
Molecular Mechanisms of Bone Metastasis: Which Targets Came from the Bench to the Bedside?


Abstract
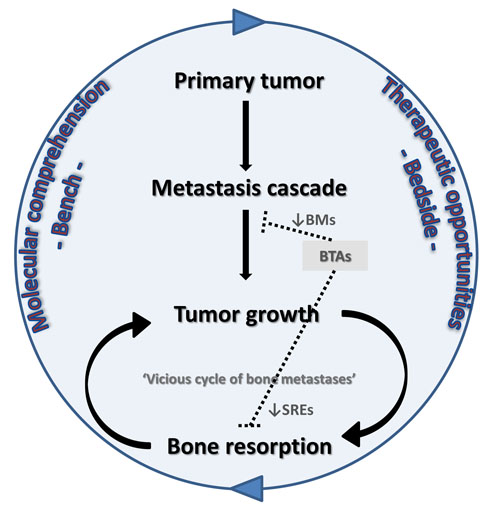
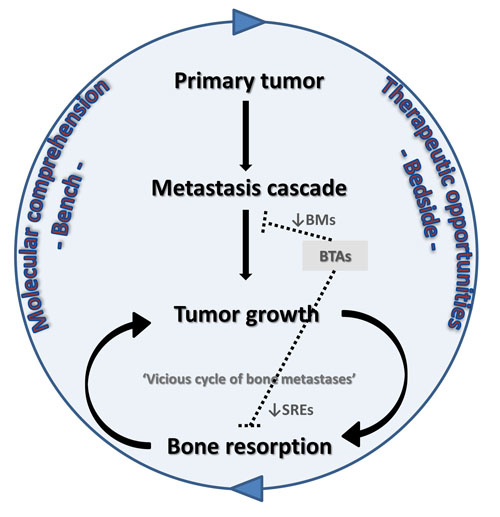
:
1. Introduction
2. Pathophysiology of Bone Metastases
2.1. The “Vicious Cycle of Bone Metastases”
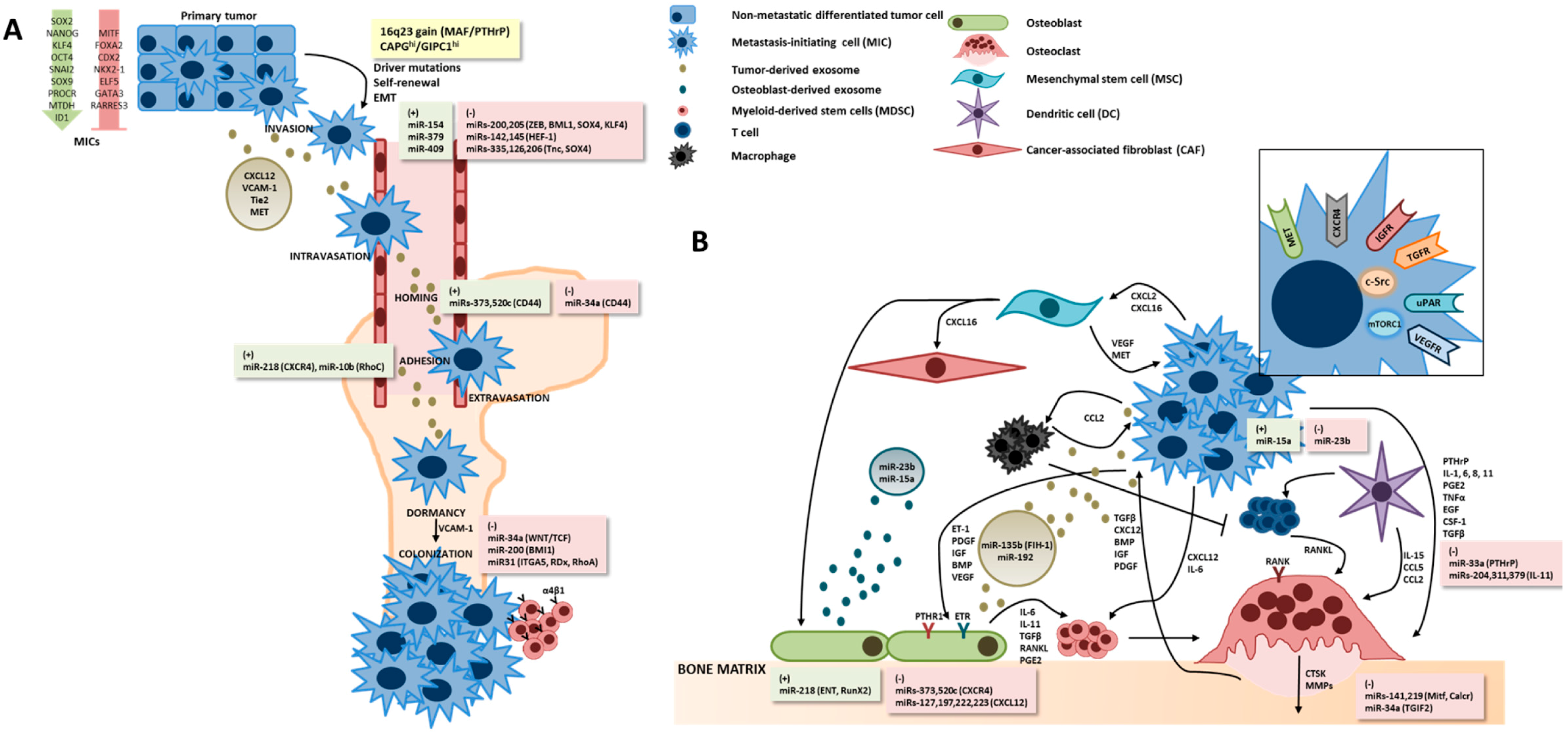
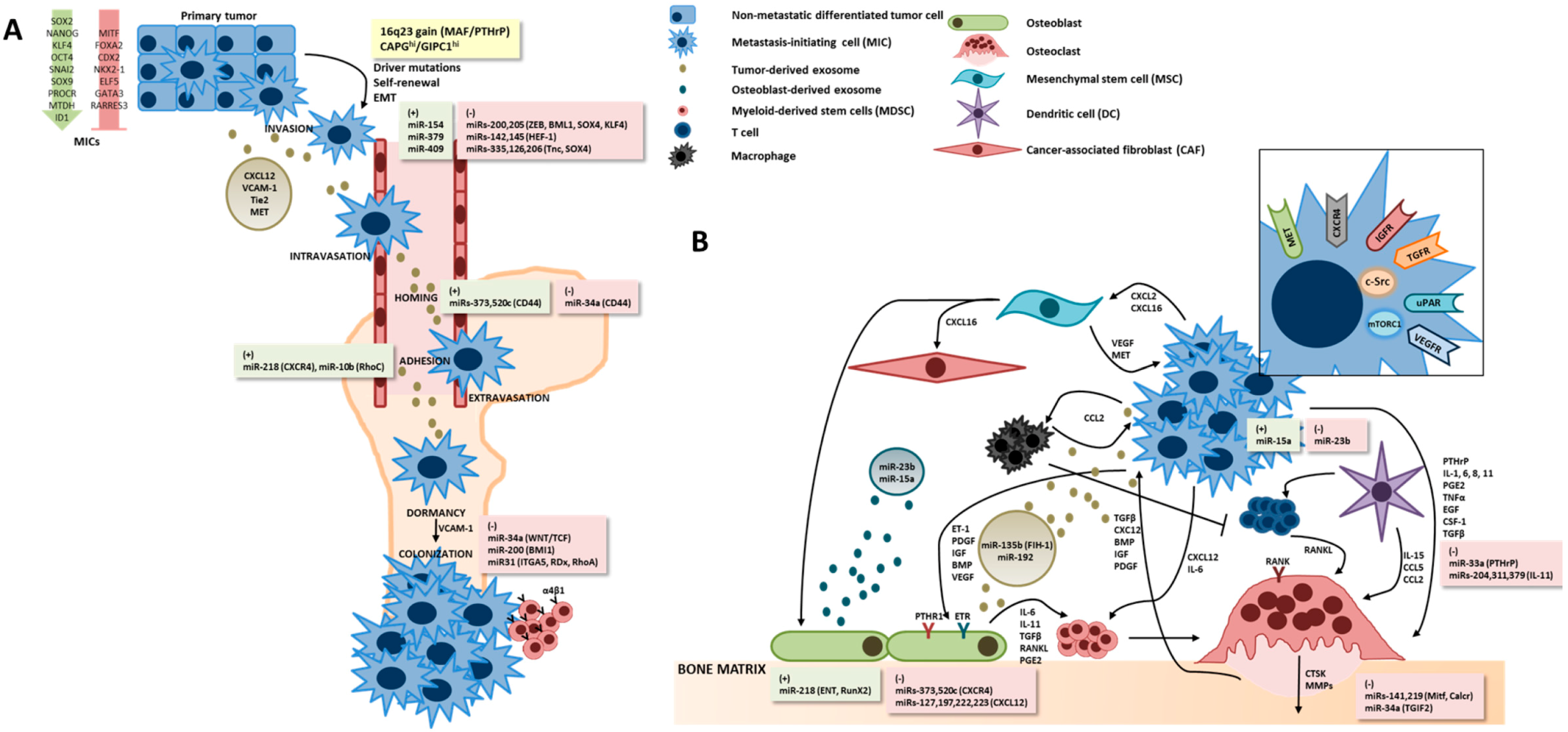
2.2. Metastasis-Initiating Cells, Chemoattraction to Bone and the “Pre-Metastatic Niche”
2.3. Genomic Signatures of Bone Metastases
3. Validated Targets
3.1. Bone Targeted Agents in Advanced Disease
3.2. Bone-Targeted Radiopharmaceuticals: Radium-223
3.3. Other Bone-Targeted Radiopharmaceuticals
3.4. Adjuvant Use of Bone Targeted Agents
4. Future Directions and Conclusions
Author Contributions
Conflicts of Interest
References
- DeVita, V.T., Jr.; Rosenberg, S.A. Two hundred years of cancer research. N. Engl. J. Med. 2012, 366, 2207–2214. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Mark, M.T.; Molina, H.; Kohsaka, S.; di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Minn, A.J.; Gupta, G.P.; Siegel, P.M.; Bos, P.D.; Shu, W.; Giri, D.D.; Viale, A.; Olshen, A.B.; Gerald, W.L.; Massague, J. Genes that mediate breast cancer metastasis to lung. Nature 2005, 436, 518–524. [Google Scholar] [CrossRef]
- Bos, P.D.; Zhang, X.H.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009. [Google Scholar] [CrossRef]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef]
- Croucher, P.I.; McDonald, M.M.; Martin, T.J. Bone metastasis: The importance of the neighbourhood. Nat. Rev. Cancer 2016, 16, 373–386. [Google Scholar] [CrossRef]
- Aapro, M.; Abrahamsson, P.A.; Body, J.J.; Coleman, R.E.; Colomer, R.; Costa, L.; Crino, L.; Dirix, L.; Gnant, M.; Gralow, J.; et al. Guidance on the use of bisphosphonates in solid tumours: Recommendations of an international expert panel. Ann. Oncol. 2008, 19, 420–432. [Google Scholar] [CrossRef]
- Lipton, A.; Fizazi, K.; Stopeck, A.T.; Henry, D.H.; Brown, J.E.; Yardley, D.A.; Richardson, G.E.; Siena, S.; Maroto, P.; Clemens, M.; et al. Superiority of denosumab to zoledronic acid for prevention of skeletal-related events: A combined analysis of 3 pivotal, randomised, phase 3 trials. Eur. J. Cancer 2012, 48, 3082–3092. [Google Scholar] [CrossRef]
- Early Breast Cancer Trialists’ Collaborative Group; Coleman, R.; Powles, T.; Paterson, A.; Gnant, M.; Anderson, S.; Diel, I.; Gralow, J.; von Minckwitz, G.; Moebus, V.; et al. Adjuvant bisphosphonate treatment in early breast cancer: Meta-analyses of individual patient data from randomised trials. Lancet 2015, 386, 1353–1361. [Google Scholar]
- Hadji, P.; Coleman, R.E.; Wilson, C.; Powles, T.J.; Clezardin, P.; Aapro, M.; Costa, L.; Body, J.J.; Markopoulos, C.; Santini, D.; et al. Adjuvant bisphosphonates in early breast cancer: Consensus guidance for clinical practice from a european panel. Ann. Oncol. 2016, 27, 379–390. [Google Scholar] [CrossRef]
- Strobl, S.; Korkmaz, B.; Devyatko, Y.; Schuetz, M.; Exner, R.; Dubsky, P.C.; Jakesz, R.; Gnant, M. Adjuvant bisphosphonates and breast cancer survival. Annu. Rev. Med. 2016, 67, 1–10. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [CrossRef]
- Steeg, P.S. Tumor metastasis: Mechanistic insights and clinical challenges. Nat. Med. 2006, 12, 895–904. [Google Scholar] [CrossRef]
- Mundy, G.R. Mechanisms of bone metastasis. Cancer 1997, 80, 1546–1556. [Google Scholar] [CrossRef]
- Mina, L.A.; Sledge, G.W., Jr. Rethinking the metastatic cascade as a therapeutic target. Nat. Rev. Clin. Oncol. 2011, 8, 325–332. [Google Scholar] [CrossRef]
- Buenrostro, D.; Mulcrone, P.L.; Owens, P.; Sterling, J.A. The bone microenvironment: A fertile soil for tumor growth. Curr. Osteoporos. Rep. 2016, 14, 151–158. [Google Scholar] [CrossRef]
- Weidle, U.H.; Birzele, F.; Kollmorgen, G.; Ruger, R. Molecular mechanisms of bone metastasis. Cancer Genom. Proteom. 2016, 13, 1–12. [Google Scholar]
- Casimiro, S.; Luis, I.; Fernandes, A.; Pires, R.; Pinto, A.; Gouveia, A.G.; Francisco, A.F.; Portela, J.; Correia, L.; Costa, L. Analysis of a bone metastasis gene expression signature in patients with bone metastasis from solid tumors. Clin. Exp. Metastasis 2012, 29, 155–164. [Google Scholar] [CrossRef]
