
Molecular Imaging of Vulnerable Atherosclerotic Plaques in Animal Models

Abstract
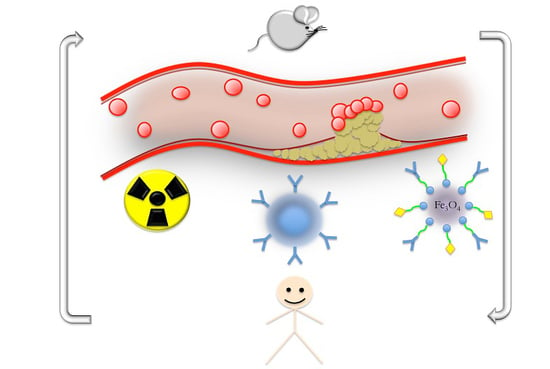
:
1. Introduction
2. Murine Models of Atherosclerosis and Molecular Imaging Targets
2.1. Murine Models of Atherosclerosis and Vulnerable Plaques
2.1.1. Murine Models of Atherosclerosis: General Considerations
2.1.2. Murine Models of Vulnerable Atherosclerotic Plaques
2.2. Molecular Biomarkers of Atherosclerotic Plaques in Humans and Murine Models
2.2.1. Leukocyte Adhesion Receptors
2.2.2. Indicators of Macrophage Infiltration
2.2.3. Indicators of Angiogenesis
2.2.4. Other Potential Molecular Targets
2.3. Emerging Applications for Molecular Imaging in Murine Models of Atherosclerosis
2.3.1. Contrast-Enhanced Ultrasound Imaging
2.3.2. Contrast-Enhanced Magnetic Resonance Imaging
2.3.3. Nuclear Imaging Techniques and X-ray Computed Tomography
2.3.4. Targeted Fluorescence Imaging
2.3.5. Photoacoustic Imaging
3. Conclusions and Future Directions
Author Contributions
Conflicts of Interest
Abbreviations
| AHA | American Heart Association |
| ApoE−/− | apolipoprotein-E-knockout |
| WTD | western type diet |
| Fbn1 | fibrillin-1 |
| MRI | magnetic resonance imaging |
| CEUS | contrast-enhanced ultrasound |
| microCT | micro-computed tomography |
| HDL | high-density lipoprotein |
| LDL | low-density lipoprotein |
| VLDL | very low-density lipoprotein |
| SMCs | smooth muscle cells |
| LDL−/− | low-density lipoprotein-knockout |
| MMP | matrix metalloproteinase |
| LPS | lipopolysaccharide |
| Ang II | angiotensin II |
| MCP-1 | monocyte chemoattractant protein-1 |
| DIO | diet-induced obese |
| DD | diabetogenic diet |
| STZ | streptozotocin |
| T1D | type 1 diabetes |
| hAR | human aldose reductase |
| T2D | type 2 diabetic |
| PCSK9 | proprotein convertase subtilisin/kexin type 9 |
| VCAM-1 | vascular cell adhesion molecule-1 |
| ICAM-1 | intercellular adhesion molecule-1 |
| VLA 4 | very late antigen-4 |
| TLR-4 | Toll-like receptor 4 |
| FR β | folate receptor β |
| CD68 | Cluster of Differentiation 68 |
| VEGF | vascular endothelial growth factor |
| VEGFRs | vascular endothelial growth factor receptors |
| VEGFR-1 | vascular endothelial growth factor receptor 1 |
| VEGFR-2 | vascular endothelial growth factor receptor 2 |
| PS | phosphatidylserine |
| A5 | annexin V |
| GPVI | glycoprotein VI |
| PET | positron emission tomography |
| SPECT | single-photon emission computed tomography |
| CT | X-ray computed tomography |
| FMT | fluorescent molecular tomography |
| PAI | photoacoustic imaging |
| HFU | high-frequency ultrasound |
| MB | microbubbles |
| GP IIb/IIIa | glycoprotein (GP) IIb/IIIa complex |
| RGD | Arg-Gly-Asp sequence |
| cRGD | cyclic RGD peptide |
| USPIO | ultrasmall superparamagnetic iron oxide nanoparticles |
| Gd-DTPA | gadolinium chelates |
| RARE | rapid-acquisition relaxation-enhanced |
| LOX-1 | Lectin-like Oxidized Low-Density Lipoprotein Receptor 1 |
| NIRF | near-infrared fluorescence |
| PC-NPs | DMSA-Fe3O4-nanoparticles |
| LyP-1 | cyclic 9-amino acid peptide |
| 99mTc | 99mTechnetium |
| PBR | peripheral benzodiazepine receptor |
| TSPO | translocator protein |
| OAT | optoacoustic tomography |
| AuNPs | gold nanoparticles |
| ICG | indocyanine green |
| eRNA | extracellular RNA |
References
- Virmani, R.; Burke, A.P.; Kolodgie, F.D.; Farb, A. Vulnerable plaque: The pathology of unstable coronary lesions. J. Interv. Cardiol. 2002, 15, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Naghavi, M.; Libby, P.; Falk, E.; Casscells, S.W.; Litovsky, S.; Rumberger, J.; Badimon, J.J.; Stefanadis, F.; Moreno, P.; Pasterkamp, G.; et al. From vulnerable plaque to vulnerable patient: A call for new definitions and risk assessment strategies: Part I. Circulation 2003, 108, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C. Natural history and histological classification of atherosclerotic lesions: An update. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1177–1178. [Google Scholar] [CrossRef] [PubMed]
- Virmani, R.; Burke, A.P.; Farb, A.; Kolodgie, F.D. Pathology of the vulnerable plaque. J. Am. Coll. Cardiol. 2006, 47, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Schapira, K.; Heeneman, S.; Daemen, M.J. Animal models to study plaque vulnerability. Curr. Pharm. Des. 2007, 13, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Faggiotto, A.; Ross, R.; Harker, L. Studies of hypercholesterolemia in the nonhuman primate. I. Changes that lead to fatty streak formation. Arteriosclerosis 1984, 4, 323–340. [Google Scholar] [CrossRef] [PubMed]
