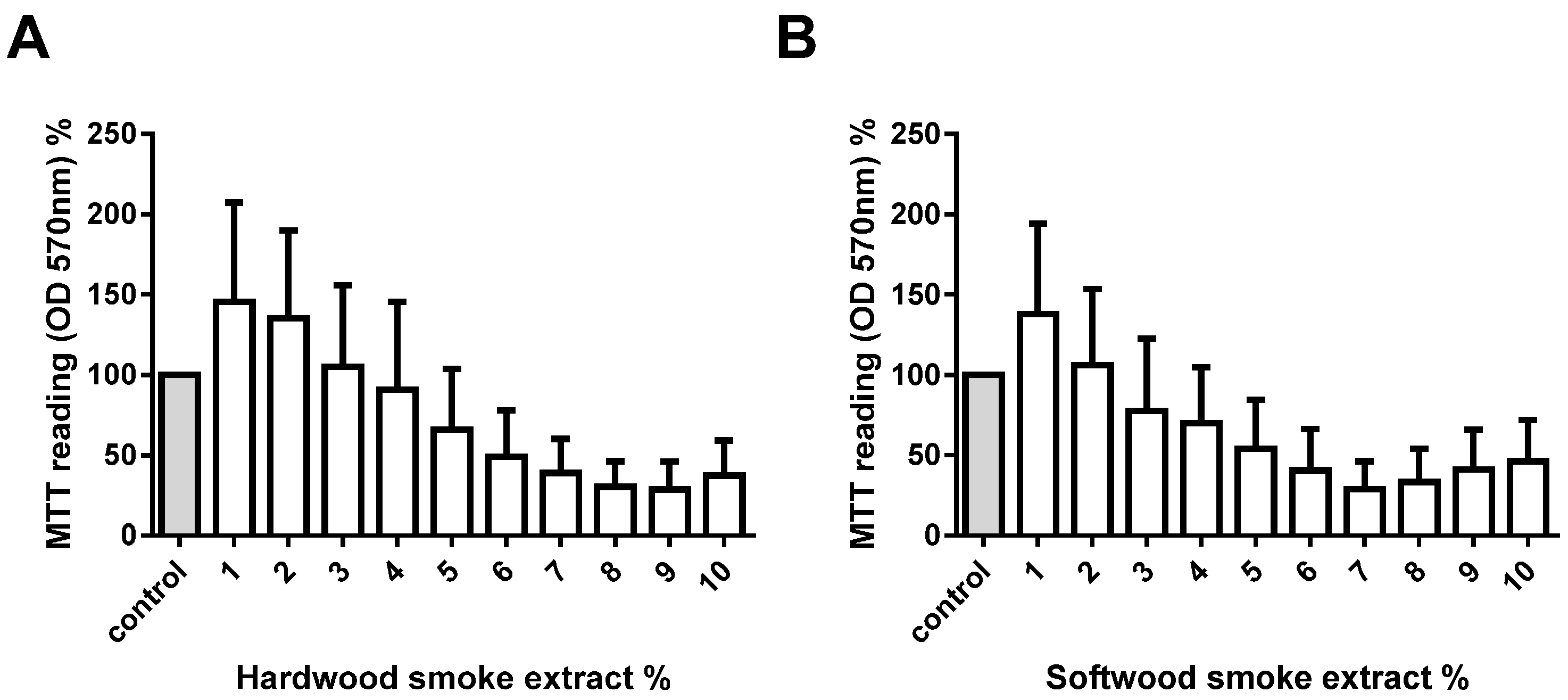
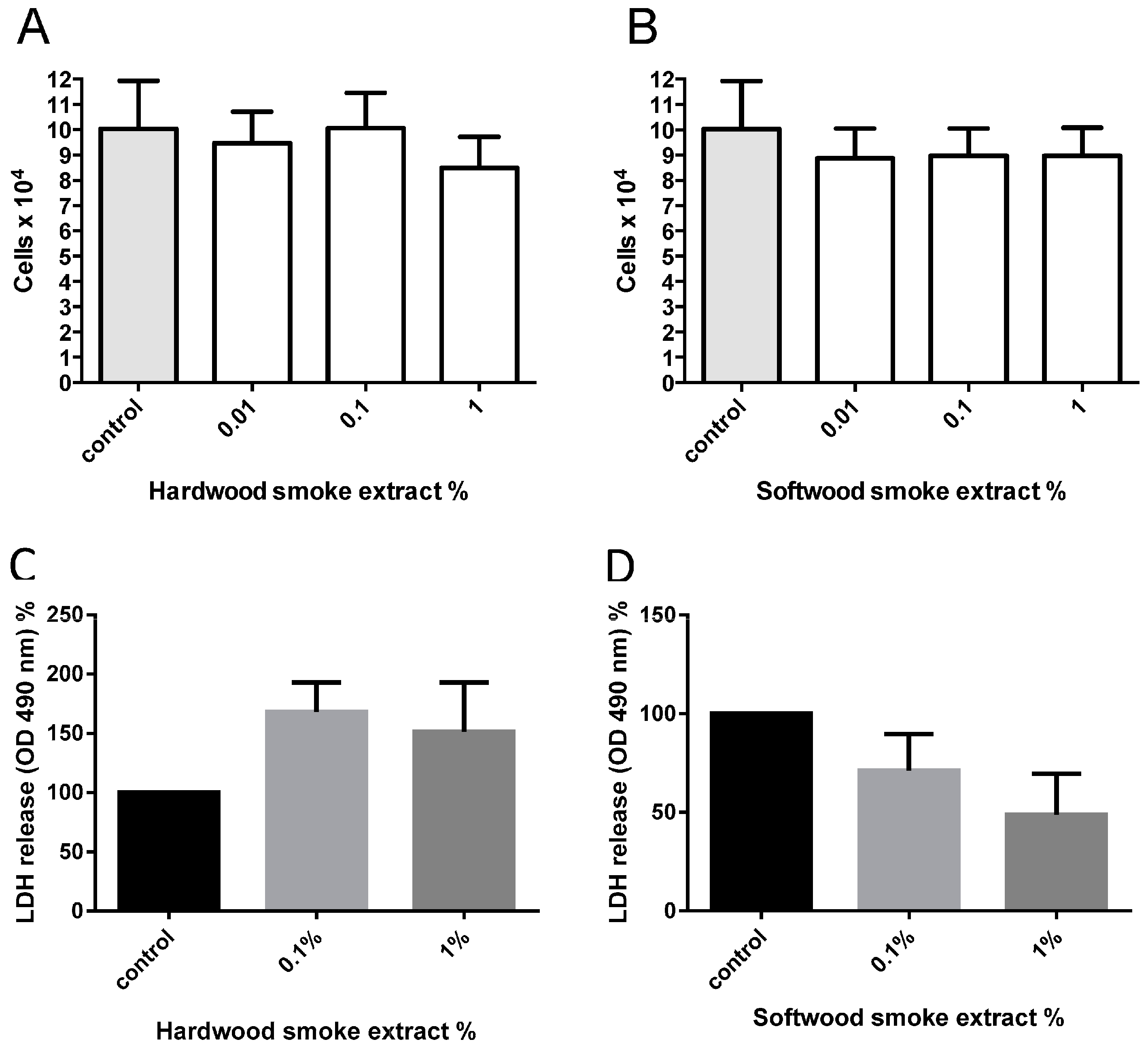
2.1. Hardwood and Softwood Smoke Extract Are Cytotoxic at Higher Concentrations
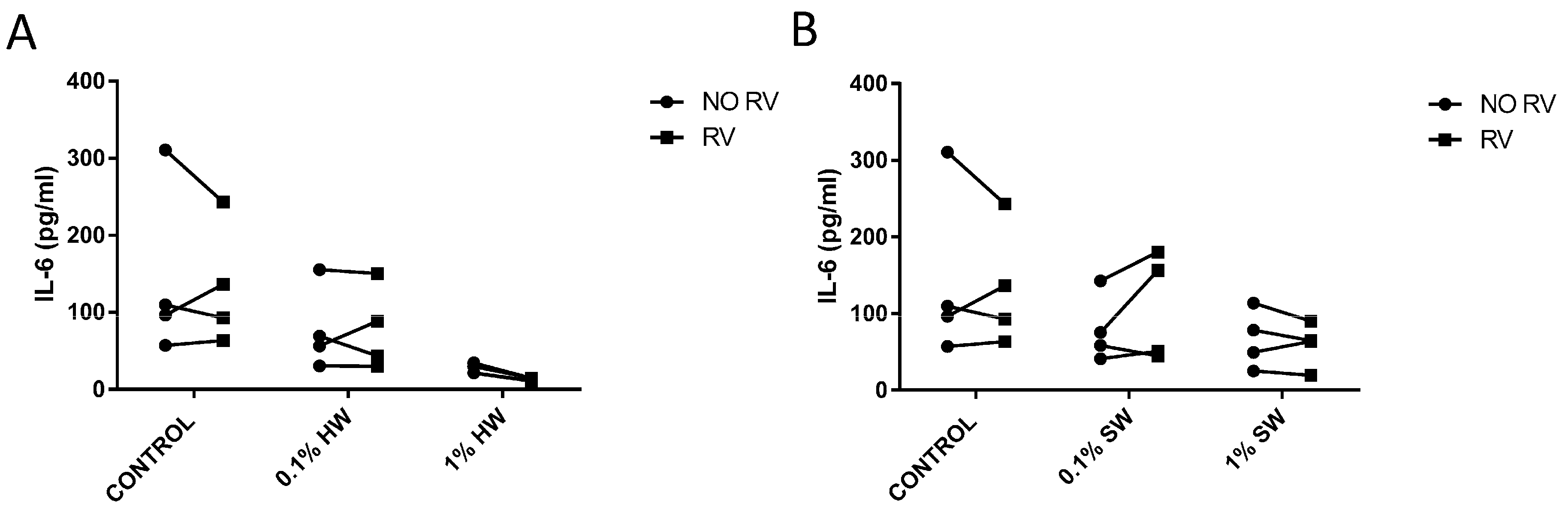
2.2. Hardwood and Softwood Smoke Extract Upregulates IL-6 and IL-8 Production at Low Concentrations
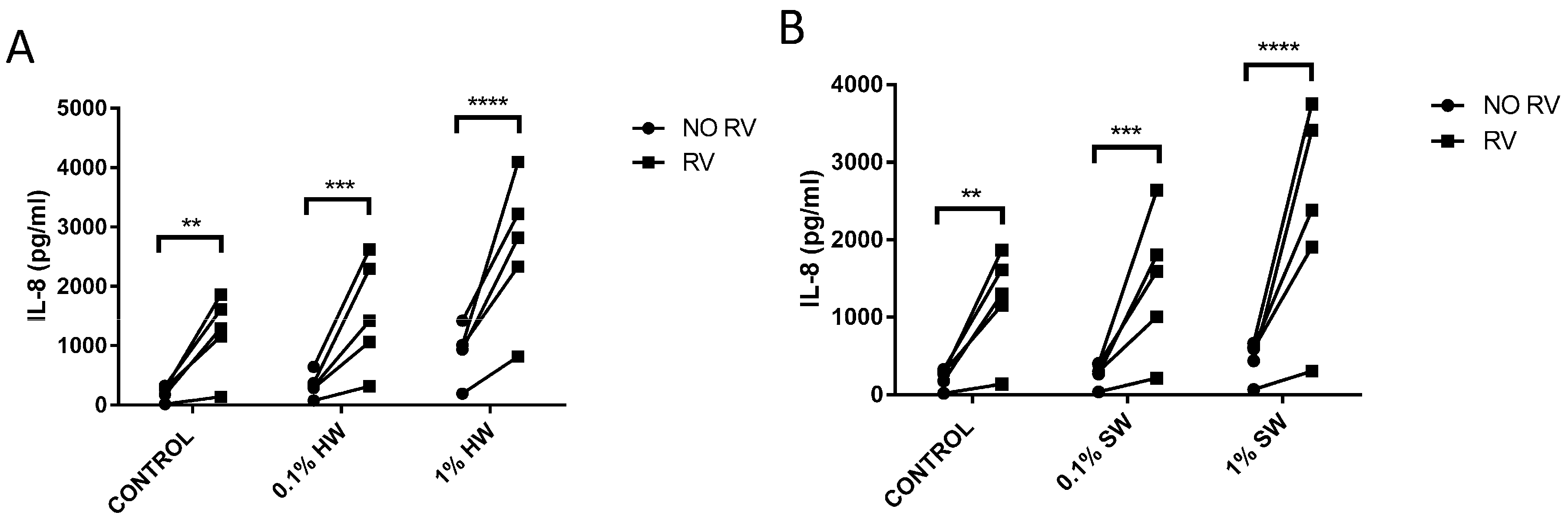
2.3. Biomass Smoke Exposure Enhances RV-16 Induced IL-8 Production
2.4. Biomass Smoke Exposure Did Not Affect RV-16 Load
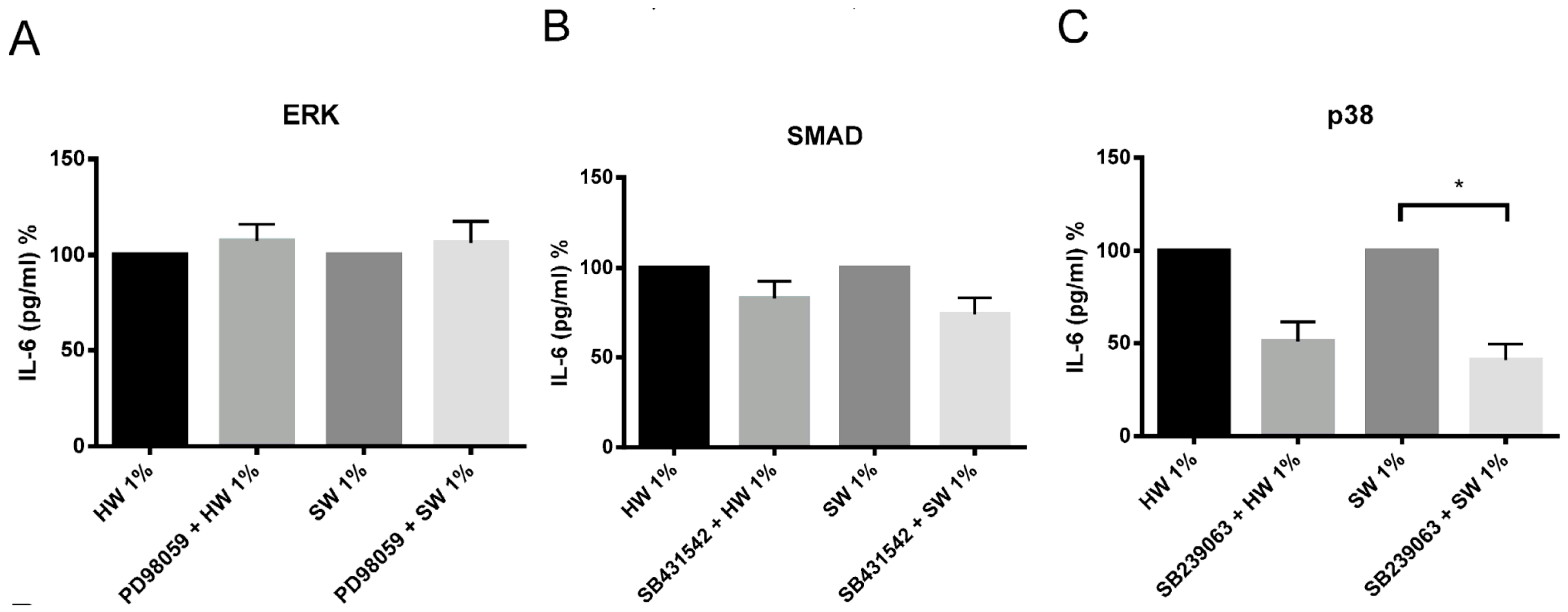
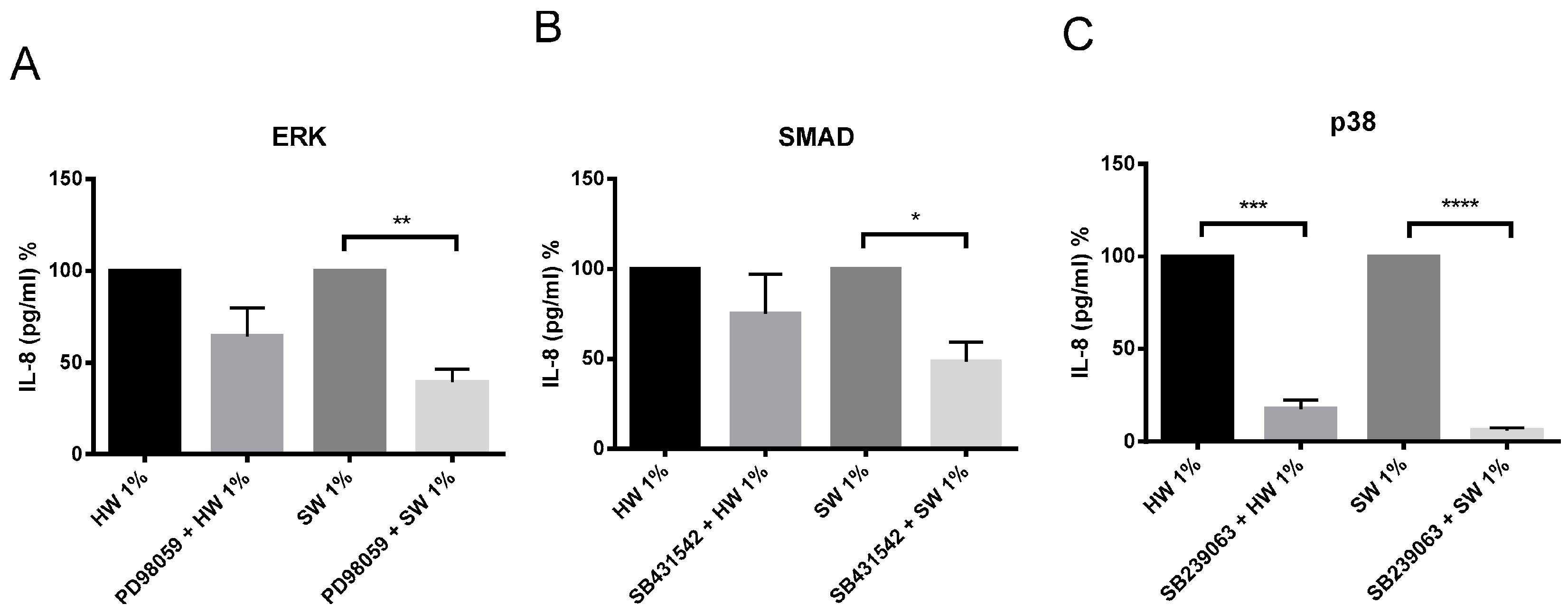
2.5. Hardwood and Softwood Smoke Induced IL-6 and IL-8 Are Attenuated by Different Signalling Pathway Inhibitors
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Isolation and Culture of Primary Human Lung Fibroblasts
4.3. Cell Culture
4.4. Biomass Smoke Extract Preparation and Stimulation
4.5. ELISA Detection of IL-6 and IL-8
4.6. Cell Viability Assay
4.7. Viral Propagation, Titration, and Stimulation
4.8. Biomass Smoke Extract and Human Rhinovirus-16 Stimulation
4.9. Signalling Pathway Inhibition Using Specific Inhibitors
4.10. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dutta, A.; Ray, M.R.; Banerjee, A. Systemic inflammatory changes and increased oxidative stress in rural indian women cooking with biomass fuels. Toxicol. Appl. Pharmacol. 