
Virtual Screening Approaches towards the Discovery of Toll-Like Receptor Modulators


Abstract
:
1. Introduction
2. Databases
2.1. ZINC Database
2.2. NCI Open Database
2.3. ASINEX Database
2.4. SPECS Database
2.5. MAYBRIDGE Database
2.6. LIFE CHEMICALS Database
2.7. ENAMINE Database
2.8. CHEMBRIDGE Database
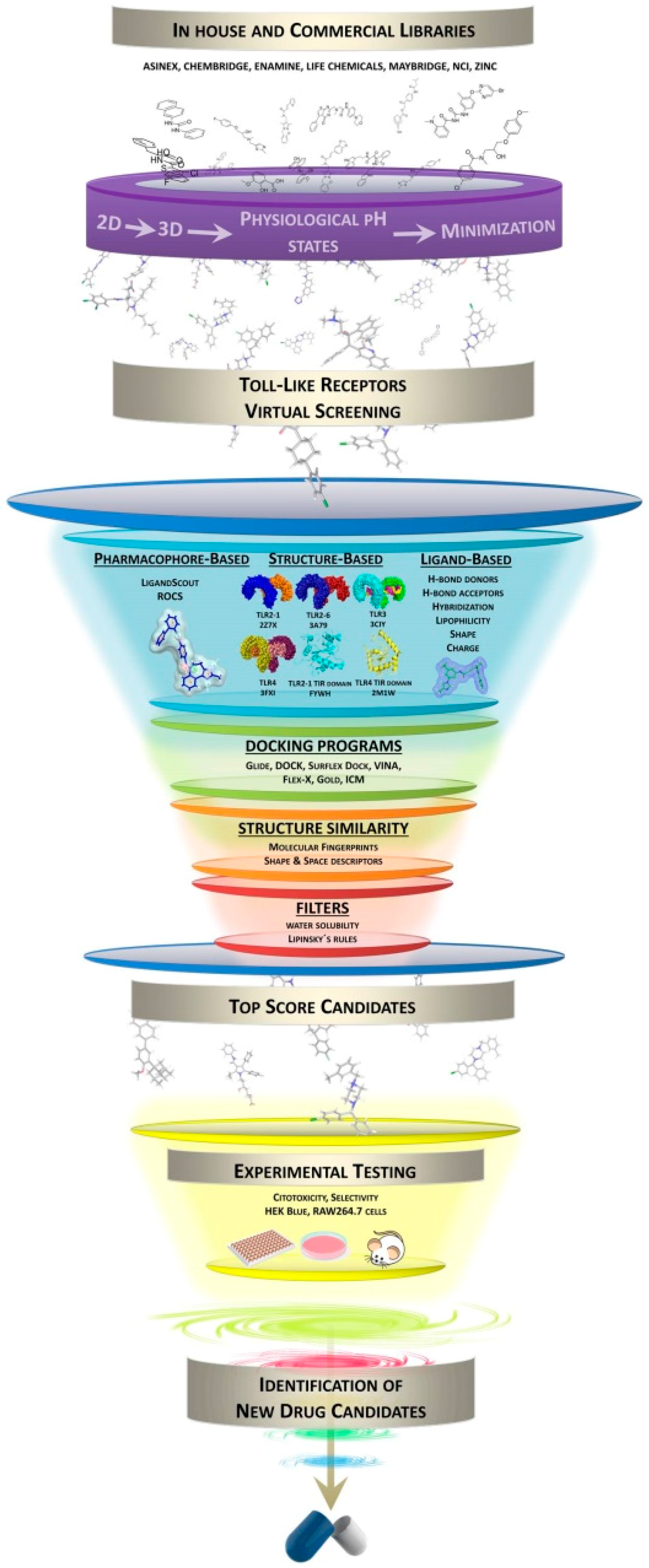
3. Virtual Screening Protocols and Techniques
3.1. Database Processing and Inclusion of Decoys
3.1.1. LigPrep
3.1.2. OMEGA
3.1.3. AutoDockTools
3.1.4. MUBD-Decoymaker
3.2. Pharmacophore Generation
3.2.1. LigandScout
3.2.2. Rapid Overlay of Chemical Structures (ROCS)
3.3. Docking Tools for Virtual Screening (VS)
3.3.1. Glide
3.3.2. AutoDock VINA
3.3.3. GOLD
3.3.4. Surflex-Dock
3.3.5. FlexX
3.3.6. ICM
3.3.7. DOCK
4. Virtual Screening in Toll-Like Receptors: TLR2, TLR3, TLR4, TLR7 and TLR8
4.1. Virtual Screening Studies in TLR2
4.2. Virtual Screening Studies in TLR3
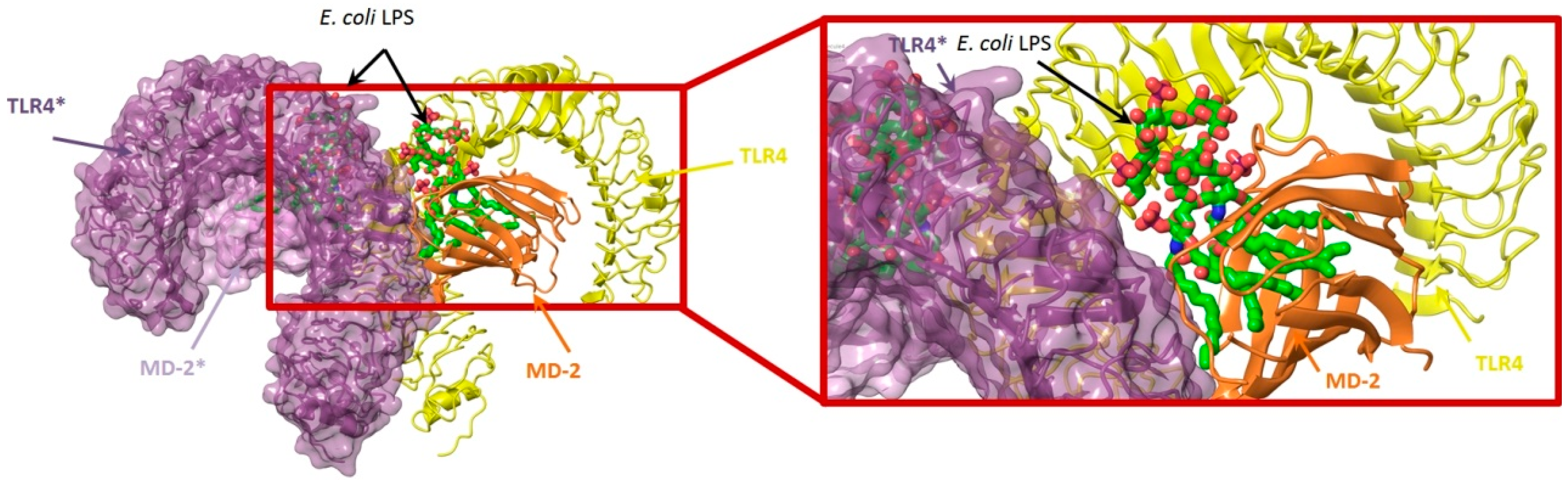
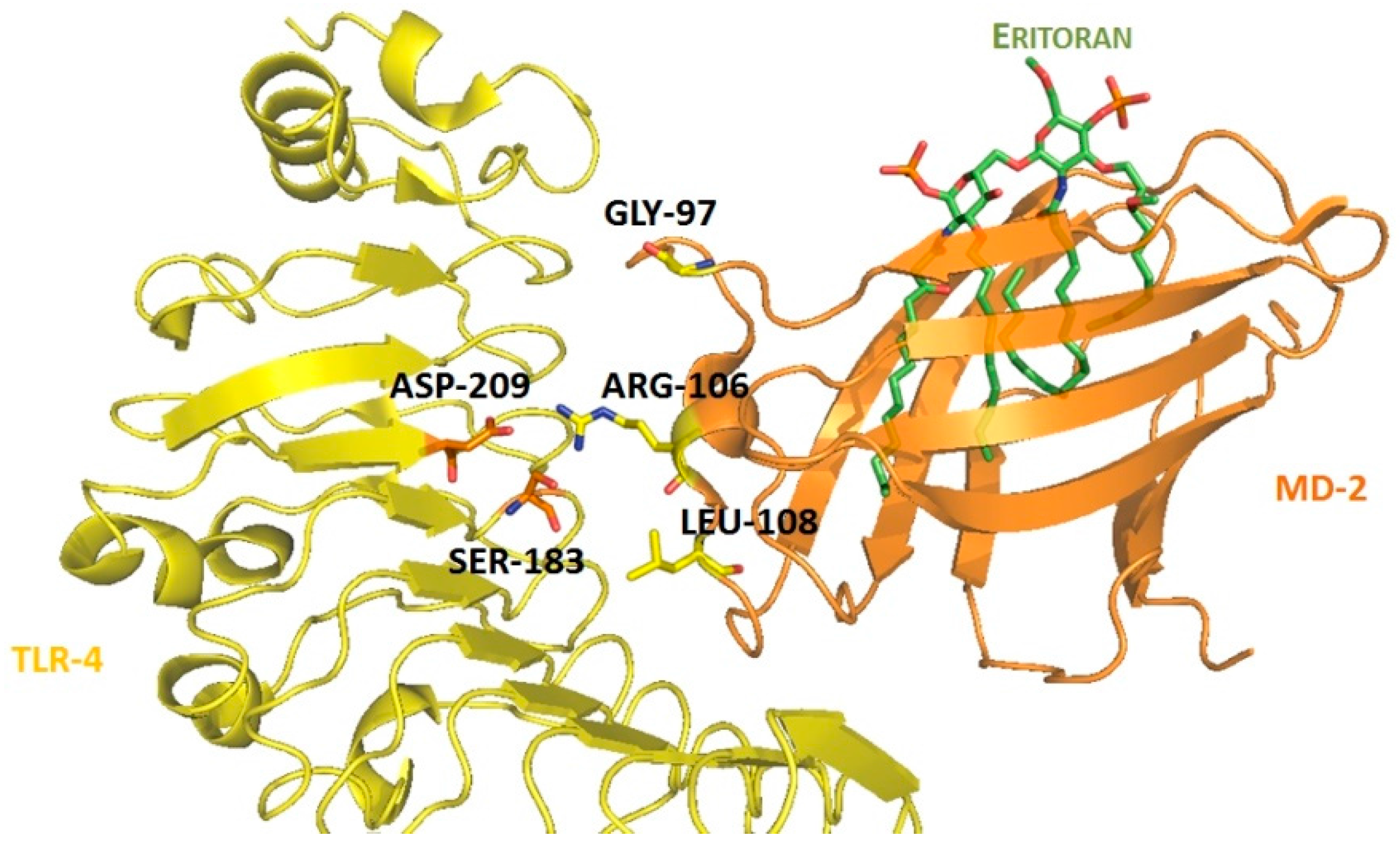
4.3. Virtual Screening Studies in TLR4
4.4. Virtual Screening Studies in TLR7
4.5. Virtual Screening Studies in TLR8
5. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D | Two-Dimensional |
| 3D | Three-Dimensional |
| 3D-QSAR | Three-Dimensional Quantitative Structure-Activity Relationships |
| ADMET | Absorption-Distribution-Metabolism-Excretion-Toxicity |
| AIDS | Acquired Immune Deficiency Syndrome |
| Akt1 | Serine-Threonine Protein Kinase 1 |
| BPMC | Biased Probability Monte Carlo |
| cLogP | Calculated logP |
| CNS | Central Nervous System |
| dsRNA | Double-Stranded RNA |
| ELISA | Enzyme-Linked Immuno-Sorbent Assay |
| EP | Extra Precision |
| FDA | Food and Drug Administration |
| GB/SA | Generalized Born/Surface Area |
| GPCR | G Protein–Coupled Receptor |
| HEK | Human Embryonic Kidney cells |
| hTLR | Human Toll-Like Receptor |
| H-bond | Hydrogen bond |
| HTS | High Throughput Screening |
| HTVS | High Throughput Virtual Screening |
| KB | Knowledge-based |
| KBP | Knowledge-based Pharmacophore |
| ICM | Internal Coordinate Mechanics |
| IL | Interleukin |
| LPS | Lipopolysaccharide |
| LRR | Leucine-Rich Repeat |
| MD Simulations | Molecular Dynamic Simulations |
| MD-2 | Myeloid Differentiation protein 2 |
| MIF | Molecular Interaction Field |
| MM | Molecular Mechanics |
| mRNA | Messenger RNA |
| MW | Molecular Weight |
| MyD88 | Myeloid Differentiation primary response gene 88 |
| NCI | National Cancer Institute |
| NF-κB-luciferase | Nuclear Factor-κB luciferase |
| NIH | National Institutes of Health |
| NMR | Nuclear Magnetic Resonance |
| NO | Nitric oxide |
| PAMP | Pathogen-Associated Molecular Pattern |
| PB/SA | Poisson-Boltzmann/Surface Area |
| PDB-ID | Protein Data Bank Identification code |
| PMF | Potential of Mean Force |
| Poly(I:C) | Polyinosinic-polycytidylic acid |
| PPI | Protein-Protein Interaction |
| pZERO-TLR1 | cloning plasmid for the expression of TLR1 TIR-deleted genes |
| RAW264.7 | Cell Line murine macrophage from blood |
| RMSD | Root-Mean-Square Deviation |
| RNA | RiboNucleic Acid |
| rotB | Rotatable Bonds |
| Ro3 | Rule of Three |
| SAR | Structure-Activity Relationship |
| SDF format | Structure Data File format |
| siRNA | Small Interfering RNA |
| SP | Standard Precision |
| SPR | Surface Plasmon Resonance |
| ssRNA | Single-Stranded RNA |
| THP-1 | Monocyte from human blood |
| TIR | Toll/Interleukin-1 Receptor |
| TIRAP | Toll-Interleukin 1 Receptor domain containing Adaptor Protein |
| TLR | Toll-Like Receptor |
| TNF-α | Tumor Necrosis Factor α |