- Ibrahim, T.; Flamini, E.; Mercatali, L.; Sacanna, E.; Serra, P.; Amadori, D. Pathogenesis of osteoblastic bone metastases from prostate cancer. Cancer 2010, 116, 1406–1418. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Trouvin, A.P.; Goeb, V. Receptor activator of nuclear factor-κB ligand and osteoprotegerin: Maintaining the balance to prevent bone loss. Clin. Interv. Aging 2010, 5, 345–354. [Google Scholar]
- Celia-Terrassa, T.; Kang, Y. Distinctive properties of metastasis-initiating cells. Genes Dev. 2016, 30, 892–908. [Google Scholar] [CrossRef]
- Pavlovic, M.; Arnal-Estape, A.; Rojo, F.; Bellmunt, A.; Tarragona, M.; Guiu, M.; Planet, E.; Garcia-Albeniz, X.; Morales, M.; Urosevic, J.; et al. Enhanced maf oncogene expression and breast cancer bone metastasis. J. Natl. Cancer Inst. 2015, 107, djv256. [Google Scholar] [CrossRef]
- Westbrook, J.A.; Cairns, D.A.; Peng, J.; Speirs, V.; Hanby, A.M.; Holen, I.; Wood, S.L.; Ottewell, P.D.; Marshall, H.; Banks, R.E.; et al. CAPG and GIPC1: Breast cancer biomarkers for bone metastasis development and treatment. J. Natl. Cancer Inst. 2016, 108, djv360. [Google Scholar] [PubMed]
- Lu, X.; Mu, E.; Wei, Y.; Riethdorf, S.; Yang, Q.; Yuan, M.; Yan, J.; Hua, Y.; Tiede, B.J.; Lu, X.; et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging α4β1-positive osteoclast progenitors. Cancer Cell 2011, 20, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Ursini-Siegel, J.; Siegel, P.M. The influence of the pre-metastatic niche on breast cancer metastasis. Cancer Lett. 2016, 380, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Vignani, F.; Bertaglia, V.; Buttigliero, C.; Tucci, M.; Scagliotti, G.V.; Di Maio, M. Skeletal metastases and impact of anticancer and bone-targeted agents in patients with castration-resistant prostate cancer. Cancer Treat. Rev. 2016, 44, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Croset, M.; Kan, C.; Clezardin, P. Tumour-derived mirnas and bone metastasis. Bonekey Rep. 2015, 4, 688. [Google Scholar] [CrossRef]
- Yin, J.J.; Mohammad, K.S.; Kakonen, S.M.; Harris, S.; Wu-Wong, J.R.; Wessale, J.L.; Padley, R.J.; Garrett, I.R.; Chirgwin, J.M.; Guise, T.A. A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proc. Natl. Acad. Sci. USA 2003, 100, 10954–10959. [Google Scholar] [CrossRef] [PubMed]
- Guise, T.A.; Mohammad, K.S.; Clines, G.; Stebbins, E.G.; Wong, D.H.; Higgins, L.S.; Vessella, R.; Corey, E.; Padalecki, S.; Suva, L.; et al. Basic mechanisms responsible for osteolytic and osteoblastic bone metastases. Clin. Cancer Res. 2006, 12, 6213s–6216s. [Google Scholar] [CrossRef] [PubMed]
- Sceneay, J.; Smyth, M.J.; Moller, A. The pre-metastatic niche: Finding common ground. Cancer Metastasis Rev. 2013, 32, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Gartland, A.; Erler, J.T. The pre-metastatic niche: Is metastasis random? Bonekey Rep. 2012, 1, 80. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, E.A.; Shiozawa, Y.; Pienta, K.J.; Taichman, R.S. The prostate cancer bone marrow niche: More than just “fertile soil”. Asian J. Androl. 2012, 14, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Webber, J.; Steadman, R.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010, 70, 9621–9630. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Ito, Y.; Ohtsuki, Y.; Ando, M.; Tsukamasa, Y.; Yamada, N.; Naoe, T.; Akao, Y. Microvesicles released from hormone-refractory prostate cancer cells facilitate mouse pre-osteoblast differentiation. J. Mol. Histol. 2012, 43, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Lorusso, G.; Ruegg, C. New insights into the mechanisms of organ-specific breast cancer metastasis. Semin. Cancer Biol. 2012, 22, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Loberg, R.; Taichman, R.S. The pivotal role of CXCL12 (SDF-1)/CXCR4 axis in bone metastasis. Cancer Metastasis Rev. 2006, 25, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordon-Cardo, C.; Guise, T.A.; Massague, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef]
- Liang, Z.; Yoon, Y.; Votaw, J.; Goodman, M.M.; Williams, L.; Shim, H. Silencing of CXCR4 blocks breast cancer metastasis. Cancer Res. 2005, 65, 967–971. [Google Scholar] [PubMed]
