
T-Tubular Electrical Defects Contribute to Blunted β-Adrenergic Response in Heart Failure

 ,
,
Abstract
: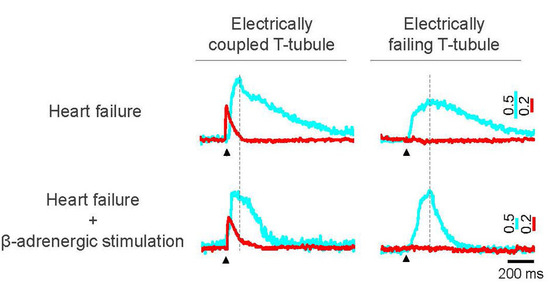
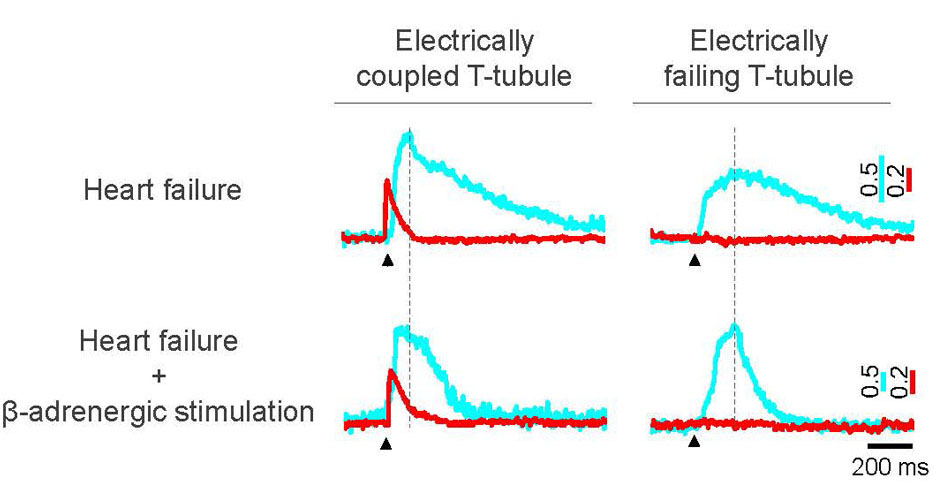{kind=link}
{kind=link}
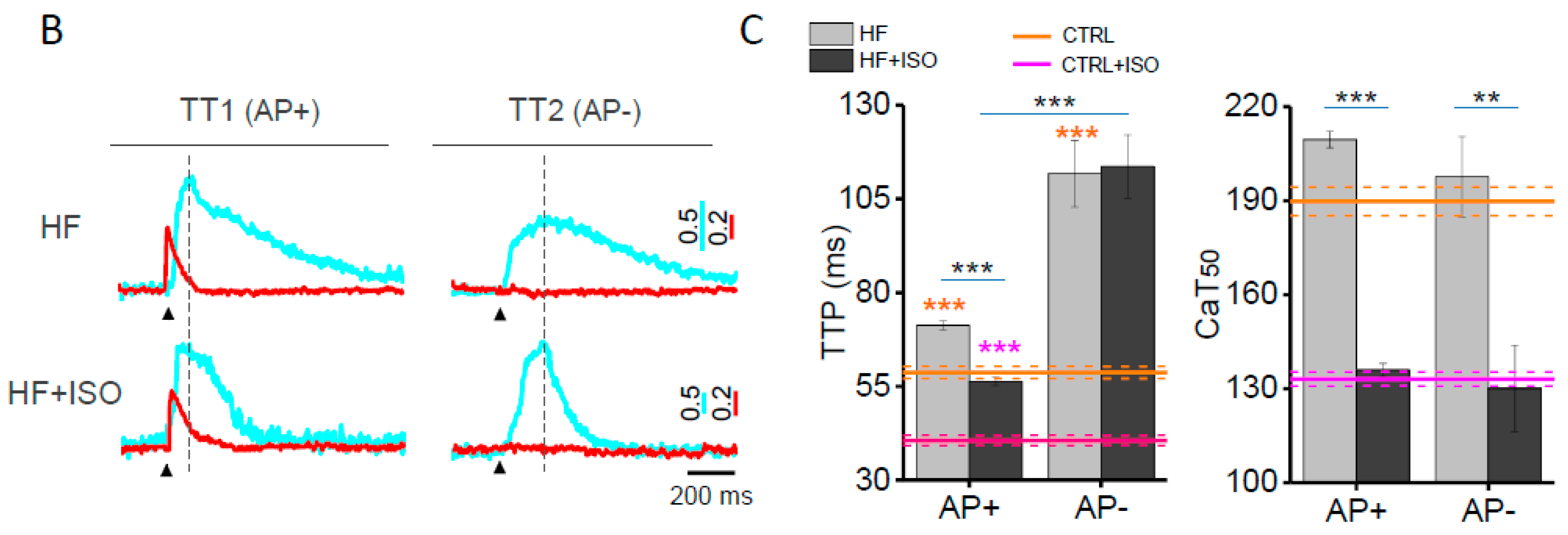{kind=link}
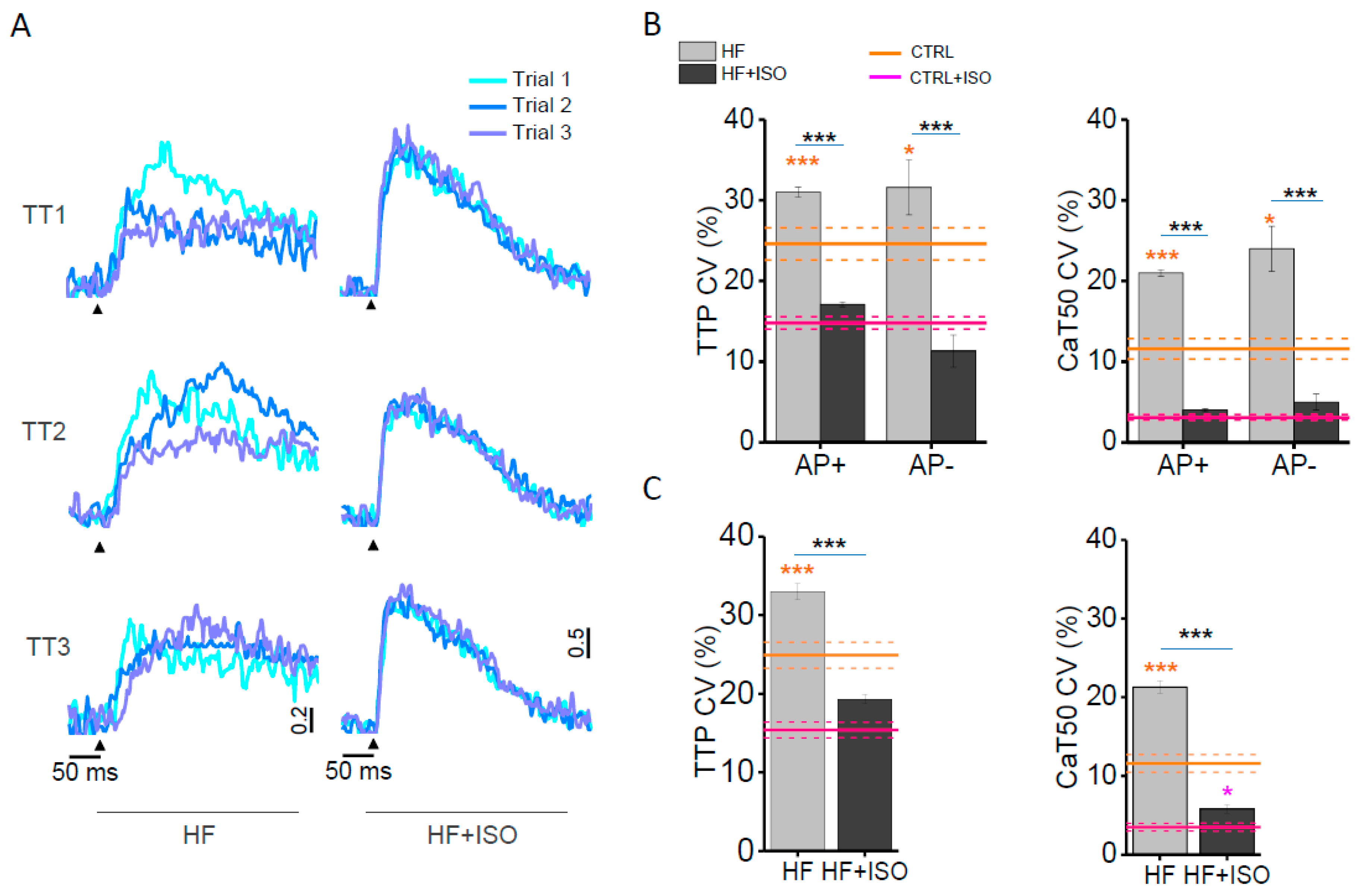{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Action Potentials and Ca2+ Transients in Failing Cells Treated with Isoproterenol
2.2. Spatio-Temporal Variability of Ca2+ Transients in Isoproterenol-Treated Failing Cardiomyocytes
2.3. Isoproterenol Effect on Ca2+ Sparks Frequency in Heart Failure
3. Discussion
4. Materials and Methods
4.1. Cardiomyocyte Preparations and Labelling
4.2. RAMP Microscope and Optical Recording
4.3. Data Analysis and Statistics
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gerhardstein, B.L.; Puri, T.S.; Chien, A.J.; Hosey, M.M. Identification of the sites phosphorylated by cyclic AMP-dependent protein kinase on the β2 subunit of L-type voltage-dependent calcium channels. Biochemistry 1999, 38, 10361–10370. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosemblit, N.; Marks, A.R. Pka phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell 2000, 101, 365–376. [Google Scholar] [CrossRef]
- Simmerman, H.K.; Jones, L.R. Phospholamban: Protein structure, mechanism of action, and role in cardiac function. Physiol. Rev. 1998, 78, 921–947. [Google Scholar] [PubMed]
- Sulakhe, P.V.; Vo, X.T. Regulation of phospholamban and troponin-I phosphorylation in the intact rat cardiomyocytes by adrenergic and cholinergic stimuli: Roles of cyclic nucleotides, calcium, protein kinases and phosphatases and depolarization. Mol. Cell. Biochem. 1995, 15, 103–126. [Google Scholar] [CrossRef]
- Kunst, G.; Kress, K.R.; Gruen, M.; Uttenweiler, D.; Gautel, M.; Fink, R.H. Myosin binding protein C, a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circ. Res. 2000, 86, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Jones, D.; Adams, R.J.; Brown, T.M.; Carnethon, M.; Dai, S.; de Simone, G.; Ferguson, T.B.; Ford, E.; Furie, K.; Gillespie, C.; et al. Executive summary: Heart disease and stroke statistics—2010 update: A report from the american heart association. Circulation 2010, 121, 948–954. [Google Scholar] [PubMed]