- Faggiotto, A.; Ross, R. Studies of hypercholesterolemia in the nonhuman primate. II. Fatty streak conversion to fibrous plaque. Arteriosclerosis 1984, 4, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Masuda, J.; Ross, R. Atherogenesis during low level hypercholesterolemia in the nonhuman primate. I. Fatty streak formation. Arteriosclerosis 1990, 10, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Masuda, J.; Ross, R. Atherogenesis during low level hypercholesterolemia in the nonhuman primate. II. Fatty streak conversion to fibrous plaque. Arteriosclerosis 1990, 10, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.J.; Sprague, E.A.; Kelley, J.L.; Valente, A.J.; Suenram, C.A. Aortic intimal monocyte recruitment in the normo and hypercholesterolemic baboon (Papio cynocephalus). An ultrastructural study: Implications in atherogenesis. Virchows Arch. A 1985, 405, 175–191. [Google Scholar] [CrossRef]
- Gerrity, R.G. The role of the monocyte in atherogenesis: I. Transition of blood-borne monocytes into foam cells in fatty lesions. Am. J. Pathol. 1981, 103, 181–190. [Google Scholar] [PubMed]
- Gerrity, R.G. The role of the monocyte in atherogenesis: II. Migration of foam cells from atherosclerotic lesions. Am. J. Pathol. 1981, 103, 191–200. [Google Scholar] [PubMed]
- Reitman, J.S.; Mahley, R.W.; Fry, D.L. Yucatan miniature swine as a model for diet-induced atherosclerosis. Atherosclerosis 1982, 43, 119–132. [Google Scholar] [CrossRef]
- Davies, P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009, 1, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Koskinas, K.C.; Feldman, C.L.; Chatzizisis, Y.S.; Coskun, A.U.; Jonas, M.; Maynard, C.; Baker, A.B.; Papafaklis, M.I.; Edelman, E.R.; Stone, P.H. Natural history of experimental coronary atherosclerosis and vascular remodeling in relation to endothelial shear stress: A serial, in vivo intravascular ultrasound study. Circulation 2010, 19, 2092–2101. [Google Scholar] [CrossRef] [PubMed]
- Prescott, M.F.; McBride, C.; Hasler-Rapacz, J.; von Linden, J.; Rapacz, J. Development of complex atherosclerotic lesions in pigs with inherited hyper-LDL cholesterolemia bearing mutant alleles for apolipoprotein B. Am. J. Pathol. 1991, 139, 139–147. [Google Scholar] [PubMed]
- Brousseau, M.E.; Hoeg, J.M. Transgenic rabbits as models for atherosclerosis research. J. Lipid Res. 1999, 3, 365–375. [Google Scholar]
- Huang, Y.; Schwender, S.W.; Rall, S.C., Jr.; Sanan, D.A.; Mahley, R.W. Apolipoprotein E2 transgenic rabbits. Modulation of the type III hyperlipoproteinemic phenotype by estrogen and occurrence of spontaneous atherosclerosis. J. Biol. Chem. 1997, 272, 22685–22694. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, M.; Ito, T.; Shiraishi, M.; Watanabe, Y. Inheritability of atherosclerosis and the role of lipoproteins as risk factors in the development of atherosclerosis in the WHHL rabbits: Risk factors related to coronary atherosclerosis are different from those related to aortic atherosclerosis. Atherosclerosis 1992, 96, 43–52. [Google Scholar] [CrossRef]
- Niimi, M.; Yang, D.; Kitajima, S.; Ning, B.; Wang, C.; Li, S.; Liu, E.; Zhang, J.; Chen, Y.E.; Fan, J. ApoE knockout rabbits: A novel model for the study of human hyperlipidemia. Atherosclerosis 2015, 245, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Busnelli, M.; Froio, A.; Bacci, M.L.; Giunti, M.; Cerrito, M.G.; Giovannoni, R.; Forni, M.; Gentilini, F.; Scagliarini, A.; Deleo, G.; et al. Pathogenetic role of hypercholesterolemia in a novel preclinical model of vascular injury in pigs. Atherosclerosis 2009, 207, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Tong, T.; Peng, Y.; Tian, J.; Gu, W.; Tang, G.; Geng, D.; Chen, Y. A modified rabbit model of carotid atherosclerotic plaque suitable for the stroke study and MRI evaluation. Int. J. Neurosci. 2011, 121, 662–669. [Google Scholar]