2012, 261, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Kurmi, O.P.; Lam, K.B.; Ayres, J.G. Indoor air pollution and the lung in low- and medium-income countries. Eur. Respir. J. 2012, 40, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Kodgule, R.; Salvi, S. Exposure to biomass smoke as a cause for airway disease in women and children. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.B.; Bruce, N.G.; Grigg, J.; Hibberd, P.L.; Kurmi, O.P.; Lam, K.B.; Mortimer, K.; Asante, K.P.; Balakrishnan, K.; Balmes, J.; et al. Respiratory risks from household air pollution in low and middle income countries. Lancet Respir. Med. 2014, 2, 823–860. [Google Scholar] [CrossRef]
- Smith, K.R.; Bruce, N.; Balakrishnan, K.; Adair-Rohani, H.; Balmes, J.; Chafe, Z.; Dherani, M.; Hosgood, H.D.; Mehta, S.; Pope, D.; et al. Millions dead: How do we know and what does it mean? Methods used in the comparative risk assessment of household air pollution. Annu. Rev. Public Health 2014, 35, 185–206. [Google Scholar] [CrossRef] [PubMed]
- Torres-Duque, C.; Maldonado, D.; Perez-Padilla, R.; Ezzati, M.; Viegi, G. Biomass fuels and respiratory diseases: A review of the evidence. Proc. Am. Thorac. Soc. 2008, 5, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; Amann, M.; Anderson, H.R.; Andrews, K.G.; Aryee, M.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the global burden of disease study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef]