| TRAM | Translocating Chain–Associated Membrane |
| UCSF | University of California-San Francisco |
| VIPER | Viral Inhibitory Peptide of TLR4 |
| VS | Virtual Screening |
| XP | Extra-precision |
References
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Botos, I.; Segal, D.M.; Davies, D.R. The structural biology of Toll-like receptors. Structure 2011, 19, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Mifsud, E.J.; Tan, A.C.; Jackson, D.C. TLR agonists as modulators of the innate immune response and their potential as agents against infectious disease. Front. Immunol. 2014, 5, 79. [Google Scholar] [CrossRef] [PubMed]
- Joosten, L.A.B.; Abdollahi-Roodsaz, S.; Dinarello, C.A.; O’Neill, L.; Netea, M.G. Toll-like receptors and chronic inflammation in rheumatic diseases: New developments. Nat. Rev. Rheumatol. 2016, 12, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Gooshe, M.; Aleyasin, A.R.; Abdolghaffari, A.H.; Rezaei, N. Toll like receptors: A new hope on the horizon to treat multiple sclerosis. Expert Rev. Clin. Immunol. 2014, 10, 1277–1279. [Google Scholar] [CrossRef] [PubMed]
- Gambuzza, M.E.; Sofo, V.; Salmeri, F.M.; Soraci, L.; Marino, S.; Bramanti, P. Toll-like receptors in Alzheimer’s disease: A therapeutic perspective. CNS Neurol. Disord. Drug Targets 2014, 13, 1542–1558. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.H.; Diven, M.A.; Huff, L.W.; Paulos, C.M. Harnessing the microbiome to enhance cancer immunotherapy. J. Immunol. Res. 2015, 2015, 368736. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S.; Medzhitov, R. Toll-like receptors and cancer. Nat. Rev. Cancer 2009, 9, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Lyne, P.D. Structure-based virtual screening: An overview. Drug Discov. Today 2002, 7, 1047–1055. [Google Scholar] [CrossRef]
- Schneider, G. Virtual screening: An endless staircase? Nat. Rev. Drug Discov. 2010, 9, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Haga, J.H.; Ichikawa, K.; Date, S. Virtual screening techniques and current computational infrastructures. Curr. Pharm. Des. 2016, 22, 3576–3584. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Cao, S.; Su, P.C.; Patel, R.; Shah, D.; Chokshi, H.B.; Szukala, R.; Johnson, M.E.; Hevener, K.E. Hit identification and optimization in virtual screening: Practical recommendations based on a critical literature analysis. J. Med. Chem. 2013, 56, 6560–6572. [Google Scholar] [CrossRef] [PubMed]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Liao, C.; Liu, Z.; Hagler, A.T.; Gu, Q.; Xu, J. Chemical structure similarity search for ligand-based virtual screening: Methods and computational resources. Curr. Drug Targets 2015, 16. Available online: http://www.eurekaselect.com/136355/article (accessed on 29 August 2016). [Google Scholar] [CrossRef]
- Sheng, C.; Dong, G.; Miao, Z.; Zhang, W.; Wang, W. State-of-the-art strategies for targeting protein-protein interactions by small-molecule inhibitors. Chem. Soc. Rev. 2015, 44, 8238–8259. [Google Scholar] [CrossRef] [PubMed]
- Krier, M.; Bret, G.; Rognan, D. Assessing the scaffold diversity of screening libraries. J. Chem. Inf. Model. 2006, 46, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Voigt, J.H.; Bienfait, B.; Wang, S.; Nicklaus, M.C. Comparison of the NCI open database with seven large chemical structural databases. J. Chem. Inf. Comput. Sci. 2001, 41, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Huggins, D.J.; Venkitaraman, A.R.; Spring, D.R. Rational methods for the selection of diverse screening compounds. ACS Chem. Biol. 2011, 6, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Monge, A.; Arrault, A.; Marot, C.; Morin-Allory, L. Managing, profiling and analyzing a library of 2.6 million compounds gathered from 32 chemical providers. Mol. Divers. 