- Tian, J.; Hu, S.; Sun, Y.; Ban, X.; Yu, H.; Dong, N.; Wu, J.; Yu, B. A novel model of atherosclerosis in rabbits using injury to arterial walls induced by ferric chloride as evaluated by optical coherence tomography as well as intravascular ultrasound and histology. J. Biomed. Biotechnol. 2012, 2012, 121867. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Feng, J.; Zhang, Y.; Sun, Y.; Lu, Y.; Jin, L.; Li, T.; Zhang, X.; Cao, R.; Wua, J. Promotion of atherosclerosis in high cholesterol diet-fed rabbits by immunization with the P277 peptide. Immunol. Lett. 2015, 170, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.G.; Wilmoth, S.K.; Gardin, J.F.; Barnes, R.W.; Sawyer, J.K. Noninvasive assessment of atherosclerosis in nonhuman primates. Adv. Exp. Med. Biol. 1985, 183, 189–195. [Google Scholar] [PubMed]
- Bloch, L.; Hansen, A.Y.; Pedersen, S.F.; Honge, J.L.; Kim, W.Y.; Hansen, E.S. Imaging of carotid artery vessel wall edema using T2-weighted cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2014, 16, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Majdouline, Y.; Ohayon, J.; Keshavarz-Motamed, Z.; Roy Cardinal, M.H.; Garcia, D.; Allard, L.; Lerouge, S.; Arsenault, F.; Soulez, G.; Cloutier, G. Endovascular shear strain elastography for the detection and characterization of the severity of atherosclerotic plaques: In vitro validation and in vivo evaluation. Ultrasound Med. Biol. 2014, 40, 890–903. [Google Scholar] [CrossRef] [PubMed]
- Teräs, M.; Kokki, T.; Durand-Schaefer, N.; Noponen, T.; Pietilä, M.; Kiss, J.; Hoppela, E.; Sipilä, H.T.; Knuuti, J. Dual-gated cardiac PET-clinical feasibility study. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.A.; Hua, N.; Phinikaridou, A.; Killiany, R.; Hamilton, J. Early in vivo discrimination of vulnerable atherosclerotic plaques that disrupt: A serial MRI study. Atherosclerosis 2016, 244, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Deng, L.; Li, D.; Wu, W.; Gong, L.; Li, Y.; Zhang, Q.; Zhang, T.; Zhang, C.; Zhang, Y. Identifying vulnerable atherosclerotic plaque in rabbits using DMSA-USPIO enhanced magnetic resonance imaging to investigate the effect of atorvastatin. PLoS ONE 2015, 10, e0125677. [Google Scholar] [CrossRef] [PubMed]
- Mouse Genome Sequencing Consortium. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–561. [Google Scholar]
- Stary, H.C.; Blankenhorn, D.H.; Chandler, A.B.; Dinsmore, R.E.; Fuster, V.; Glagov, S.; Insull, W.; Rosenfeld, M.E.; Schwartz, C.J.; Wagner, W.D.; et al. A definition of the intima of human arteries and of its atherosclerosis-prone regions. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1992, 85, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C.; Chandler, A.B.; Dinsmore, R.E.; Fuster, V.; Glagov, S.; Insull, W., Jr.; Rosenfeld, M.E.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1995, 92, 1355–1374. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.E.; Polinsky, P.; Virmani, R.; Kauser, K.; Rubanyi, G.; Schwartz, S.M. Advanced atherosclerotic lesions in the innominate artery of the ApoE knockout mouse. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2587–2592. [Google Scholar] [CrossRef]
- Von der Thüsen, J.H.; van Berkel, T.J.; Biessen, E.A. Induction of rapid atherogenesis by perivascular carotid collar placement in apolipoprotein E-deficient and low-density lipoprotein receptor-deficient mice. Circulation 2001, 103, 1164–1170. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.; Ivanov, V.; Ivanova, S.; Kalinovsky, T.; Rath, M.; Niedzwiecki, A. Evolution of angiotensin II-mediated atherosclerosis in ApoE−/− mice. Mol. Med. Rep. 2010, 3, 565–570. [Google Scholar] [PubMed]
- Chen, Y.C.; Bui, A.V.; Diesch, J.; Manasseh, R.; Hausding, C.; Rivera, J.; Haviv, I.; Agrotis, A.; Htun, N.M.; Jowett, J.; et al. A novel mouse model of atherosclerotic plaque instability for drug testing and mech-anistic/therapeutic discoveries using gene and microRNA expression profiling. Circ. Res. 2013, 113, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Chiwata, T.; Aragane, K.; Fujinami, K.; Kojima, K.; Ishibashi, S.; Yamada, N.; Kusunoki, J. Direct effect of an acyl-CoA: Cholesterol acyltransferase inhibitor, F-1394, on atherosclerosis in apolipoprotein E and low density lipoprotein receptor double knockout mice. Br. J. Pharmacol. 2001, 133, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Reardon, C.A.; Blachowicz, L.; Lukens, J.; Nissenbaum, M.; Getz, G.S. Genetic background selectively influences innominate artery atherosclerosis: Immune deficiency as probe. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.; Stefanovic, N.; Pete, J.; Calkin, A.C.; Giunti, S.; Thallas-Bonke, V.; Jandeleit-Dahm, K.A.; Allen, T.J.; Kola, I.; Cooper, M.E.; et al. Lack of the antioxidant enzyme glutathione peroxidase-1 accelerates atherosclerosis in diabetic apolipoprotein E-deficient mice. Circulation 2007, 115, 2178–2187. [Google Scholar] [CrossRef] [PubMed]