- Gurley, E.S.; Homaira, N.; Salje, H.; Ram, P.K.; Haque, R.; Petri, W.; Bresee, J.; Moss, W.J.; Breysse, P.; Luby, S.P.; et al. Indoor exposure to particulate matter and the incidence of acute lower respiratory infections among children: A birth cohort study in urban bangladesh. Indoor Air 2013, 23, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Dherani, M.; Pope, D.; Mascarenhas, M.; Smith, K.R.; Weber, M.; Bruce, N. Indoor air pollution from unprocessed solid fuel use and pneumonia risk in children aged under five years: A systematic review and meta-analysis. Bull. World Health Organ. 2008, 86, 390C–398C. [Google Scholar]
- Upadhyay, A.K.; Singh, A.; Kumar, K.; Singh, A. Impact of indoor air pollution from the use of solid fuels on the incidence of life threatening respiratory illnesses in children in India. BMC Public Health 2015, 15, 300. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Kinney, P.; Chillrud, S.; Jack, D. A systematic review of innate immunomodulatory effects of household air pollution secondary to the burning of biomass fuels. Ann. Glob. Health 2015, 81, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Souza, N.; Dolganov, G.; Dubin, R.; Sachs, L.A.; Sassina, L.; Sporer, H.; Yagi, S.; Schnurr, D.; Boushey, H.A.; Widdicombe, J.H. Resistance of differentiated human airway epithelium to infection by rhinovirus. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L373–L381. [Google Scholar] [CrossRef] [PubMed]