2006, 10, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Le Guilloux, V.; Colliandre, L.; Bourg, S.; Guenegou, G.; Dubois-Chevalier, J.; Morin-Allory, L. Visual characterization and diversity quantification of chemical libraries: 1. Creation of delimited reference chemical subspaces. J. Chem. Inf. Model. 2011, 51, 1762–1774. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- ZINC 2016. Available online: http://zinc15.docking.org (accessed on 29 August 2016).
- Irwin, J.J.; Shoichet, B.K. Zinc—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. Zinc: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. Zinc 15—Ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, T.T. IBM Internal Report, 17th November; IBM Company: Armonk, NY, USA, 1957. [Google Scholar]
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Development Therapeutics Program. Available online: https://dtp.cancer.gov/ (accessed on 29 August 2016).
- Downloadable Structure Files of NCI Open Database Compounds. Available online: https://cactus.nci.nih.gov/download/nci/ (accessed on 29 August 2016).
- Asinex. Available online: http://www.asinex.com (accessed on 29 August 2016).
- Guan, Y.; Omueti-Ayoade, K.; Mutha, S.K.; Hergenrother, P.J.; Tapping, R.I. Identification of novel synthetic Toll-like receptor 2 agonists by high throughput screening. J. Biol. Chem. 2010, 285, 23755–23762. [Google Scholar] [CrossRef] [PubMed]
- Specs. Available online: http://www.specs.net (accessed on 29 August 2016).
- Maybridge. Available online: http://www.maybridge.com (accessed on 29 August 2016).
- Butina, D. Unsupervised data base clustering based on daylight’s fingerprint and tanimoto similarity: A fast and automated way to cluster small and large data sets. J. Chem. Inf. Comput. Sci. 1999, 39, 747–750. [Google Scholar] [CrossRef]
- Lifechemicals. Available online: http://www.lifechemicals.com (accessed on 29 August 2016).
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Gregori-Puigjane, E.; Mestres, J. Shed: Shannon entropy descriptors from topological feature distributions. J. Chem. Inf. Model. 2006, 46, 1615–1622. [Google Scholar] [CrossRef] [PubMed]
- Mestres, J.; Martín-Couce, L.; Gregori-Puigjané, E.; Cases, M.; Boyer, S. Ligand-based approach to in silico pharmacology: Nuclear receptor profiling. J. Chem. Inf. Model. 2006, 46, 2725–2736. [Google Scholar] [CrossRef] [PubMed]
- Chemotargets. Available online: http://www.chemotargets.com (accessed on 29 August 2016).
- Enamine. Available online: http://www.enamine.net (accessed on 29 August 2016).
- ChemBridge. Available online: http://www.chembridge.com (accessed on 29 August 2016).
- ChemBridge Online Chemical Store. Available online: http://www.hit2lead.com (accessed on 29 August 2016).
- Schrödinger LLC. Schrödinger Release 2015–3: Ligprep; Version 3.5; Schrödinger LLC: New York, NY, USA, 2015. [Google Scholar]
- Maestro: A Powerful, All-Purpose Molecular Modeling Environment. Available online: http://www.schrodinger.com/maestro (accessed on 29 August 2016).
- Hawkins, P.C.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with omega: Algorithm and validation using high quality structures from the protein databank and cambridge structural database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Daylight. Available online: http://www.daylight.com (accessed on 29 August 2016).