- Sun, Y.X.; Wang, J.; Shelburne, C.E.; Lopatin, D.E.; Chinnaiyan, A.M.; Rubin, M.A.; Pienta, K.J.; Taichman, R.S. Expression of CXCR4 and CXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. J. Cell. Biochem. 2003, 89, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Schneider, A.; Jung, Y.; Wang, J.; Dai, J.; Wang, J.; Cook, K.; Osman, N.I.; Koh-Paige, A.J.; Shim, H.; et al. Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J. Bone Miner. Res. 2005, 20, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Fang, M.; Wang, J.; Cooper, C.R.; Pienta, K.J.; Taichman, R.S. Expression and activation of αvβ3 integrins by SDF-1/CXC12 increases the aggressiveness of prostate cancer cells. Prostate 2007, 67, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Chinni, S.R.; Sivalogan, S.; Dong, Z.; Filho, J.C.; Deng, X.; Bonfil, R.D.; Cher, M.L. CXCL12/CXCR4 signaling activates AKT-1 and MMP-9 expression in prostate cancer cells: The role of bone microenvironment-associated CXCL12. Prostate 2006, 66, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, J.; Sun, Y.; Song, W.; Nor, J.E.; Wang, C.Y.; Taichman, R.S. Diverse signaling pathways through the SDF-1/CXCR4 chemokine axis in prostate cancer cell lines leads to altered patterns of cytokine secretion and angiogenesis. Cell Signal. 2005, 17, 1578–1592. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Shiozawa, Y.; Jung, Y.; Kim, J.K.; Pedersen, E.; Mishra, A.; Zalucha, J.L.; Wang, J.; Keller, E.T.; Pienta, K.J.; et al. Disseminated prostate cancer cells can instruct hematopoietic stem and progenitor cells to regulate bone phenotype. Mol. Cancer Res. 2012, 10, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Taichman, R.S.; Patel, L.R.; Bedenis, R.; Wang, J.; Weidner, S.; Schumann, T.; Yumoto, K.; Berry, J.E.; Shiozawa, Y.; Pienta, K.J. Gas6 receptor status is associated with dormancy and bone metastatic tumor formation. PLoS ONE 2013, 8, e61873. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, C.; Gao, X.; Welte, T.; Muscarella, A.M.; Tian, L.; Zhao, H.; Zhao, Z.; Du, S.; Tao, J.; et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell 2015, 27, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Van’t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Bueno-de-Mesquita, J.M.; Linn, S.C.; Keijzer, R.; Wesseling, J.; Nuyten, D.S.; van Krimpen, C.; Meijers, C.; de Graaf, P.W.; Bos, M.M.; Hart, A.A.; et al. Validation of 70-gene prognosis signature in node-negative breast cancer. Breast Cancer Res. Treat. 2009, 117, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Buyse, M.; Loi, S.; van’t Veer, L.; Viale, G.; Delorenzi, M.; Glas, A.M.; d’Assignies, M.S.; Bergh, J.; Lidereau, R.; Ellis, P.; et al. Validation and clinical utility of a 70-gene prognostic signature for women with node-negative breast cancer. J. Natl. Cancer Inst. 2006, 98, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Van de Vijver, M.J.; He, Y.D.; Van’t Veer, L.J.; Dai, H.; Hart, A.A.; Voskuil, D.W.; Schreiber, G.J.; Peterse, J.L.; Roberts, C.; Marton, M.J.; et al. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 2002, 347, 1999–2009. [Google Scholar] [CrossRef] [PubMed]
- Knauer, M.; Mook, S.; Rutgers, E.J.; Bender, R.A.; Hauptmann, M.; van de Vijver, M.J.; Koornstra, R.H.; Bueno-de-Mesquita, J.M.; Linn, S.C.; Van’t Veer, L.J. The predictive value of the 70-gene signature for adjuvant chemotherapy in early breast cancer. Breast Cancer Res. Treat. 2010, 120, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Mook, S.; Knauer, M.; Bueno-de-Mesquita, J.M.; Retel, V.P.; Wesseling, J.; Linn, S.C.; van’t Veer, L.J.; Rutgers, E.J. Metastatic potential of T1 breast cancer can be predicted by the 70-gene mammaprint signature. Ann. Surg. Oncol. 2010, 17, 1406–1413. [Google Scholar] [CrossRef] [PubMed]