- Ghildyal, R.; Dagher, H.; Donninger, H.; de Silva, D.; Li, X.; Freezer, N.J.; Wilson, J.W.; Bardin, P.G. Rhinovirus infects primary human airway fibroblasts and induces a neutrophil chemokine and a permeability factor. J. Med. Virol. 2005, 75, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Gern, J.E.; Martin, M.S.; Anklam, K.A.; Shen, K.; Roberg, K.A.; Carlson-Dakes, K.T.; Adler, K.; Gilbertson-White, S.; Hamilton, R.; Shult, P.A.; et al. Relationships among specific viral pathogens, virus-induced interleukin-8, and respiratory symptoms in infancy. Pediatr. Allergy Immunol. 2002, 13, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lau, C.; Wiehler, S.; Pow, A.; Mazzulli, T.; Gutierrez, C.; Proud, D.; Chow, C.W. Syk is downstream of intercellular adhesion molecule-1 and mediates human rhinovirus activation of p38 MAPK in airway epithelial cells. J. Immunol. 2006, 177, 6859–6870. [Google Scholar] [CrossRef] [PubMed]
- Claude, J.A.; Grimm, A.; Savage, H.P.; Pinkerton, K.E. Perinatal exposure to environmental tobacco smoke (ETS) enhances susceptibility to viral and secondary bacterial infections. Int. J. Environ. Res. Public Health 2012, 9, 3954–3964. [Google Scholar] [CrossRef] [PubMed]