- Takeuchi, O.; Kawai, T.; Muhlradt, P.F.; Morr, M.; Radolf, J.D.; Zychlinsky, A.; Takeda, K.; Akira, S. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol. 2001, 13, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Olson, A.J. Using autodock for ligand-receptor docking. Curr. Protoc. Bioinform. 2008. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and autodocktools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Dornhofer, A.A.; Langer, T. Efficient overlay of small organic molecules using 3D pharmacophores. J. Comput. Aided Mol. Des. 2006, 20, 773–788. [Google Scholar] [CrossRef] [PubMed]
- Guner, O.; Clement, O.; Kurogi, Y. Pharmacophore modeling and three dimensional database searching for drug design using catalyst: Recent advances. Curr. Med. Chem. 2004, 11, 2991–3005. [Google Scholar] [CrossRef] [PubMed]
- Labute, P.; Williams, C.; Feher, M.; Sourial, E.; Schmidt, J.M. Flexible alignment of small molecules. J. Med. Chem. 2001, 44, 1483–1490. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. Phase: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. Phase: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. Ligandscout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Murgueitio, M.S.; Henneke, P.; Glossmann, H.; Santos-Sierra, S.; Wolber, G. Prospective virtual screening in a sparse data scenario: Design of small-molecule TLR2 antagonists. Chem. Med. Chem. 2014, 9, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Goodford, P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Cambridge Crystallographic Data Centre. Gold Suite v. 5.1; Cambridge Crystallographic Data Centre: Cambridge, UK, 2012. [Google Scholar]
- Pei, F.; Jin, H.; Zhou, X.; Xia, J.; Sun, L.; Liu, Z.; Zhang, L. Enrichment assessment of multiple virtual screening strategies for Toll-like receptor 8 agonists based on a maximal unbiased benchmarking data set. Chem. Biol. Drug. Des. 2015, 86, 1226–1241. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.; Skillman, A.G.; Nicholls, A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef] [PubMed]
- OpenEye Scientific. Available online: http://www.eyesopen.com/rocs (accessed on 29 August 2016).
- Swann, S.L.; Brown, S.P.; Muchmore, S.W.; Patel, H.; Merta, P.; Locklear, J.; Hajduk, P.J. A unified, probabilistic framework for structure- and ligand-based virtual screening. J. Med. Chem. 2011, 54, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Muchmore, S.W.; Debe, D.A.; Metz, J.T.; Brown, S.P.; Martin, Y.C.; Hajduk, P.J. Application of belief theory to similarity data fusion for use in analog searching and lead hopping. J. Chem. Inf. Model. 2008, 48, 941–948. [Google Scholar] [CrossRef] [PubMed]
- McGaughey, G.B.; Sheridan, R.P.; Bayly, C.I.; Culberson, J.C.; Kreatsoulas, C.; Lindsley, S.; Maiorov, V.; Truchon, J.F.; Cornell, W.D. Comparison of topological, shape, and docking methods in virtual screening. J. Chem. Inf. Model. 2007, 47, 1504–1519. [Google Scholar] [CrossRef] [PubMed]
- Švajger, U.; Horvat, Ž.; Knez, D.; Rožman, P.; Turk, S.; Gobec, S. New antagonists of Toll-like receptor 7 discovered through 3D ligand-based virtual screening. Med. Chem. Res. 2014, 24, 362–371. [Google Scholar] [CrossRef]
- Sousa, S.F.; Ribeiro, A.J.; Coimbra, J.T.; Neves, R.P.; Martins, S.A.; Moorthy, N.S.; Fernandes, P.A.; Ramos, M.J. Protein-ligand docking in the new millennium—A retrospective of 10 years in the field. Curr. Med. Chem. 2013, 20, 2296–2314. [Google Scholar] [CrossRef] [PubMed]
- Moroni, E.; Paladino, A.; Colombo, G. The dynamics of drug discovery. Curr. Top. Med. Chem. 2015, 15, 2043–2055. [Google Scholar] [CrossRef] [PubMed]
- Spyrakis, F.; Cavasotto, C.N. Open challenges in structure-based virtual screening: Receptor modeling, target flexibility consideration and active site water molecules description. Arch. Biochem. Biophys. 2015, 583, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Halperin, I.; Ma, B.; Wolfson, H.; Nussinov, R. Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins 2002, 47, 409–443. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Bohm, H.J. The development of a simple empirical scoring function to estimate the binding constant for a protein-ligand complex of known three-dimensional structure. J. Comput. Aided Mol. Des. 1994, 8, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G.; Appelt, K.; Freer, S.T.; Villafranca, J.E. Empirical free energy calculations of ligand-protein crystallographic complexes. I. Knowledge-based ligand-protein interaction potentials applied to the prediction of human immunodeficiency virus 1 protease binding affinity. Protein Eng. 1995, 8, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Hendlich, M.; Klebe, G. Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol. 2000, 295, 337–356. [Google Scholar] [CrossRef] [PubMed]
- AutoDock Vina. Available online: http://vina.scripps.edu (accessed on 29 August 2016).
- The Official UCSF DOCK Web-Site. Available online: http://dock.compbio.ucsf.edu (accessed on 29 August 2016).
- BioSolveIT GmbH. Available online: http://www.biosolveit.de/flexx/index.Html (accessed on 29 August 2016).
- Glide: A Complete Solution for Ligand-Receptor Docking. Available online: http://www.schrodinger.com/glide (accessed on 29 August 2016).
- GOLD. Available online: http://www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold (accessed on 29 August 2016).
- Molsoft. Available online: http://www.molsoft.com (accessed on 29 August 2016).
- CERTARA. Available online: http://www.certara.com/products/molmod/sybyl-x/sbd (accessed on 29 August 2016).
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Autodock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- MGLTools. Available online: http://mgltools.scripps.edu (accessed on 29 August 2016).