- Mook, S.; Schmidt, M.K.; Weigelt, B.; Kreike, B.; Eekhout, I.; van de Vijver, M.J.; Glas, A.M.; Floore, A.; Rutgers, E.J.; Van’t Veer, L.J. The 70-gene prognosis signature predicts early metastasis in breast cancer patients between 55 and 70 years of age. Ann. Oncol. 2010, 21, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Drukker, C.A.; van Tinteren, H.; Schmidt, M.K.; Rutgers, E.J.; Bernards, R.; van de Vijver, M.J.; Van’t Veer, L.J. Long-term impact of the 70-gene signature on breast cancer outcome. Breast Cancer Res. Treat. 2014, 143, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 2004, 351, 2817–2826. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Tang, G.; Shak, S.; Kim, C.; Baker, J.; Kim, W.; Cronin, M.; Baehner, F.L.; Watson, D.; Bryant, J.; et al. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J. Clin. Oncol. 2006, 24, 3726–3734. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.J.; Gray, R.; Badve, S.; Childs, B.H.; Yoshizawa, C.; Rowley, S.; Shak, S.; Baehner, F.L.; Ravdin, P.M.; Davidson, N.E.; et al. Prognostic utility of the 21-gene assay in hormone receptor-positive operable breast cancer compared with classical clinicopathologic features. J. Clin. Oncol. 2008, 26, 4063–4071. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.S.; Mumby, P.B.; Norton, J.; Rychlik, K.; Smerage, J.; Kash, J.; Chew, H.K.; Gaynor, E.R.; Hayes, D.F.; Epstein, A.; et al. Prospective multicenter study of the impact of the 21-gene recurrence score assay on medical oncologist and patient adjuvant breast cancer treatment selection. J. Clin. Oncol. 2010, 28, 1671–1676. [Google Scholar] [CrossRef] [PubMed]
- Albain, K.S.; Barlow, W.E.; Shak, S.; Hortobagyi, G.N.; Livingston, R.B.; Yeh, I.T.; Ravdin, P.; Bugarini, R.; Baehner, F.L.; Davidson, N.E.; et al. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: A retrospective analysis of a randomised trial. Lancet Oncol. 2010, 11, 55–65. [Google Scholar] [CrossRef]
- Minn, A.J.; Gupta, G.P.; Padua, D.; Bos, P.; Nguyen, D.X.; Nuyten, D.; Kreike, B.; Zhang, Y.; Wang, Y.; Ishwaran, H.; et al. Lung metastasis genes couple breast tumor size and metastatic spread. Proc. Natl. Acad. Sci. USA 2007, 104, 6740–6745. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.J.; Kang, Y.; Serganova, I.; Gupta, G.P.; Giri, D.D.; Doubrovin, M.; Ponomarev, V.; Gerald, W.L.; Blasberg, R.; Massague, J. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J. Clin. Investig. 2005, 115, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Smid, M.; Wang, Y.; Klijn, J.G.; Sieuwerts, A.M.; Zhang, Y.; Atkins, D.; Martens, J.W.; Foekens, J.A. Genes associated with breast cancer metastatic to bone. J. Clin. Oncol. 2006, 24, 2261–2267. [Google Scholar] [CrossRef] [PubMed]
- Savci-Heijink, C.D.; Halfwerk, H.; Koster, J.; van de Vijver, M.J. A novel gene expression signature for bone metastasis in breast carcinomas. Breast Cancer Res. Treat. 2016, 156, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Fazilaty, H.; Mehdipour, P. Genetics of breast cancer bone metastasis: A sequential multistep pattern. Clin. Exp. Metastasis 2014, 31, 595–612. [Google Scholar] [CrossRef] [PubMed]
- Saad, F.; Gleason, D.M.; Murray, R.; Tchekmedyian, S.; Venner, P.; Lacombe, L.; Chin, J.L.; Vinholes, J.J.; Goas, J.A.; Zheng, M.; et al. Long-term efficacy of zoledronic acid for the prevention of skeletal complications in patients with metastatic hormone-refractory prostate cancer. J. Natl. Cancer Inst. 2004, 96, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E. Skeletal complications of malignancy. Cancer 1997, 80, 1588–1594. [Google Scholar] [CrossRef]
- Coleman, R.E. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin. Cancer Res. 2006, 12, 6243s–6249s. [Google Scholar] [CrossRef] [PubMed]
- Roelofs, A.J.; Thompson, K.; Gordon, S.; Rogers, M.J. Molecular mechanisms of action of bisphosphonates: Current status. Clin. Cancer Res. 2006, 12, 6222s–6230s. [Google Scholar] [CrossRef] [PubMed]
- Gnant, M.; Dubsky, P.; Fitzal, F.; Blaha, P.; Schoppmann, S.; Steger, G.; Marth, C.; Samonigg, H.; Huttner, K.; Fohler, H.; et al. Maintaining bone density in patients undergoing treatment for breast cancer: Is there an adjuvant benefit? Clin. Breast Cancer 2009, 9 (Suppl. S1), S18–S27. [Google Scholar] [CrossRef] [PubMed]