- Du Preez, K.; Mandalakas, A.M.; Kirchner, H.L.; Grewal, H.M.; Schaaf, H.S.; van Wyk, S.S.; Hesseling, A.C. Environmental tobacco smoke exposure increases mycobacterium tuberculosis infection risk in children. Int. J. Tuberc. Lung Dis. 2011, 15, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Kariya, C.; Chu, H.W.; Huang, J.; Leitner, H.; Martin, R.J.; Day, B.J. Mycoplasma pneumoniae infection and environmental tobacco smoke inhibit lung glutathione adaptive responses and increase oxidative stress. Infect. Immun. 2008, 76, 4455–4462. [Google Scholar] [CrossRef] [PubMed]
- Hudy, M.H.; Traves, S.L.; Wiehler, S.; Proud, D. Cigarette smoke modulates rhinovirus-induced airway epithelial cell chemokine production. Eur. Respir. J. 2010, 35, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Logan, J.; Chen, L.; Gangell, C.; Sly, P.D.; Fantino, E.; Liu, K. Brief exposure to cigarette smoke impairs airway epithelial cell innate anti-viral defence. Toxicol. In Vitro 2014, 28, 1430–1435. [Google Scholar] [CrossRef] [PubMed]
- Eddleston, J.; Lee, R.U.; Doerner, A.M.; Herschbach, J.; Zuraw, B.L. Cigarette smoke decreases innate responses of epithelial cells to rhinovirus infection. Am. J. Respir. Cell Mol. Biol. 2011, 44, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Groskreutz, D.J.; Monick, M.M.; Babor, E.C.; Nyunoya, T.; Varga, S.M.; Look, D.C.; Hunninghake, G.W. Cigarette smoke alters respiratory syncytial virus-induced apoptosis and replication. Am. J. Respir. Cell