- Wang, R.; Lai, L.; Wang, S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comput. Aided Mol. Des. 2002, 16, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Fang, X.; Lu, Y.; Wang, S. The pdbbind database: Collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J. Med. Chem. 2004, 47, 2977–2980. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Fang, X.; Lu, Y.; Yang, C.Y.; Wang, S. The PDBbind database: Methodologies and updates. J. Med. Chem. 2005, 48, 4111–4119. [Google Scholar] [CrossRef] [PubMed]
- Baxter, J. Local optima avoidance in depot location. J. Oper. Res. Soc. 1981, 32, 815–819. [Google Scholar] [CrossRef]
- Blum, C.; Blesa Aguilera, M.J.; Roli, A.; Sampels, M. Hybrid Metaheuristics: An Emerging Approach to Optimization; Springer Publishing Company: Berlin, Germany, 2008; p. 290. [Google Scholar]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using gold. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43–53. [Google Scholar] [CrossRef]
- Muegge, I.; Martin, Y.C. A general and fast scoring function for protein-ligand interactions: A simplified potential approach. J. Med. Chem. 1999, 42, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Mooij, W.T.; Verdonk, M.L. General and targeted statistical potentials for protein-ligand interactions. Proteins 2005, 61, 272–287. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stutzle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with plants. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Verkhivker, G.M.; Bouzida, D.; Gehlhaar, D.K.; Rejto, P.A.; Arthurs, S.; Colson, A.B.; Freer, S.T.; Larson, V.; Luty, B.A.; Marrone, T.; et al. Deciphering common failures in molecular docking of ligand-protein complexes. J. Comput. Aided Mol. Des. 2000, 14, 731–751. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I. A knowledge-based scoring function for protein-ligand interactions: Probing the reference state. Perspect. Drug Discov. Des. 2000, 20, 99–114. [Google Scholar] [CrossRef]
- Gohlke, H.; Hendlich, M.; Klebe, G. Predicting binding modes, binding affinities and “hot spots” for protein-ligand complexes using a knowledge-based scoring function. Perspect. Drug Discov. Des. 2000, 20, 115–144. [Google Scholar] [CrossRef]
- Liebeschuetz, J.W.; Cole, J.C.; Korb, O. Pose prediction and virtual screening performance of gold scoring functions in a standardized test. J. Comput. Aided Mol. Des. 2012, 26, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N. Surflex-dock 2.1: Robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J. Comput. Aided Mol. Des. 2007, 21, 281–306. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, J.; Welch, W.; Jain, A.N. Automatic identification and representation of protein binding sites for molecular docking. Protein Sci. 1997, 6, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Welch, W.; Ruppert, J.; Jain, A.N. Hammerhead: Fast, fully automated docking of flexible ligands to protein binding sites. Chem. Biol. Drug Des. 1996, 3, 449–462. [Google Scholar] [CrossRef]
- Pham, T.A.; Jain, A.N. Parameter estimation for scoring protein-ligand interactions using negative training data. J. Med. Chem. 2006, 49, 5856–5868. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed]
- Bohm, H.J. Prediction of binding constants of protein ligands: A fast method for the prioritization of hits obtained from de novo design or 3D database search programs. J. Comput. Aided Mol. Des. 1998, 12, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Bohm, H.J. The computer program LUDI: A new method for the de novo design of enzyme inhibitors. J. Comput. Aided Mol. Des. 1992, 6, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Brylinski, M. Nonlinear scoring functions for similarity-based ligand docking and binding affinity prediction. J. Chem. Inf. Model. 2013, 53, 3097–3112. [Google Scholar] [CrossRef] [PubMed]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 1994, 235, 983–1002. [Google Scholar] [CrossRef] [PubMed]
- Bursulaya, B.D.; Totrov, M.; Abagyan, R.; Brooks, C.L. Comparative study of several algorithms for flexible ligand docking. J. Comput. Aided Mol. Des. 2003, 17, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Neves, M.A.; Totrov, M.; Abagyan, R. Docking and scoring with ICM: The benchmarking results and strategies for improvement. J. Comput. Aided Mol. Des. 2012, 26, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Totrov, M.; Abagyan, R. Derivation of sensitive discrimination potential for virtual ligand screening. In Proceedings of the Third Annual International Conference on Computational Molecular Biology, Lyon, France, 11–14 April 1999; pp. 312–320.
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. Dock 6: Combining techniques to model RNA-small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Ewing, T.J.A.; Makino, S.; Skillman, A.G.; Kuntz, I.D. Dock 4.0: Search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001, 15, 411–428. [Google Scholar] [CrossRef] [PubMed]
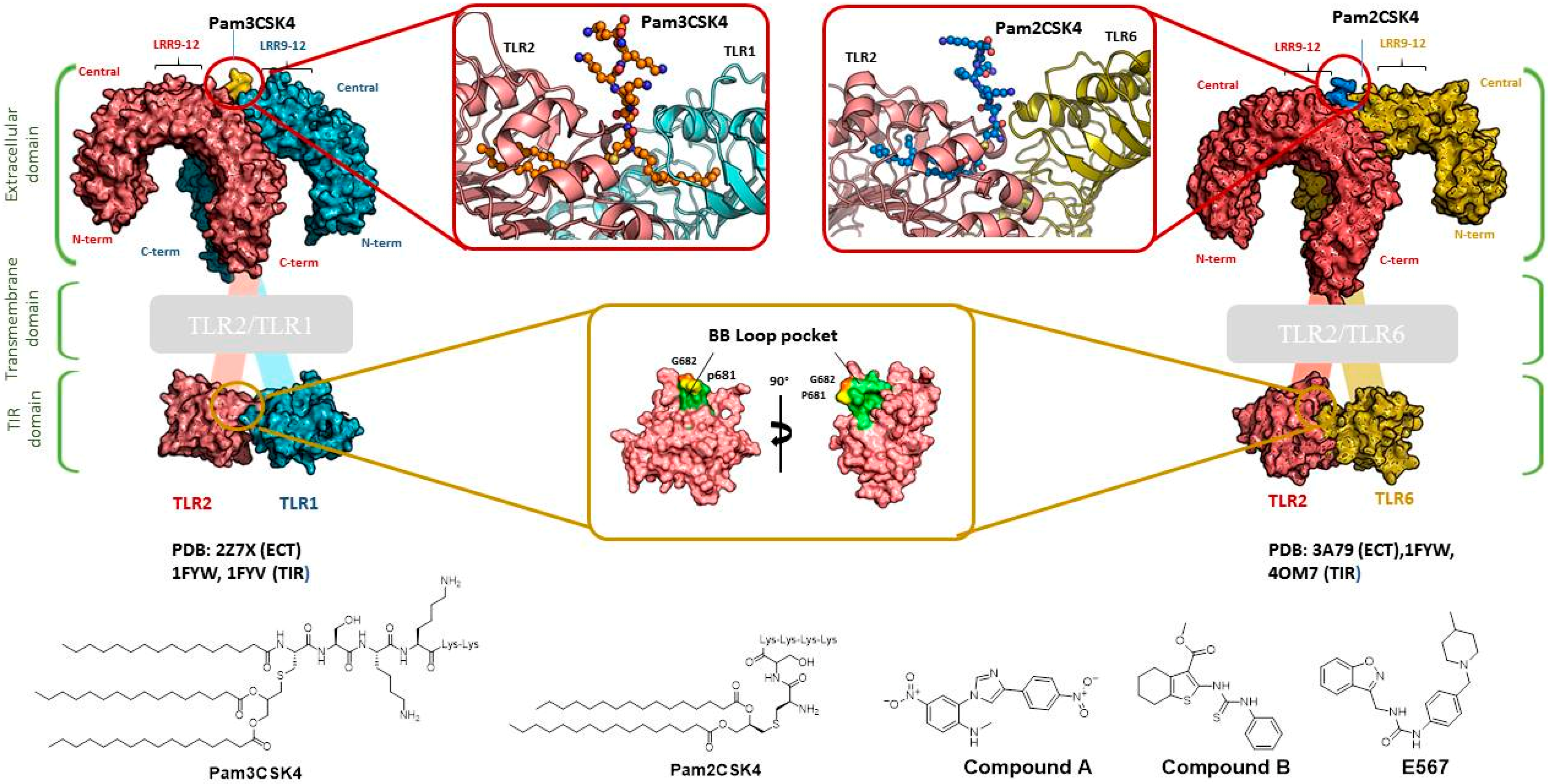
- Jin, M.S.; Kim, S.E.; Heo, J.Y.; Lee, M.E.; Kim, H.M.; Paik, S.G.; Lee, H.; Lee, J.O. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 2007, 130, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.Y.; Nan, X.; Jin, M.S.; Youn, S.J.; Ryu, Y.H.; Mah, S.; Han, S.H.; Lee, H.; Paik, S.G.; Lee, J.O. Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 2009, 31, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Liu, L.J.; Dong, Z.Q.; Lu, L.; Wang, M.; Leung, C.H.; Ma, D.L.; Wang, Y. Structure-based discovery of an immunomodulatory inhibitor of TLR1-TLR2 heterodimerization from a natural product-like database. Chem. Commun. 2015, 51, 11178–11181. [Google Scholar] [CrossRef] [PubMed]