- Stopeck, A.T.; Lipton, A.; Body, J.J.; Steger, G.G.; Tonkin, K.; de Boer, R.H.; Lichinitser, M.; Fujiwara, Y.; Yardley, D.A.; Viniegra, M.; et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: A randomized, double-blind study. J. Clin. Oncol. 2010, 28, 5132–5139. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Carducci, M.; Smith, M.; Damiao, R.; Brown, J.; Karsh, L.; Milecki, P.; Shore, N.; Rader, M.; Wang, H.; et al. Denosumab vs. zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: A randomised, double-blind study. Lancet 2011, 377, 813–822. [Google Scholar] [CrossRef]
- Henry, D.H.; Costa, L.; Goldwasser, F.; Hirsh, V.; Hungria, V.; Prausova, J.; Scagliotti, G.V.; Sleeboom, H.; Spencer, A.; Vadhan-Raj, S.; et al. Randomized, double-blind study of denosumab vs. zoledronic acid in the treatment of bone metastases in patients with advanced cancer (excluding breast and prostate cancer) or multiple myeloma. J. Clin. Oncol. 2011, 29, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Kohno, N.; Aogi, K.; Minami, H.; Nakamura, S.; Asaga, T.; Iino, Y.; Watanabe, T.; Goessl, C.; Ohashi, Y.; Takashima, S. Zoledronic acid significantly reduces skeletal complications compared with placebo in japanese women with bone metastases from breast cancer: A randomized, placebo-controlled trial. J. Clin. Oncol. 2005, 23, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.S.; Gordon, D.; Tchekmedyian, S.; Yanagihara, R.; Hirsh, V.; Krzakowski, M.; Pawlicki, M.; de Souza, P.; Zheng, M.; Urbanowitz, G.; et al. Zoledronic acid vs. placebo in the treatment of skeletal metastases in patients with lung cancer and other solid tumors: A phase III, double-blind, randomized trial—The zoledronic acid lung cancer and other solid tumors study group. J. Clin. Oncol. 2003, 21, 3150–3157. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.H.; Stockler, M.R.; Pavlakis, N. Bisphosphonates and other bone agents for breast cancer. Cochrane Library 2012, CD003474. [Google Scholar] [CrossRef]
- Barrett-Lee, P.; Casbard, A.; Abraham, J.; Hood, K.; Coleman, R.; Simmonds, P.; Timmins, H.; Wheatley, D.; Grieve, R.; Griffiths, G.; et al. Oral ibandronic acid vs. intravenous zoledronic acid in treatment of bone metastases from breast cancer: A randomised, open label, non-inferiority phase 3 trial. Lancet Oncol. 2014, 15, 114–122. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fossa, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Elomaa, I.; Blomqvist, C.; Porkka, L.; Lamberg-Allardt, C.; Borgstrom, G.H. Treatment of skeletal disease in breast cancer: A controlled clodronate trial. Bone 1987, 8 (Suppl. S1), S53–S56. [Google Scholar] [PubMed]
- Paterson, A.H.; Powles, T.J.; Kanis, J.A.; McCloskey, E.; Hanson, J.; Ashley, S. Double-blind controlled trial of oral clodronate in patients with bone metastases from breast cancer. J. Clin. Oncol. 1993, 11, 59–65. [Google Scholar] [PubMed]
- Van Holten-Verzantvoort, A.T.; Kroon, H.M.; Bijvoet, O.L.; Cleton, F.J.; Beex, L.V.; Blijham, G.; Hermans, J.; Neijt, J.P.; Papapoulos, S.E.; Sleeboom, H.P.; et al. Palliative pamidronate treatment in patients with bone metastases from breast cancer. J. Clin. Oncol. 1993, 11, 491–498. [Google Scholar] [PubMed]
- Hortobagyi, G.N.; Theriault, R.L.; Porter, L.; Blayney, D.; Lipton, A.; Sinoff, C.; Wheeler, H.; Simeone, J.F.; Seaman, J.; Knight, R.D. Efficacy of pamidronate in reducing skeletal complications in patients with breast cancer and lytic bone metastases. N. Engl. J. Med. 1996, 335, 1785–1791. [Google Scholar] [CrossRef] [PubMed]
- Theriault, R.L.; Lipton, A.; Hortobagyi, G.N.; Leff, R.; Gluck, S.; Stewart, J.F.; Costello, S.; Kennedy, I.; Simeone, J.; Seaman, J.J.; et al. Pamidronate reduces skeletal morbidity in women with advanced breast cancer and lytic bone lesions: A randomized, placebo-controlled trial. J. Clin. Oncol. 1999, 17, 846–854. [Google Scholar] [PubMed]
- Rosen, L.S.; Gordon, D.; Kaminski, M.; Howell, A.; Belch, A.; Mackey, J.; Apffelstaedt, J.; Hussein, M.; Coleman, R.E.; Reitsma, D.J.; et al. Zoledronic acid vs. pamidronate in the treatment of skeletal metastases in patients with breast cancer or osteolytic lesions of multiple myeloma: A phase III, double-blind, comparative trial. Cancer J. 2001, 7, 377–387. [Google Scholar] [PubMed]