- Novak, P.; Li, C.; Shevchuk, A.I.; Stepanyan, R.; Caldwell, M.; Hughes, S.; Smart, T.G.; Gorelik, J.; Ostanin, V.P.; Lab, M.J.; et al. Nanoscale live-cell imaging using hopping probe ion conductance microscopy. Nat. Methods 2009, 6, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Shenasa, H.; Calderone, A.; Vermeulen, M.; Paradis, P.; Stephens, H.; Cardinal, R.; de Champlain, J.; Rouleau, J.L. Chronic doxorubicin induced cardiomyopathy in rabbits: Mechanical, intracellular action potential, and beta adrenergic characteristics of the failing myocardium. Cardiovasc. Res. 1990, 24, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.; Lauterbach, M.A.; Kohl, T.; Westphal, V.; Williams, G.S.; Steinbrecher, J.H.; Streich, J.H.; Korff, B.; Tuan, H.T.; Hagen, B.; et al. Stimulated emission depletion live-cell super-resolution imaging shows proliferative remodeling of T-tubule membrane structures after myocardial infarction. Circ. Res. 2012, 111, 402–414. [Google Scholar] [CrossRef] [PubMed]
- CIBIS-II Investigators and Committees. The cardiac insufficiency bisoprolol study II (CIBIS-II): A randomised trial. Lancet 1999, 353, 9–13. [Google Scholar]
- Bristow, M.R.; Ginsburg, R.; Umans, V.; Fowler, M.; Minobe, W.; Rasmussen, R.; Zera, P.; Menlove, R.; Shah, P.; Jamieson, S.; et al. Beta 1- and beta 2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: Coupling of both receptor subtypes to muscle contraction and selective beta 1-receptor down-regulation in heart failure. Circ. Res. 1986, 59, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Bristow, M.R.; Minobe, W.A.; Raynolds, M.V.; Port, J.D.; Rasmussen, R.; Ray, P.E.; Feldman, A.M. Reduced beta 1 receptor messenger RNA abundance in the failing human heart. J. Clin. Investig. 1993, 92, 2737–2745. [Google Scholar] [CrossRef] [PubMed]
- Brodde, O.E. Beta-adrenoceptors in cardiac disease. Pharmacol. Ther. 1993, 60, 405–430. [Google Scholar] [CrossRef]
- Swynghedauw, B. Molecular mechanisms of myocardial remodeling. Physiol. Rev. 1999, 79, 215–262. [Google Scholar] [PubMed]
- Ferrantini, C.; Crocini, C.; Coppini, R.; Vanzi, F.; Tesi, C.; Cerbai, E.; Poggesi, C.; Pavone, F.S.; Sacconi, L. The transverse-axial tubular system of cardiomyocytes. Cell. Mol. Life Sci. CMLS 2013, 70, 4695–4710. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. Beta2-adrenergic receptor redistribution in heart failure changes camp compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Cannell, M.B.; Crossman, D.J.; Soeller, C. Effect of changes in action potential spike configuration, junctional sarcoplasmic reticulum micro-architecture and altered T-tubule structure in human heart failure. J. Muscle Res. Cell Motil. 2006, 27, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Lyon, A.R.; MacLeod, K.T.; Zhang, Y.; Garcia, E.; Kanda, G.K.; Lab, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc. Natl. Acad. Sci. USA 2009, 106, 6854–6859. [Google Scholar] [CrossRef] [PubMed]