- Arnautova, Y.A.; Abagyan, R.A.; Totrov, M. Development of a new physics-based internal coordinate mechanics force field and its application to protein loop modeling. Proteins 2011, 79, 477–498. [Google Scholar] [CrossRef] [PubMed]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Cuevas, C.D.; Ross, S.R. Toll-like receptor 2-mediated innate immune responses against Junin virus in mice lead to antiviral adaptive immune responses during systemic infection and do not affect viral replication in the brain. J. Virol. 2014, 88, 7703–7714. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Wang, X.; Zhang, S.Y.; Yin, H. Discovery of small molecule inhibitors of the TLR1-TLR2 complex. Angew. Chem. Int. Ed. 2012, 51, 12246–12249. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Cerny, A.M.; Bowen, G.; Chan, M.; Knipe, D.M.; Kurt-Jones, E.A.; Finberg, R.W. Discovery of a novel TLR2 signaling inhibitor with anti-viral activity. Antivir. Res. 2010, 87, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.; Laird, M.H.W.; Schwarz, R.S.; Greene, S.; Dyson, T.; Snyder, G.A.; Xiao, T.S.; Chauhan, J.; Fletcher, S.; Toshchakov, V.Y.; et al. Inhibition of TLR2 signaling by small molecule inhibitors targeting a pocket within the TLR2 TIR domain. Proc. Natl. Acad. Sci. USA 2015, 112, 5455–5460. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Kopp, E.B.; Medzhitov, R. The toll-receptor family and control of innate immunity. Curr. Opin. Immunol. 1999, 11, 13–18. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tao, X.; Shen, B.; Horng, T.; Medzhitov, R.; Manley, J.L.; Tong, L. Structural basis for signal transduction by the toll/interleukin-1 receptor domains. Nature 2000, 408, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Kuntz, I.D. Structure-based strategies for drug design and discovery. Science 1992, 257, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L.; MacKerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. Charmm: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Gautam, J.K.; Ashish; Comeau, L.D.; Krueger, J.K.; Smith, M.F., Jr. Structural and functional evidence for the role of the TLR2 DD loop in TLR1/TLR2 heterodimerization and signaling. J. Biol. Chem. 2006, 281, 30132–30142. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.K.; Botos, I.; Hall, P.R.; Askins, J.; Shiloach, J.; Segal, D.M.; Davies, D.R. The molecular structure of the Toll-like receptor 3 ligand-binding domain. Proc. Natl. Acad. Sci. USA 2005, 102, 10976–10980. [Google Scholar] [CrossRef] [PubMed]
- Barton, G.M.; Medzhitov, R. Toll-like receptor signaling pathways. Science 2003, 300, 1524–1525. [Google Scholar] [CrossRef] [PubMed]
- Assmann, T.S.; Brondani, L.D.A.; Bouças, A.P.; Canani, L.H.; Crispim, D. Toll-like receptor 3 (TLR3) and the development of type 1 diabetes mellitus. Arch. Endocrinol. Metab. 2015, 59, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Amarante, M.K.; Watanabe, M.A. Toll-like receptor 3: Involvement with exogenous and endogenous RNA. Int. Rev. Immunol. 2010, 29, 557–573. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Jouanguy, E.; Ugolini, S.; Smahi, A.; Elain, G.; Romero, P.; Segal, D.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007, 317, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Town, T.; Alexopoulou, L.; Anderson, J.F.; Fikrig, E.; Flavell, R.A. Toll-like receptor 3 mediates west nile virus entry into the brain causing lethal encephalitis. Nat. Med. 2004, 10, 1366–1373. [Google Scholar] [CrossRef] [PubMed]
- Gowen, B.B.; Hoopes, J.D.; Wong, M.H.; Jung, K.H.; Isakson, K.C.; Alexopoulou, L.; Flavell, R.A.; Sidwell, R.W. TLR3 deletion limits mortality and disease severity due to phlebovirus infection. J. Immunol. 2006, 177, 6301–6307. [Google Scholar] [CrossRef] [PubMed]
- Hutchens, M.; Luker, K.E.; Sottile, P.; Sonstein, J.; Lukacs, N.W.; Nunez, G.; Curtis, J.L.; Luker, G.D. TLR3 increases disease morbidity and mortality from vaccinia infection. J. Immunol. 2008, 180, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Pashine, A.; Valiante, N.M.; Ulmer, J.B. Targeting the innate immune response with improved vaccine adjuvants. Nat. Med. 2005, 11, S63–S68. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Matsumoto, M.; Funami, K.; Akazawa, T.; Seya, T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat. Immunol. 2003, 4, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Wang, X.; Yin, H. Small-molecule inhibitors of the TLR3/dsRNA complex. J. Am. Chem. Soc. 2011, 133, 3764–3767. [Google Scholar] [CrossRef] [PubMed]
- Heitmeier, M.R.; Scarim, A.L.; Corbett, J.A. Double-stranded RNA-induced inducible nitric-oxide synthase expression and interleukin-1 release by murine macrophages requires NF-κB activation. J. Biol. Chem. 1998, 273, 15301–15307. [Google Scholar] [CrossRef] [PubMed]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in TLR4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. TLR4 as the mammalian endotoxin sensor. Curr. Top. Microbiol. Immunol. 2002, 270, 109–120. [Google Scholar] [PubMed]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.S.; Lee, H.; Lee, J.O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Ohto, U.; Fukase, K.; Miyake, K.; Shimizu, T. Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2. Proc. Natl. Acad. Sci. USA 2012, 109, 7421–7426. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, A.; Holst, O.; Di Lorenzo, F.; Callaghan, M.; Nurisso, A.; D’Errico, G.; Zamyatina, A.; Peri, F.; Berisio, R.; Jerala, R.; et al. Chemistry of lipid a: At the heart of innate immunity. Chemistry 2015, 7, 500–519. [Google Scholar] [CrossRef] [PubMed]
- Klett, J.; Reeves, J.; Oberhauser, N.; Pérez-Regidor, L.; Martín-Santamaría, S. Modulation of Toll-like receptor 4. Insights from X-ray crystallography and molecular modeling. Curr. Top. Med. Chem. 2014, 14, 2672–2683. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Jarvis, R.A.; Patrick, E.A. Clustering using a similarity measure based on shared near neighbors. IEEE Trans. Comput. 1973, 22, 1025–1034. [Google Scholar] [CrossRef]
- Willett, J. Similarity and Clustering in Chemical Information Systems; John Wiley & Sons, Inc.: New York, NY, USA, 1987; 266p. [Google Scholar]
- Todeschini, R.; Consonni, V.; Xiang, H.; Holliday, J.; Buscema, M.; Willett, P. Similarity coefficients for binary chemoinformatics data: Overview and extended comparison using simulated and real data sets. J. Chem. Inf. Model. 2012, 52, 2884–2901. [Google Scholar] [CrossRef] [PubMed]