- Elomaa, I.; Kylmala, T.; Tammela, T.; Viitanen, J.; Ottelin, J.; Ruutu, M.; Jauhiainen, K.; Ala-Opas, M.; Roos, L.; Seppanen, J.; et al. Effect of oral clodronate on bone pain. Int. Urol. Nephrol. 1992, 24, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Kylmala, T.; Taube, T.; Tammela, T.L.; Risteli, L.; Risteli, J.; Elomaa, I. Concomitant IV and oral clodronate in the relief of bone pain—A double-blind placebo-controlled study in patients with prostate cancer. Br. J. Cancer 1997, 76, 939–942. [Google Scholar] [CrossRef] [PubMed]
- Ernst, D.S.; Tannock, I.F.; Venner, P.M.; Winquist, E.W.; Reyno, L.; Walker, H.; Ding, C.; Elliot, W.; Parulekar, W. Randomized, placebo-controlled trial of mitoxantrone/prednisone and clodronate vs. mitoxantrone/prednisone alone in patients with hormone-refractory prostate cancer (HRPC) and pain. Proc. Am. Soc. Clin. Oncol. 2002, 21, 177a. [Google Scholar]
- Lipton, A.; Small, E.J.; Saad, A. The new bisphosphonate, zometa decreases skeletal complications in both lytic and blastic lesions: A comparision to pamidronate. Cancer Investig. 2002, 20, 45–54. [Google Scholar] [CrossRef]
- Saad, F.; Gleason, D.M.; Murray, R.; Tchekmedyian, S.; Venner, P.; Lacombe, L.; Chin, J.L.; Vinholes, J.J.; Goas, J.A.; Chen, B.; et al. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J. Natl. Cancer Inst. 2002, 94, 1458–1468. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Halabi, S.; Ryan, C.J.; Hussain, A.; Vogelzang, N.; Stadler, W.; Hauke, R.J.; Monk, J.P.; Saylor, P.; Bhoopalam, N.; et al. Randomized controlled trial of early zoledronic acid in men with castration-sensitive prostate cancer and bone metastases: Results of CALGB 90202 (alliance). J. Clin. Oncol. 2014, 32, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Costa, L. Which bisphosphonate to treat bone metastases? Lancet Oncol. 2014, 15, 15–16. [Google Scholar] [CrossRef]
- Rosen, L.S.; Gordon, D.; Tchekmedyian, N.S.; Yanagihara, R.; Hirsh, V.; Krzakowski, M.; Pawlicki, M.; de Souza, P.; Zheng, M.; Urbanowitz, G.; et al. Long-term efficacy and safety of zoledronic acid in the treatment of skeletal metastases in patients with nonsmall cell lung carcinoma and other solid tumors: A randomized, phase III, double-blind, placebo-controlled trial. Cancer 2004, 100, 2613–2621. [Google Scholar] [CrossRef] [PubMed]
- Roodman, G.D. Pathogenesis of myeloma bone disease. J. Cell. Biochem. 2010, 109, 283–291. [Google Scholar] [PubMed]
- Berenson, J.R.; Lichtenstein, A.; Porter, L.; Dimopoulos, M.A.; Bordoni, R.; George, S.; Lipton, A.; Keller, A.; Ballester, O.; Kovacs, M.; et al. Long-term pamidronate treatment of advanced multiple myeloma patients reduces skeletal events. Myeloma aredia study group. J. Clin. Oncol. 1998, 16, 593–602. [Google Scholar] [PubMed]
- Gralow, J.R.; Biermann, J.S.; Farooki, A.; Fornier, M.N.; Gagel, R.F.; Kumar, R.N.; Shapiro, C.L.; Shields, A.; Smith, M.R.; Srinivas, S.; et al. NCCN task force report: Bone health in cancer care. J. Natl. Compr. Cancer Netw. 2009, 7 (Suppl. S3), S1–S32. [Google Scholar]
- Sartor, O.; Coleman, R.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fossa, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; et al. Effect of radium-223 dichloride on symptomatic skeletal events in patients with castration-resistant prostate cancer and bone metastases: Results from a phase 3, double-blind, randomised trial. Lancet Oncol. 2014, 15, 738–746. [Google Scholar] [CrossRef]
- Jong, J.M.; Oprea-Lager, D.E.; Hooft, L.; de Klerk, J.M.; Bloemendal, H.J.; Verheul, H.M.; Hoekstra, O.S.; van den Eertwegh, A.J. Radiopharmaceuticals for palliation of bone pain in patients with castration-resistant prostate cancer metastatic to bone: A systematic review. Eur. Urol. 2015, 70, 416–426. [Google Scholar] [CrossRef]
- Sartor, O. Radiopharmaceuticals: A path forward. Eur. Urol. 2016, 70, 427–428. [Google Scholar] [CrossRef]
- Clezardin, P. Mechanisms of action of bisphosphonates in oncology: A scientific concept evolving from antiresorptive to anticancer activities. Bonekey Rep. 2013, 2, 267. [Google Scholar] [CrossRef]
- Gnant, M.; Mlineritsch, B.; Schippinger, W.; Luschin-Ebengreuth, G.; Postlberger, S.; Menzel, C.; Jakesz, R.; Seifert, M.; Hubalek, M.; Bjelic-Radisic, V.; et al. Endocrine therapy plus zoledronic acid in premenopausal breast cancer. N. Engl. J. Med. 2009, 360, 679–691. [Google Scholar] [CrossRef]