- Sacconi, L.; Ferrantini, C.; Lotti, J.; Coppini, R.; Yan, P.; Loew, L.M.; Tesi, C.; Cerbai, E.; Poggesi, C.; Pavone, F.S. Action potential propagation in transverse-axial tubular system is impaired in heart failure. Proc. Natl. Acad. Sci. USA 2012, 109, 5815–5819. [Google Scholar] [CrossRef] [PubMed]
- Crocini, C.; Coppini, R.; Ferrantini, C.; Yan, P.; Loew, L.M.; Tesi, C.; Cerbai, E.; Poggesi, C.; Pavone, F.S.; Sacconi, L. Defects in T-tubular electrical activity underlie local alterations of calcium release in heart failure. Proc. Natl. Acad. Sci. USA 2014, 111, 15196–15201. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Zhao, Y.T.; Guo, Y.B.; Xu, S.M.; Bai, S.H.; Lakatta, E.G.; Cheng, H.; Hao, X.M.; Wang, S.Q. β-adrenergic signaling accelerates and synchronizes cardiac ryanodine receptor response to a single L-type Ca2+ channel. Proc. Natl. Acad. Sci. USA 2009, 106, 18028–18033. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Lederer, W.J.; Cannell, M.B. Calcium sparks: Elementary events underlying excitation-contraction coupling in heart muscle. Science 1993, 262, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Franzini-Armstrong, C.; Protasi, F.; Ramesh, V. Shape, size, and distribution of Ca(2+) release units and couplons in skeletal and cardiac muscles. Biophys. J. 1999, 77, 1528–1539. [Google Scholar] [CrossRef]
- Soeller, C.; Crossman, D.; Gilbert, R.; Cannell, M.B. Analysis of ryanodine receptor clusters in rat and human cardiac myocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 14958–14963. [Google Scholar] [CrossRef] [PubMed]
- Lindegger, N.; Niggli, E. Paradoxical SR Ca2+ release in guinea-pig cardiac myocytes after beta-adrenergic stimulation revealed by two-photon photolysis of caged Ca2+. J. Physiol. 2005, 565, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Yano, M.; Ohkusa, T.; Kohno, M.; Hisaoka, T.; Tanigawa, T.; Kobayashi, S.; Kohno, M.; Matsuzaki, M. Altered interaction of FKBP12.6 with ryanodine receptor as a cause of abnormal Ca(2+) release in heart failure. Cardiovasc. Res. 2000, 48, 323–331. [Google Scholar] [CrossRef]