- Salim, N.; Holliday, J.; Willett, P. Combination of fingerprint-based similarity coefficients using data fusion. J. Chem. Inform. Comput. Sci. 2003, 43, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Švajger, U.; Brus, B.; Turk, S.; Sova, M.; Hodnik, V.; Anderluh, G.; Gobec, S. Novel Toll-like receptor 4 (TLR4) antagonists identified by structure- and ligand-based virtual screening. Eur. J. Med. Chem. 2013, 70, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Joce, C.; Stahl, J.A.; Shridhar, M.; Hutchinson, M.R.; Watkins, L.R.; Fedichev, P.O.; Yin, H. Application of a novel in silico high-throughput screen to identify selective inhibitors for protein-protein interactions. Bioorg. Med. Chem. Lett. 2010, 20, 5411–5413. [Google Scholar] [CrossRef] [PubMed]
- Mahita, J.; Harini, K.; Rao Pichika, M.; Sowdhamini, R. An in silico approach towards the identification of novel inhibitors of the TLR-4 signaling pathway. J. Biomol. Struct. Dyn. 2015, 1–18. [Google Scholar]
- Fontaine, F.; Bolton, E.; Borodina, Y.; Bryant, S. Fast 3D shape screening of large chemical databases through alignment-recycling. Chem. Cent. J. 2007, 1, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Salzberg, S.L.; Warnow, T. Algorithms in Bioinformatics. In Proceedings of the 9th International Workshop, WABI 2009, Philadelphia, PA, USA, 12–13 September 2009; Springer: New York, NY, USA, 2009. [Google Scholar]
- Schrödinger, LLC. Small-Molecule Drug Discovery Suite 2016–1: Qikprop; Version 4.7; Schrödinger, LLC: New York, NY, USA, 2016. [Google Scholar]
- Duan, J.A.; Dixon, S.L.; Lowrie, J.F.; Sherman, W. Analysis and comparison of 2D fingerprints: Insights into database screening performance using eight fingerprint methods. J. Mol. Graph. Model. 2010, 29, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Sastry, M.; Lowrie, J.F.; Dixon, S.L.; Sherman, W. Large-scale systematic analysis of 2D fingerprint methods and parameters to improve virtual screening enrichments. J. Chem. Inf. Model. 2010, 50, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Farid, R.; Day, T.J.; Friesner, R.A.; Pearlstein, R.A. New insights about herg blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.J.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug. Des. 2006, 67, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Hacker, H.; Mischak, H.; Miethke, T.; Liptay, S.; Schmid, R.; Sparwasser, T.; Heeg, K.; Lipford, G.B.; Wagner, H. CpG-DNA-specific activation of antigen-presenting cells requires stress kinase activity and is preceded by non-specific endocytosis and endosomal maturation. EMBO J. 1998, 17, 6230–6240. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Guenthner-Biller, M.; Bourquin, C.; Ablasser, A.; Schlee, M.; Uematsu, S.; Noronha, A.; Manoharan, M.; Akira, S.; de Fougerolles, A.; et al. Sequence-specific potent induction of IFN-α by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med. 2005, 11, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Blasius, A.L.; Beutler, B. Intracellular Toll-like receptors. Immunity 2010, 32, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Eberle, F.; Sirin, M.; Binder, M.; Dalpke, A.H. Bacterial RNA is recognized by different sets of immunoreceptors. Eur. J. Immunol. 2009, 39, 2537–2547. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Kanzler, H.; Barrat, F.J.; Hessel, E.M.; Coffman, R.L. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat. Med. 2007, 13, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.L.; Gerster, J.F.; Owens, M.L.; Slade, H.B.; Tomai, M.A. Imiquimod applied topically: A novel immune response modifier and new class of drug. Int. J. Immunopharmacol. 1999, 21, 1–14. [Google Scholar] [CrossRef]
- Shukla, N.M.; Malladi, S.S.; Day, V.; David, S.A. Preliminary evaluation of a 3H imidazoquinoline library as dual TLR7/TLR8 antagonists. Bioorg. Med. Chem. 2011, 19, 3801–3811. [Google Scholar] [CrossRef] [PubMed]
- Ohto, U.; Tanji, H.; Shimizu, T. Structure and function of Toll-like receptor 8. Microbes Infect. 2014, 16, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdorfer, B.; Giese, T.; Endres, S.; Hartmann, G. Quantitative expression of Toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.T.; Williams, B.R.G. Activation of the mammalian immune system by siRNAs. Nat. Biotechnol. 2005, 23, 1399–1405. [Google Scholar] [CrossRef] [PubMed]
- Philbin, V.J.; Dowling, D.J.; Gallington, L.C.; Cortes, G.; Tan, Z.; Suter, E.E.; Chi, K.W.; Shuckett, A.; Stoler-Barak, L.; Tomai, M.; et al. Imidazoquinoline Toll-like receptor 8 agonists activate human newborn monocytes and dendritic cells through adenosine-refractory and caspase-1-dependent pathways. J. Allergy Clin. Immunol. 2012, 130, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Tanji, H.; Ohto, U.; Shibata, T.; Miyake, K.; Shimizu, T. Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science 2013, 339, 1426–1429. [Google Scholar] [CrossRef] [PubMed]
- Yoo, E.; Salunke, D.B.; Sil, D.; Guo, X.; Salyer, A.C.; Hermanson, A.R.; Kumar, M.; Malladi, S.S.; Balakrishna, R.; Thompson, W.H.; et al. Determinants of activity at human Toll-like receptors 7 and 8: Quantitative structure-activity relationship (QSAR) of diverse heterocyclic scaffolds. J. Med. Chem. 2014, 57, 7955–7970. [Google Scholar] [CrossRef] [PubMed]
- Kokatla, H.P.; Sil, D.; Tanji, H.; Ohto, U.; Malladi, S.S.; Fox, L.M.; Shimizu, T.; David, S.A. Structure-based design of novel human Toll-like receptor 8 agonists. Chem. Med. Chem. 2014, 9, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Kokatla, H.P.; Yoo, E.; Salunke, D.B.; Sil, D.; Ng, C.F.; Balakrishna, R.; Malladi, S.S.; Fox, L.M.; David, S.A. Toll-like receptor-8 agonistic activities in C2, C4, and C8 modified thiazolo[4,5-c]quinolines. Org. Biomol. Chem. 2013, 11, 1179–1198. [Google Scholar] [CrossRef] [PubMed]