- Gnant, M.; Mlineritsch, B.; Stoeger, H.; Luschin-Ebengreuth, G.; Knauer, M.; Moik, M.; Jakesz, R.; Seifert, M.; Taucher, S.; Bjelic-Radisic, V.; et al. Zoledronic acid combined with adjuvant endocrine therapy of tamoxifen vs. anastrozol plus ovarian function suppression in premenopausal early breast cancer: Final analysis of the austrian breast and colorectal cancer study group trial 12. Ann. Oncol. 2015, 26, 313–320. [Google Scholar] [CrossRef]
- Gralow, J.; Barlow, W.E.; Paterson, A.; Lew, D.; Stopeck, A.; Hayes, D.F.; Hershman, D.L.; Schubert, M.; Clemons, M.J.; van Poznak, C.H.; et al. Phase III trial of bisphosphonates as adjuvant therapy in primary breast cancer: SWOG/Alliance/ECOG-ACRIN/NCIC clinical trials group/NRG oncology study S0307. J. Clin. Oncol. 2015, 33, 503. [Google Scholar] [CrossRef]
- Gnant, M.; Pfeiler, G.; Dubsky, P.C.; Hubalek, M.; Greil, R.; Jakesz, R.; Wette, V.; Balic, M.; Haslbauer, F.; Melbinger, E.; et al. Adjuvant denosumab in breast cancer (ABCSG-18): A multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2015, 386, 433–443. [Google Scholar] [CrossRef]
- Gnant, M.; Pfeiler, G.; Dubsky, P.; Hubalek, M.; Greil, R.; Jakesz, R.; Wette, V.; Balic, M.; Haslbauer, F.; Melbinger-Zeinitzer, E.; et al. The impact of adjuvant denosumab on disease-free survival: Results from 3425 postmenopausal patients of the ABCSG-18 trial. Cancer Res. 2016, 76, S2-02. [Google Scholar] [CrossRef]
- Wirth, M.; Tammela, T.; Cicalese, V.; Gomez Veiga, F.; Delaere, K.; Miller, K.; Tubaro, A.; Schulze, M.; Debruyne, F.; Huland, H.; et al. Prevention of bone metastases in patients with high-risk nonmetastatic prostate cancer treated with zoledronic acid: Efficacy and safety results of the zometa european study (ZEUS). Eur. Urol. 2015, 67, 482–491. [Google Scholar] [CrossRef]
- Denham, J.W.; Joseph, D.; Lamb, D.S.; Spry, N.A.; Duchesne, G.; Matthews, J.; Atkinson, C.; Tai, K.H.; Christie, D.; Kenny, L.; et al. Short-term androgen suppression and radiotherapy vs. intermediate-term androgen suppression and radiotherapy, with or without zoledronic acid, in men with locally advanced prostate cancer (TROG 03.04 RADAR): An open-label, randomised, phase 3 factorial trial. Lancet Oncol. 2014, 15, 1076–1089. [Google Scholar]
- Hiraga, T. Targeted agents in preclinical and early clinical development for the treatment of cancer bone metastases. Expert Opin. Investig. Drugs 2016, 25, 319–334. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Agent and Dose | Drug Class and Target | EMA Label | FDA Label | Time to First SRE (if Not Otherwise Specified) | Overall Survival | Refs. |
|---|---|---|---|---|---|---|
| Denosumab; 120 mg, SC, every 4 weeks | Fully human monoclonal antibody; Anti-RANKL | Label; Prevention of SREs (pathological fracture, radiation to bone, spinal cord compression or surgery to bone) in adults with bone metastases from solid tumors. | Label; Prevention of skeletal-related events in patients with bone metastases from solid tumors; excludes bone metastases from MM. | Breast (vs. ZA): NR vs. 26.4 months; HR 0.82 (95% CI, 0.71 to 0.95), p = 0.01; | Overall (vs. ZA): No benefit. | [8,73,74,75] |
| Prostate (vs. ZA): 20.7 vs. 17.1 months; HR 0.82 (95% CI 0.71–0.95), p < 0.001; | ||||||
| Other solid tumors and MM (vs. ZA): 20.6 vs. 16.3 months; HR 0.84 (95% CI 0.71–0.98), p < 0.001; | ||||||
| Overall (vs. ZA): 27.7 vs. 19.5 months; HR 0.83 (95% CI 0.76–0.90), p < 0.001. | ||||||
| ZA; 4 mg, IV, every 3 to 4 weeks | Amino-bisphosphonat; farnesyl diphosphate synthase inhibitor | Prevention of skeletal-related events (pathological fractures, spinal compression, radiation or surgery to bone, or tumor-induced hypercalcemia) in adult patients with advanced malignancies involving bone. | Patients with MM and patients with documented bone metastases from solid tumors, in conjunction with standard antineoplastic therapy. Prostate cancer should have progressed after treatment with at least one hormonal therapy. | Breast (vs. placebo): NR vs. 360 days; HR 0.56 (95% CI 0.36–0.87); p = 0.009 | Overall (any BP vs. control): HR 1.01 (95% CI 0.92–1.11); p = 0.87; No benefit. | [68,76,77,78] |