- Gomez, A.M.; Valdivia, H.H.; Cheng, H.; Lederer, M.R.; Santana, L.F.; Cannell, M.B.; McCune, S.A.; Altschuld, R.A.; Lederer, W.J. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science 1997, 276, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Shannon, T.R.; Pogwizd, S.M.; Bers, D.M. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ. Res. 2003, 93, 592–594. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac sarcoplasmic reticulum calcium leak: Basis and roles in cardiac dysfunction. Annu. Rev. Physiol. 2014, 76, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Meissner, G. Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annu. Rev. Physiol. 1994, 56, 485–508. [Google Scholar] [CrossRef] [PubMed]
- Curran, J.; Brown, K.H.; Santiago, D.J.; Pogwizd, S.; Bers, D.M.; Shannon, T.R. Spontaneous ca waves in ventricular myocytes from failing hearts depend on Ca(2+)-calmodulin-dependent protein kinase II. J. Mol. Cell. Cardiol. 2010, 49, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Ai, X.; Curran, J.W.; Shannon, T.R.; Bers, D.M.; Pogwizd, S.M. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 2005, 97, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Terentyev, D.; Gyorke, I.; Belevych, A.E.; Terentyeva, R.; Sridhar, A.; Nishijima, Y.; de Blanco, E.C.; Khanna, S.; Sen, C.K.; Cardounel, A.J.; et al. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ. Res. 2008, 103, 1466–1472. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kranias, E.G.; Mignery, G.A.; Bers, D.M. Protein kinase a phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ. Res. 2002, 90, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Louch, W.E.; Bito, V.; Heinzel, F.R.; Macianskiene, R.; Vanhaecke, J.; Flameng, W.; Mubagwa, K.; Sipido, K.R. Reduced synchrony of Ca2+ release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc. Res. 2004, 62, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Louch, W.E.; Mork, H.K.; Sexton, J.; Stromme, T.A.; Laake, P.; Sjaastad, I.; Sejersted, O.M. T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J. Physiol. 2006, 574, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Frisk, M.; Ruud, M.; Espe, E.K.; Aronsen, J.M.; Roe, A.T.; Zhang, L.; Norseng, P.A.; Sejersted, O.M.; Christensen, G.A.; Sjaastad, I.; et al. Elevated ventricular wall stress disrupts cardiomyocyte t-tubule structure and calcium homeostasis. Cardiovasc. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Crocini, C.; Coppini, R.; Ferrantini, C.; Pavone, F.S.; Sacconi, L. Functional cardiac imaging by random access microscopy. Front. Physiol. 2014, 5, 403. [Google Scholar] [CrossRef] [PubMed]