- Kokatla, H.P.; Sil, D.; Malladi, S.S.; Balakrishna, R.; Hermanson, A.R.; Fox, L.M.; Wang, X.; Dixit, A.; David, S.A. Exquisite selectivity for human Toll-like receptor 8 in substituted furo[2,3-c]quinolines. J. Med. Chem. 2013, 56, 6871–6885. [Google Scholar] [CrossRef] [PubMed]
- Salunke, D.B.; Yoo, E.; Shukla, N.M.; Balakrishna, R.; Malladi, S.S.; Serafin, K.J.; Day, V.W.; Wang, X.; David, S.A. Structure–activity relationships in human Toll-like receptor 8-active 2,3-diamino-furo[2,3-c]pyridines. J. Med. Chem. 2012, 55, 8137–8151. [Google Scholar] [CrossRef] [PubMed]
- Schiaffo, C.E.; Shi, C.; Xiong, Z.; Olin, M.; Ohlfest, J.R.; Aldrich, C.C.; Ferguson, D.M. Structure–activity relationship analysis of imidazoquinolines with Toll-like receptors 7 and 8 selectivity and enhanced cytokine induction. J. Med. Chem. 2014, 57, 339–347. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Program | Ligand Flexibility Method | Scoring Function | Society | |||
|---|---|---|---|---|---|---|
| Type | Algorithm | Type | Name | Name (Availability) | Website | |
| AutoDock VINA | ST | Iterated Local Search global optimizer | Hybrid E/K-based | - | The Scripps Research Institute, la Jolla (Free) | [76] |
| DOCK | SYS | Incremental construction Anchor-and-Grow Algorithm | FF-based | DOCK 3.5 score | University of California San Francisco (Free) | [77] |
| FlexX | SYS | Incremental Reconstruction Algorithm | E | SCORE1 | BioSolveIT (Commercial) | [78] |
| Glide | SYS | Exhaustive Search Algorithm | E | GlideScore | Schrödinger (Commercial) | [79] |
| GOLD | ST | Genetic Algorithm | FF_based E K-based E | GoldScore ChemScore ASP ChemPLP | University of Sheffield, GlaxoSmithKline plc and CCDC (Commercial) | [80] |
| ICM | ST | Pseudo-Brownian sampling and local minimization | E K-based | ICMScore PMF | MolSoft (Commercial) | [81] |
| Surflex-Dock | SYS | Incremental Reconstruction Algorithm Whole Molecule Approach | E | Re-parameterized Hammerhead | Tripos (Commercial) | [82] |
| TLR2/TLR1 | TLR2/TLR6 |
|---|---|
 |  |
| 3D structure from PDB-ID 2Z7X | 3D structure from PDB-ID 3A79 |
 |  |
| ZINC: ZINC12899676 [121] TLR2-TLR1 heterodimerization inhibitor | ENAMINE: Z416323354 [56] MolPort a: MolPort-009-315-475 TLR2/1 & TLR2/6 inhibitor |
 |  |
| MolPort a: Molport-001-796-266 [56] TLR2/1 & TLR2/6 inhibitor | MolPort a: MolPort-009-737-181 [56] TLR2/1 & TLR2/6 inhibitor with a decrease of cell viability |
 |  |
| ZINC: ZINC1676936 [56] NCI: Plated 2007: 44661TLR2/1 & TLR2/6 inhibitor | MolPort a: MolPort-002-914-354 [56] TLR2/1 & TLR2/6 inhibitor |
 |  |
| ZINC: ZINC398557 [56] NCI: Plated 2007: 205636 MolPort a: MolPort-001-835-401 TLR2/1 & TLR2/6 inhibitor | C29 [127] TLR2 TIR domain inhibitor |
 |  |
| ZINC: ZINC1758666 [56] NCI: Plated 2007: 17379 TLR2/1 & TLR2/6 inhibitor | C29L (O-vanillin) [127] TLR2 TIR domain inhibitor |
 | |
| ZINC: ZINC585632 [56] TLR2/1 & TLR2/6 inhibitor | |
| TLR3 | TLR7 | ||
|---|---|---|---|
 | No X-ray crystallographic structure available | ||
| 3D structure from PDB-ID 3CIY | - | ||
 |  |  |  |
| (R) Compound 4a [146] TLR3 inhibitor | ENAMINE: T5528092 [146] TLR3 inhibitor | Query 1 (Imiquimod) [65] | ZINC: ZINC1667204 [65] TLR7 inhibitor |
 |  |  |  |
| T5631009 [146] TLR3 inhibitor | ENAMINE: T5630975 [146] TLR3 inhibitor | Query 2 [65] | ZINC: ZINC39698 [65] TLR7 inhibitor |
 |  |  |  |
| T0519-9149 [146] TLR3 inhibitor | ENAMINE: T5626448 [146] TLR3 inhibitor | ZINC: ZINC12382420 [65] TLR7 inhibitor | ZINC: ZINC36416 [65] TLR7 inhibitor |
 |  |  | |
| ENAMINE: T5643856 [146] TLR3 inhibitor | ENAMINE: T5260630 [146] TLR3 inhibitor | ZINC: ZINC4756232 [65] TLR7 inhibitor | |
 |  |  | |
| ENAMINE: T55994342 [146] TLR3 inhibitor | ENAMINE: T0505-4844 [146] TLR3 inhibitor | ZINC: ZINC8686004 [65] TLR7 inhibitor | |
| TLR4 | ||
|---|---|---|
 | ||
| 3D structure from PDB-ID 3FXI | ||
 |  |  |
| ENAMINE: T5342126 [161] TLR4 inhibitor | ZINC: ZINC04272679 [162] Predicted TLR4 inhibitor | ZINC: ZINC00611718 [162] Predicted TLR4 inhibitor |
 |  |  |
| ENAMINE: T6071187 [161] MD-2 inhibitor | ZINC: ZINC04272561 [162] Predicted TLR4 inhibitor | ZINC: ZINC48141941 [162] Predicted TLR4 inhibitor |
 |  |  |
| ENAMINE: T5339238 ZINC: ZINC25778142 [160] TLR4 inhibitor | ZINC: ZINC09535665 [162] Predicted TLR4 inhibitor | ENAMINE: T6969316 ZINC: ZINC51408124 [160] TLR4 activity not determined (solubility problems) |
 |  |  |
| ENAMINE: T5458371 ZINC: ZINC49563556 [160] TLR4 inhibitor | ZINC: ZINC70039563 [162] Predicted TLR4 inhibitor | ZINC: ZINC464832 [160] TLR4 activity not determined (cytotoxicity on HEK293 cells) |
 |  |  |
| ENAMINE: T5315798 ZINC: ZINC3415865 [160] TLR4 inhibitor | ZINC: ZINC29450369 [162] Predicted TLR4 inhibitor | ENAMINE: T6417643 ZINC: ZINC26905159 [160] Predicted TLR4 inhibitor but not active |
 |  |  |
| ZINC: ZINC64951618 [162] Predicted TLR4 inhibitor | ZINC: ZINC41124663 [162] Predicted TLR4 inhibitor | ENAMINE: T6280209 ZINC: ZINC32525142 [160] Predicted TLR4 inhibitor but not active |
 |  |  |
| ZINC: ZINC64951738 [162] Predicted TLR4 inhibitor | ZINC: ZINC08687988 [162] Predicted TLR4 inhibitor | ENAMINE: T6279749 ZINC: ZINC32524933 [160] Predicted TLR4 inhibitor but not active |
 | ||
| ZINC: ZINC72278680 [162] Predicted TLR4 inhibitor | ||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Regidor, L.; Zarioh, M.; Ortega, L.; Martín-Santamaría, S. Virtual Screening Approaches towards the Discovery of Toll-Like Receptor Modulators. Int. J. Mol. Sci. 2016, 17, 1508. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091508
Pérez-Regidor L, Zarioh M, Ortega L, Martín-Santamaría S. Virtual Screening Approaches towards the Discovery of Toll-Like Receptor Modulators. International Journal of Molecular Sciences. 2016; 17(9):1508. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091508
Chicago/Turabian StylePérez-Regidor, Lucía, Malik Zarioh, Laura Ortega, and Sonsoles Martín-Santamaría. 2016. "Virtual Screening Approaches towards the Discovery of Toll-Like Receptor Modulators" International Journal of Molecular Sciences 17, no. 9: 1508. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091508