- Van der Donckt, C.; van Herck, J.L.; Schrijvers, D.M.; Vanhoutte, G.; Verhoye, M.; Blockx, I.; van der Linden, A.; Bauters, D.; Lijnen, H.R.; Sluimer, J.C.; et al. Elastin fragmentation in atherosclerotic mice leads to intraplaque neovascularization, plaque rupture, myocardial infarction, stroke and sudden death. Eur. Heart J. 2015, 36, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- De Wilde, D.; Trachet, B.; van der Donckt, C.; Vandeghinste, B.; Descamps, B.; Vanhove, C.; de Meyer, G.R.; Segers, P. Vulnerable plaque detection and quantification with gold particle-enhanced computed tomography in atherosclerotic mouse models. Mol. Imaging 2015, 14, 9–19. [Google Scholar]
- Paigen, B.; Plump, A.S.; Rubin, E.M. The mouse as a model for human cardiovascular disease and hyperlipidemia. Curr. Opin. Lipidol. 1994, 5, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Moghadasian, M.H. Experimental atherosclerosis: A historical overview. Life Sci. 2002, 70, 855–865. [Google Scholar] [CrossRef]
- Jawien, J.; Nastalek, P.; Korbut, R. Mouse models of experimental atherosclerosis. J. Physiol. Pharmacol. 2004, 55, 503–517. [Google Scholar] [PubMed]
- Getz, G.S.; Reardon, C.A. Animal Models of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1104–1115. [Google Scholar] [CrossRef] [PubMed]
- Whitman, S.C. A practical approach to using mice in atherosclerosis research. Clin. Biochem. Rev. 2004, 25, 81–93. [Google Scholar] [PubMed]
- Li, X.; Liu, Y.; Zhang, H.; Ren, L.; Li, Q.; Li, N. Animal models for the atherosclerosis research: A review. Protein Cell 2011, 2, 189–201. [Google Scholar]
- Paigen, B.; Ishida, B.Y.; Verstuyft, J.; Winters, R.B.; Albee, D. Atherosclerosis susceptibility differences among progenitors of recombinant inbred strains of mice. Arteriosclerosis 1990, 10, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Andalibi, A.; deBeer, F.C.; Fogelman, A.M.; Lusis, A.J. Genetic control of inflammatory gene induction and NF κB-like transcription factor activation in response to an atherogenic diet in mice. J. Clin. Investig. 1993, 91, 2572–2579. [Google Scholar] [CrossRef] [PubMed]
- Moghadasian, M.H.; McManus, B.M.; Pritchard, P.H.; Frohlich, J.J. “Tall oil”-derived phytosterols reduce atherosclerosis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Föger, B.; Chase, M.; Amar, M.J.; Vaisman, B.L.; Shamburek, R.D.; Paigen, B.; Fruchart-Najib, J.; Paiz, J.A.; Koch, C.A.; Hoyt, R.F.; et al. Cholesteryl ester transfer protein corrects dysfunctional high density lipoproteins and reduces aortic atherosclerosis in lecithin cholesterol acyltransferase transgenic mice. J. Biol. Chem. 1999, 274, 36912–36920. [Google Scholar] [CrossRef] [PubMed]
- Moghadasian, M.H.; McManus, B.M.; Godin, D.V.; Rodrigues, B.; Frohlich, J.J. Proatherogenic and antiatherogenic effects of probucol and phytosterols in apolipoprotein E-deficient mice: Possible mechanisms of action. Circulation 1999, 99, 1733–1739. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Plump, A.S.; Raines, E.W.; Breslow, J.L.; Ross, R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler. Thromb. Vasc. Biol. 1994, 14, 133–140. [Google Scholar] [CrossRef]
- Piedrahita, J.A.; Zhang, S.H.; Hagaman, J.R.; Oliver, P.M.; Maeda, N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc. Natl. Acad. Sci. USA 1992, 89, 4471–4475. [Google Scholar] [CrossRef] [PubMed]
- Plump, A.S.; Smith, J.D.; Hayek, T.; Aalto-Setala, K.; Walsh, A.; Verstuyft, J.G.; Rubin, E.M.; Breslow, J.L. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell 1992, 71, 343–353. [Google Scholar] [CrossRef]
- Meir, K.S.; Leitersdorf, E. Atherosclerosis in the apolipoprotein E-deficient mouse. A decade of progress. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Libby, P.; Schonbeck, U.; Yan, Z.Q. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ. Res. 2002, 91, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Pereira, T.M.C.; Nogueira, B.V.; Lima, L.C.F.; Porto, M.L.; Arruda, J.A.; Vasquez, E.C.; Meyrelles, S.S. Cardiac and vascular changes in elderly atherosclerotic mice: The influence of gender. Lipids Health Dis. 2010, 9, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Heistad, D.D. Unstable coronary artery plaques. N. Engl. J. Med. 2003, 349, 2285–2287. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C. The sequence of cell and matrix changes in atherosclerotic lesions of coronary arteries in the first forty years of life. Eur. Heart J. 1990, 11, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Jonasson, L.; Holm, J.; Skalli, O.; Bondjers, G.; Hansson, G.K. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis 1986, 6, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, S.; Goldstein, J.L.; Brown, M.S.; Herz, J.; Burns, D.K. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J. Clin. Investig. 1994, 93, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Reddick, R.L.; Zhang, S.H.; Maeda, N. Atherosclerosis in mice lacking ApoE: Evaluation of lesional development and progression. Arterioscler. Thromb. Vasc. Biol. 1994, 14, 141–147. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, W.; Zhang, J.; Lu, Y.; Wu, W.; Yan, H.; Wang, Y. Hyperlipidemia and atherosclerotic lesion development in Ldlr-deficient mice on a long-term high-fat diet. PLoS ONE 2012, 7, e35835. [Google Scholar]
- Williams, H.; Johnson, J.L.; Carson, K.G.; Jackson, C.L. Characteristics of intact and ruptured atherosclerotic plaques in brachiocephalic arteries of apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Getz, G.S. Mouse model of unstable atherosclerotic plaque? Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2503–2505. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.P.; Kolodgie, F.D.; Farb, A.; Weber, D.K.; Malcom, G.T.; Smialek, J.; Virmani, R. Healed plaque ruptures and sudden coronary death. Circulation 2001, 103, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Carson, K.; Williams, H.; Karanam, S.; Newby, A.; Angelini, G.; George, S.; Jackson, C. Plaque rupture after short periods of fat feeding in the apolipoprotein E-knockout mouse: Model characterization and effects of pravastatin treatment. Circulation 2005, 111, 1422–1430. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Jackson, C.L. Atherosclerotic plaque rupture in the apolipoprotein E knockout mouse. Atherosclerosis 2001, 154, 399–406. [Google Scholar] [CrossRef]
- Calara, F.; Silvestre, M.; Casanada, F.; Yuan, N.; Napoli, C.; Palinski, W. Spontaneous plaque rupture and secondary thrombosis in apolipoprotein E-deficient and LDL receptor-deficient mice. J. Pathol. 2001, 195, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Van Vlijmen, B.J.; van den Maagdenberg, A.M.; Gijbels, M.J.; van der Boom, H.; HogenEsch, H.; Frants, R.R.; Hofker, M.H.; Havekes, L.M. Diet-induced hyperlipoproteinemia and atherosclerosis in apolipoprotein E3-Leiden transgenic mice. J. Clin. Investig. 1994, 93, 1403–1410. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J. Acute coronary thrombosis—The role of plaque disruption and its initiation and prevention. Eur. Heart J. 1995, 16, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Van der Wal, A.C.; Becker, A.E.; van der Loos, C.M.; Das, P.K. Site of intimal rupture or erosion of throm-bosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation 1994, 89, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Reddick, R.L.; Zhang, S.H.; Maeda, N. Aortic atherosclerotic plaque injury in apolipoprotein E deficient mice. Atherosclerosis 1998, 140, 297–305. [Google Scholar] [CrossRef]
- Bentzon, J.F.; Sondergaard, C.S.; Kassem, M.; Falk, E. Smooth muscle cells healing atherosclerotic plaque disruptions are of local, not blood, origin in apolipoprotein E knockout mice. Circulation 2007, 116, 2053–2061. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Tempel, D.; van Haperen, R.; van der Baan, A.; Grosved, F.; Daemen, M.J.A.P.; Krams, R.; de Crom, R. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation 2006, 113, 2744–2753. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Kuzuya, M.; Nakamura, K.; Cheng, X.W.; Shibata, T.; Sato, K.; Iguchi, A. A simple method of plaque rupture induction in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Aono, J.; Suzuki, J.; Iwai, M.; Horiucki, M.; Nagai, T.; Nishimura, K.; Inoue, K.; Ogimoto, A.; Okayama, H.; Higaki, J. Deletion of the Angiotensin II Type 1a Receptor Prevents Atherosclerotic Plaque Rupture in Apolipoprotein E−/− Mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.X.; Shen, L.H.; Nie, P.; Yuan, W.; Hu, L.H.; Li, D.D.; Chen, X.J.; Zhang, X.K.; He, B. Endogenous renovascular hypertension combined with low shear stress induces plaque rupture in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2372–2379. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.C.F.; Porto, M.L.; Campagnaro, B.P.; Tonini, C.L.; Nogueira, B.V.; Pereira, T.M.C.; Vasquez, E.C.; Meyrelles, S.S. Mononuclear cell therapy reverts cuff-induced thrombosis in apolipoprotein E-deficient mice. Lipids Health Dis. 2012, 11, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.M.; Galis, Z.S.; Rosenfeld, M.E.; Falk, E. Plaque rupture in humans and mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Falk, E.; Schwartz, S.M.; Galis, Z.S.; Rosenfeld, M.E. Neointimal cracks (plaque rupture?) and thrombosis in wrapped arteries without flow. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Von der Thusen, J.H.; van Vlijmen, B.J.M.; Hoeben, R.C.; Kockx, M.M.; Havekes, L.M.; van Berkel, T.J.C.; Biessen, E.A.L. Induction of atherosclerotic plaque rupture in apolipoprotein E−/− mice after adenovirus-mediated transfer of p53. Circulation 2002, 105, 2064–2070. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Gojova, A.; Marchiol-Fournigault, C.; Esposito, B.; Kamate, C.; Merval, R.; Fradelizi, D.; Tedgui, A. Inhibition of transforming growth factor- signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ. Res. 2001, 89, 930–934. [Google Scholar] [CrossRef] [PubMed]
- Lutgens, E.; Gijbels, M.; Smook, M.; Heeringa, P.; Gotwals, P.; Koteliansky, V.E.; Daemen, M.J. Transforming growth factor-β mediates balance between inflammation and fibrosis during plaque progression. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Lutgens, E.; de Frutos, P.G.; Apericio, C.; Moons, L.; Daemen, M.; Collen, D.; Carmeliet, P. Gas6−/−/ApoE−/− mice develop a collagen-rich, disorganized plaque phenotype, prone to intraplaque hemorrhage. Circulation 2000, 102, 38. [Google Scholar]
- Angelillo-Scherrer, A.; de Frutos, P.; Aparicio, C.; Melis, E.; Savi, P.; Lupu, F.; Arnout, J.; Dewerchin, M.; Hoylaerts, M.; Herbert, J.; et al. Deficiency or inhibition of Gas6 causes platelet dys-function and protects mice against thrombosis. Nat. Med. 2001, 7, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Braun, A.; Trigatti, B.L.; Post, M.J.; Sato, K.; Simons, M.; Edelberg, J.M.; Rosenberg, R.D.; Schrenzel, M.; Krieger, M. Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ. Res. 2002, 90, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Gough, P.J.; Gomez, I.G.; Wille, P.T.; Raines, E.W. Macrophage expression of active MMP-9 induces acute plaque disruption in ApoE-deficient mice. J. Clin. Investig. 2006, 116, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Gao, Q.; Zhu, F.; Guo, C.; Wang, Q.; Gao, F.; Zhang, L. Th17 cells and IL-17 are involved in the disruption of vulnerable plaques triggered by short-term combination stimulation in apolipoprotein E-knockout mice. Cell. Mol. Immunol. 2013, 10, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Pellegrin, M.; Mazzolai, L. Angiotensin II as an inducer of atherosclerosis: Evidence from mouse studies. J. Clin. Exp. Cardiol. 2013. [Google Scholar] [CrossRef]
- Billet, S.; Aguilar, F.; Baudry, C.; Clauser, E. Role of angiotensin II AT1 receptor activation in cardiovascular diseases. Kidney Int. 2008, 74, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Lemarié, C.A.; Schiffrin, E.L. The angiotensin II type 2 receptor in cardiovascular disease. J. Renin Angiotensin Aldosterone Syst. 2010, 11, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, A.; Manning, M.W.; Cassis, L.A. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J. Clin. Investig. 2000, 105, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Weiss, D.; Kools, J.J.; Taylor, W.R. Angiotensin II-induced hypertension accelerates the development of atherosclerosis in ApoE-deficient mice. Circulation 2001, 103, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Mazzolai, L.; Duchosal, M.A.; Korber, M.; Bouzourene, K.; Aubert, J.F.; Hao, H.; Vallet, V.; Brunner, H.R.; Nussberger, J.; Gabbiani, G.; et al. Endogenous angiotensin II induces atherosclerotic plaque vulnerability and elicits a Th1 response in ApoE−/− mice. Hypertension 2004, 44, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Kitamoto, S.; Ishibashi, M.; Usui, M.; Inoue, S.; Hiasa, K.; Zhao, Q.; Nishida, K.; Takeshita, A.; Egashira, K. Monocyte chemoattractant protein-1 is an essential in ammatory mediator in angiotensin II-induced progression of established atherosclerosis in hypercholesterolemic mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Simon, G.; Abraham, G. Angiotensin II administration as an experimental model of hypertension. In Hypertension: Pathophysiology, Diagnosis, and Management, 2nd ed.; Laragh, J.H., Brenner, B.M., Eds.; Raven Press Ltd.: New York, NY, USA, 1995; pp. 1423–1435. [Google Scholar]