- Crocini, C.; Ferrantini, C.; Scardigli, M.; Coppini, R.; Mazzoni, L.; Lazzeri, E.; Pioner, J.M.; Scellini, B.; Guo, A.; Song, L.S.; et al. Novel insights on the relationship between T-tubular defects and contractile dysfunction in a mouse model of hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2015, 91, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Piacentino, V., 3rd; Furukawa, S.; Goldman, B.; Margulies, K.B.; Houser, S.R. L-type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ. Res. 2002, 91, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Shaw, S.A.; Naami, R.; Vuong, C.L.; Basheer, W.A.; Guo, X.; Hong, T. Isoproterenol promotes rapid ryanodine receptor movement to bridging integrator 1 (BIN1)-organized dyads. Circulation 2016, 133, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Farman, G.P.; Tachampa, K.; Mateja, R.; Cazorla, O.; Lacampagne, A.; de Tombe, P.P. Blebbistatin: Use as inhibitor of muscle contraction. Pflug. Arch. Eur. J. Physiol. 2008, 455, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Howarth, F.C.; Boyett, M.R.; White, E. Rapid effects of cytochalasin-D on contraction and intracellular calcium in single rat ventricular myocytes. Pflug. Arch. Eur. J. Physiol. 1998, 436, 804–806. [Google Scholar] [CrossRef] [PubMed]
- Zipfel, W.R.; Williams, R.M.; Webb, W.W. Nonlinear magic: Multiphoton microscopy in the biosciences. Nat. Biotechnol. 2003, 21, 1369–1377. [Google Scholar] [CrossRef] [PubMed]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crocini, C.; Coppini, R.; Ferrantini, C.; Yan, P.; Loew, L.M.; Poggesi, C.; Cerbai, E.; Pavone, F.S.; Sacconi, L. T-Tubular Electrical Defects Contribute to Blunted β-Adrenergic Response in Heart Failure. Int. J. Mol. Sci. 2016, 17, 1471. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091471
Crocini C, Coppini R, Ferrantini C, Yan P, Loew LM, Poggesi C, Cerbai E, Pavone FS, Sacconi L. T-Tubular Electrical Defects Contribute to Blunted β-Adrenergic Response in Heart Failure. International Journal of Molecular Sciences. 2016; 17(9):1471. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091471
Chicago/Turabian StyleCrocini, Claudia, Raffaele Coppini, Cecilia Ferrantini, Ping Yan, Leslie M. Loew, Corrado Poggesi, Elisabetta Cerbai, Francesco S. Pavone, and Leonardo Sacconi. 2016. "T-Tubular Electrical Defects Contribute to Blunted β-Adrenergic Response in Heart Failure" International Journal of Molecular Sciences 17, no. 9: 1471. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091471