Mol. Biol. 2009, 41, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Proud, D.; Hudy, M.H.; Wiehler, S.; Zaheer, R.S.; Amin, M.A.; Pelikan, J.B.; Tacon, C.E.; Tonsaker, T.O.; Walker, B.L.; Kooi, C.; et al. Cigarette smoke modulates expression of human rhinovirus-induced airway epithelial host defense genes. PLoS ONE 2012, 7, e40762. [Google Scholar] [CrossRef] [PubMed]
- Regalado, J.; Pérez-Padilla, R.; Sansores, R.; Páramo Ramirez, J.I.; Brauer, M.; Paré, P.; Vedal, S. The effect of biomass burning on respiratory symptoms and lung function in rural Mexican women. Am. J. Respir. Crit. Care Med. 2006, 174, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Fine, P.M.; Cass, G.R.; Simoneit, B.R. Chemical characterization of fine particle emissions from fireplace combustion of woods grown in the Northeastern United States. Environ. Sci. Technol. 2001, 35, 2665–2675. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.Y.; Zhang, W.; Atkiston, S. The characterisation of polycyclic aromatic hydrocarbons emissions from burning of different firewood species in Australia. Environ. Pollut. 2003, 124, 283–289. [Google Scholar] [CrossRef]
- Krimmer, D.I.; Burgess, J.K.; Wooi, T.K.; Black, J.L.; Oliver, B.G. Matrix proteins from smoke-exposed fibroblasts are pro-proliferative. Am. J. Respir. Cell Mol. Biol. 2012, 46, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Oliver, B.G.; Johnston, S.L.; Baraket, M.; Burgess, J.K.; King, N.J.; Roth, M.; Lim, S.; Black, J.L. Increased proinflammatory responses from asthmatic human airway smooth muscle cells in response to rhinovirus infection. Respir. Res. 2006, 7, 71. [Google Scholar] [CrossRef] [PubMed]
- Griego, S.D.; Weston, C.B.; Adams, J.L.; Tal-Singer, R.; Dillon, S.B. Role of p38 mitogen-activated protein kinase in rhinovirus-induced cytokine production by bronchial epithelial cells. J. Immunol. 2000, 165, 5211–5220. [Google Scholar] [CrossRef] [PubMed]
- Pelaia, G.; Cuda, G.; Vatrella, A.; Fratto, D.; Grembiale, R.D.; Tagliaferri, P.; Maselli, R.; Costanzo, F.S.; Marsico, S.A. Effects of transforming growth factor-β and budesonide on mitogen-activated protein kinase activation and apoptosis in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2003, 29, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Ip, W.K.; Wong, C.K.; Lam, C.W. Interleukin (IL)-4 and IL-13 up-regulate monocyte chemoattractant protein-1 expression in human bronchial epithelial cells: Involvement of p38 mitogen-activated protein kinase, extracellular signal-regulated kinase 1/2 and janus kinase-2 but not c-Jun NH2-terminal kinase 1/2 signalling pathways. Clin. Exp. Immunol. 2006, 145, 162–172. [Google Scholar] [PubMed]