- Candido, R.; Jandeleit-Dahm, K.A.; Cao, Z.; Nesteroff, S.P.; Burns, W.C.; Twigg, S.M.; Dilley, R.J.; Cooper, M.E.; Allen, T.J. Prevention of accelerated atherosclerosis by angiotensin-converting enzyme inhibition in diabetic apolipoprotein E-deficient mice. Circulation 2002, 106, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Howard, B.V.; Rodriguez, B.L.; Bennett, P.H.; Harris, M.I.; Hamman, R.; Kuller, L.H.; Pearson, T.A.; Wylie-Rosett, J. Prevention conference VI: Diabetes and cardiovascular disease: Writing Group I: Epidemiology. Circulation 2002, 105, e132–e137. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.A.; Creager, M.A.; Libby, P. Diabetes and atherosclerosis: Epidemiology, pathophysiology, and management. JAMA 2002, 287, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, S.; Oinuma, T.; Yamada, T. A comparative study of cultured smooth muscle cell proliferation and injury, utilizing glycated low density lipoproteins with slight oxidation, auto-oxidation, or extensive oxidation. J. Atheroscler. Thromb. 2000, 7, 132–137. [Google Scholar] [CrossRef]
- Schreyer, S.A.; Wilson, D.L.; Leboeuf, R.C. C57BL/6 mice fed high fat diets as models for diabetes-accelerated atherosclerosis. Atherosclerosis 1998, 136, 17–24. [Google Scholar] [CrossRef]
- King, V.L.; Hatch, N.W.; Chan, H.W.; deBeer, M.C.; deBeer, F.C.; Tannock, L.R. A murine model of obesity with accelerated atherosclerosis. Obesity 2010, 18, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Cannizzo, B.; Lujàn, A.; Estrella, N.; Lembo, C.; Cruzado, M.; Castro, C. Insulin resistance promotes early atherosclerosis via increased proin ammatory proteins and oxidative stress in fructose-fed ApoE-KO mice. Exp. Diabetes Res. 2012, 2012, 941304. [Google Scholar] [CrossRef] [PubMed]
- Merat, S.; Casanada, F.; Sutphin, M.; Palinski, W.; Reaven, P.D. Western-type diets induce insulin resistance and hyperinsulinemia in LDL receptor-deficient mice but do not increase aortic atherosclerosis compared with normoinsulinemic mice in which similar plasma cholesterol levels are achieved by a fructose-rich diet. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Schreyer, S.A.; Lystig, T.C.; Vick, C.M.; LeBoeuf, R.C. Mice deficient in apolipoprotein E but not LDL receptors are resistant to accelerated atherosclerosis associated with obesity. Atherosclerosis 2003, 171, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Kunjathoor, V.V.; Wilson, D.L.; LeBoeuf, R.C. Increased atherosclerosis in streptozotocin-induced diabetic mice. J. Clin. Investig. 1996, 97, 1767–1773. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Raman, K.G.; Lee, K.J.; Lu, Y.; Ferran, L.J., Jr.; Chow, W.S.; Stern, D.; Schmidt, A.M. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat. Med. 1998, 4, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Calkin, A.C.; Forbes, J.M.; Smith, C.M.; Lassila, M.; Cooper, M.E.; Jandeleit-Dahm, K.A.; Allen, T.J. Rosiglitazone attenuates atherosclerosis in a model of insulin insufficiency independent of its metabolic effects. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1903–9. [Google Scholar] [CrossRef] [PubMed]
- Zuccollo, A.; Shi, C.; Mastroianni, R.; Maitland-Toolan, K.A.; Weisbrod, R.M.; Zang, M.; Xu, S.; Jiang, B.; Oliver-Krasinski, J.M.; Cayatte, A.J.; et al. The thromboxane A2 receptor antagonist S18886 prevents enhanced atherogenesis caused by diabetes mellitus. Circulation 2005, 112, 3001–3008. [Google Scholar] [CrossRef] [PubMed]