| Prostate (vs. placebo): 488 vs. 321 days; HR 0.68 (95% CI 0.51–0.91); p = 0.009 | ||||||
| Lung and other solid tumors (vs. placebo): 230 vs. 155 days; HR 0.70 (95% CI NR); p = 0.006 | ||||||
| Ibandronic acid; 50 mg, PO, daily | Amino-bisphosphonat; farnesyl diphosphate synthase inhibitor | Prevention of skeletal events (pathological fractures, bone complications requiring radiotherapy or surgery) in patients with breast cancer and bone metastases. | Off-label | Breast (vs. ZA): annual rate ratio for SREs 1.148 (95% CI 0.967–1.362); did not demonstrate non-inferiority to ZA. | Overall (any BP vs. control): HR 1.01 (95% CI 0.92–1.11); p = 0.87; No benefit. | [79] |
| Ra-223; 50 kBq per kilogram, every 4 weeks for 6 administrations | Alpha-emitter; DNA damage | Treatment of adults with castration-resistant prostate cancer, symptomatic bone metastases and no known visceral metastases. | Treatment of patients with castration-resistant prostate cancer, symptomatic bone metastases and no known visceral metastatic disease | Prostate (vs. placebo): 15.6 months vs. 9.8 months; 0.66 (95% CI, 0.52–0.83); p < 0.001 | Prostate (vs. placebo): 14.0 vs. 11.2 months; HR 0.70 (95% CI 0.58–0.83); p < 0.001 | [80] |
| Agent | Group of Patients; Number of pts | EMA/FDA Label | Bone Recurrence | Disease Recurrence | Cancer Mortality | Refs. |
|---|---|---|---|---|---|---|
| Breast cancer | ||||||
| Bisphophonates | Overall; n = 18,766 | Off-label | HR 0.83 (95% CI 0.73–0.94); p = 0.004 | HR 0.94 (95% CI 0.87–1.01); p = 0.08 | HR 0.91 (95% CI 0.83–0.99); p = 0.04 | [9] |
| Postmenopausal; n = 7388 | HR 0.72 (95% CI 0.60–0.86); p = 0.0002 | HR 0.86 (95% CI 0.78–0.94); p = 0.002 | HR 0.82 (95% CI 0.73–0.93); p = 0.002 | |||
| Denosumab | Postmenopausal; n = 3425 | Off-label | - | (any recurrence or death) HR 0.82 (95% CI 0.66–1.00); p = 0.0515 | - | [105,106] |
| Prostate cancer | ||||||
| Zoledronic acid | High risk disease; 1393 | Off-label | 14.7% vs. 13.2% in the control group; HR 1.075 (95% CI 0.81–1.44); p = 0.62 | - | 116 vs. 122 deaths in the control group; log-rank p = 0.76 | [107] |
| Locally advanced disease treated with RT and ADT ± ZA | See text. | [108] | ||||
| Target | Agent | NCT | Combination Therapy/Comparator | Tumor Type | Bone-Specific Endpoints | Trial Status |
|---|---|---|---|---|---|---|
| Cathepesin K | Odanacatib | NCT00691899 | None/Placebo | Prostate | Bone metastasis-free survival | Withdrawn |
| NCT00692458 | None/Placebo | Breast | Development of bone metastasis | Withdrawn | ||
| c-Src | Dasatinib | NCT00744497 | Docetaxel, prednisone/Placebo | Prostate | Time to first SRE; reduction of NTX from baseline | Completed |
| Endothelines | Atrasentan | NCT00036543 | None/Placebo | Prostate | None | Completed |
| NCT00134056 | Docetaxel, prednisone/placebo | Prostate | None | Ongoing | ||
| Zibotentan | NCT00554229 | None/Placebo | Prostate | Incidence of SRE; New bone metastases | Completed | |
| mTOR | Everolimus | NCT00863655 | Exemestane/Placebo | Breast | None | Completed |
| MET/VEGFR | Cabozantinib | NCT01605227 | None/Prednisone | Prostate | Bone scan response | Completed |
| NCT01522443 | None/Mitoxantrone, prednisone | Prostate | Bone scan response | Terminated |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casimiro, S.; Ferreira, A.R.; Mansinho, A.; Alho, I.; Costa, L. Molecular Mechanisms of Bone Metastasis: Which Targets Came from the Bench to the Bedside? Int. J. Mol. Sci. 2016, 17, 1415. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091415
Casimiro S, Ferreira AR, Mansinho A, Alho I, Costa L. Molecular Mechanisms of Bone Metastasis: Which Targets Came from the Bench to the Bedside? International Journal of Molecular Sciences. 2016; 17(9):1415. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091415
Chicago/Turabian StyleCasimiro, Sandra, Arlindo R. Ferreira, André Mansinho, Irina Alho, and Luis Costa. 2016. "Molecular Mechanisms of Bone Metastasis: Which Targets Came from the Bench to the Bedside?" International Journal of Molecular Sciences 17, no. 9: 1415. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091415