- Johnson, P.R.; Burgess, J.K.; Ge, Q.; Poniris, M.; Boustany, S.; Twigg, S.M.; Black, J.L. Connective tissue growth factor induces extracellular matrix in asthmatic airway smooth muscle. Am. J. Respir. Crit. Care Med. 2006, 173, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Gencay, M.M.; Bubendorf, L.; Burgess, J.K.; Parson, H.; Robinson, B.W.; Tamm, M.; Black, J.L.; Roth, M. ERK1/2 and p38 Map kinase control MMP-2, MT1-MMP, and TIMP action and affect cell migration: A comparison between mesothelioma and mesothelial cells. J. Cell. Physiol. 2006, 207, 540–552. [Google Scholar] [CrossRef] [PubMed]







© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capistrano, S.J.; Zakarya, R.; Chen, H.; Oliver, B.G. Biomass Smoke Exposure Enhances Rhinovirus-Induced Inflammation in Primary Lung Fibroblasts. Int. J. Mol. Sci. 2016, 17, 1403. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091403
Capistrano SJ, Zakarya R, Chen H, Oliver BG. Biomass Smoke Exposure Enhances Rhinovirus-Induced Inflammation in Primary Lung Fibroblasts. International Journal of Molecular Sciences. 2016; 17(9):1403. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091403
Chicago/Turabian StyleCapistrano, Sarah J., Razia Zakarya, Hui Chen, and Brian G. Oliver. 2016. "Biomass Smoke Exposure Enhances Rhinovirus-Induced Inflammation in Primary Lung Fibroblasts" International Journal of Molecular Sciences 17, no. 9: 1403. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091403