- Veerman, K.J.; Venegas-Pino, D.E.; Shi, Y.; Khan, M.I.; Gerstein, H.C.; Werstuck, G.H. Hyperglycaemia is associated with impaired vasa vasorum neovascularization and accelerated atherosclerosis in apolipoprotein-E deficient mice. Atherosclerosis 2013, 227, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Vikramadithyan, R.K.; Hu, Y.; Noh, H.L.; Liang, C.P.; Hallam, K.; Tall, A.R.; Ramasamy, R.; Goldberg, I.J. Human aldose reductase expression accelerates diabetic atherosclerosis in transgenic mice. J. Clin. Investig. 2005, 115, 2434–2443. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.Y.; Ma, Z.; Segar, L. Spontaneously diabetic Ins2+/Akita:ApoE-deficient mice exhibit exaggerated hyperc-holesterolemia and atherosclerosis. Am. J. Physiol. Endocrinol. Metabol. 2011, 301, E145–E154. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Pridgen, B.; King, N.; Xu, J.; Breslow, J.L. Hyperglycemic Ins2AkitaLdlr−/− mice show severely elevated lipid levels and increased atherosclerosis: A model of type 1 diabetic macrovascular disease. J. Lipid Res. 2011, 52, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Engelbertsen, D.; To, F.; Dunér, P.; Kotova, O.; Söderberg, I.; Alm, R.; Gomez, M.F.; Nilsson, J.; Bengtsson, E. Increased inflammation in atherosclerotic lesions of diabetic Akita-LDLr−/− mice compared to nondiabetic LDLr−/−mice. Exp. Diabetes Res. 2012, 2012, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Shang, Y.Y.; Zhang, S.; Zhong, M.; Wang, X.P.; Deng, J.T.; Pan, J.; Zhang, Y.; Zhang, W. Silence of TRIB3 suppresses atherosclerosis and stabilizes plaques in diabetic ApoE−/−/LDL receptor−/− mice. Diabetes 2012, 61, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.K.; Wu, T.J.; Chin, J.; Mitnaul, L.J.; Hernandez, M.; Cai, T.Q.; Ren, N.; Waters, M.G.; Wright, S.D.; Cheng, K. Increased hypercholesterolemia and atherosclerosis in mice lacking both ApoE and leptin receptor. Atherosclerosis 2005, 181, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Wendt, T.; Harja, E.; Bucciarelli, L.; Qu, W.; Lu, Y.; Rong, L.L.; Jenkins, D.G.; Stein, G.; Schmidt, A.M.; Yan, S.F. RAGE modulates vascular inflammation and atherosclerosis in a murine model of type 2 diabetes. Atherosclerosis 2006, 185, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Vliegenthart, R.; Oudkerk, M.; Hofman, A.; Oei, H.H.; van Dijck, W.; van Rooij, F.J.; Witteman, J.C. Coronary calcification improves cardiovascular risk prediction in the elderly. Circulation 2005, 112, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Janssen, C.H.; Kuijpers, D.; Vliegenthart, R.; Overbosch, J.; van Dijkman, P.R.; Zijlstra, F.; Oudkerk, M. Coronary artery calcification score by multislice computed tomography predicts the outcome of dobutamine cardiovascular magnetic resonance imaging. Eur. Radiol. 2005, 15, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, F.; Sakakura, K.; Yahagi, K.; Joner, M.; Virmani, R. Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler. Thromb. Vasc. Biol. 2014, 34, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.C.; Tintut, Y.; Lyman, A.; Mack, W.; Demer, L.L.; Hsiai, T.K. Mechanical response of a calcified plaque model to fluid shear force. Ann. Biomed. Eng. 2006, 34, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, N.; Kelly-Arnold, A.; Vengrenyuk, Y.; Laudier, D.; Fallon, J.T.; Virmani, R.; Cardoso, L.; Weinbaum, S. A mechanistic analysis of the role of microcalcifications in atherosclerotic plaque stability: Potential implications for plaque rupture. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Kelly-Arnold, A.; Maldonado, N.; Laudier, D.; Aikawa, E.; Cardoso, L.; Weinbaum, S. Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proc. Natl. Acad. Sci. USA 2013, 110, 10741–10746. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.H.; Xie, P.Z.; Fishbein, M.C.; Kreuzer, J.; Drake, T.A.; Demer, L.L.; Lusis, A.J. Pathology of atheromatous lesions in inbred and genetically engineered mice: Genetic determination of arterial calcification. Arterioscler. Thromb. 1994, 14, 1480–1497. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.H.; Fishbein, M.C.; Demer, L.L.; Lusis, A.J. Genetic determination of cartilaginous metaplasia in mouse aorta. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 2265–2272. [Google Scholar] [CrossRef] [PubMed]
- Rattazzi, M.; Bennett, B.J.; Bea, F.; Kirk, E.A.; Ricks, J.L.; Speer, M.; Schwartz, S.M.; Giachelli, C.M.; Rosenfeld, M.E. Calcification of advanced atherosclerotic lesions in the innominate arteries of ApoE-deficient mice: Potential role of chondrocyte-like cells. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1420–1425. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Rittling, S.R.; Okamoto, H.; Inobe, M.; Jia, N.; Shimizu, T.; Akino, M.; Sugawara, T.; Morimoto, J.; Kimura, C.; et al. Osteopontin deficiency attenuates atherosclerosis in female apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Speer, M.Y.; Chien, Y.C.; Quan, M.; Yang, H.Y.; Vali, H.; McKee, M.D.; Giachelli, C.M. Smooth muscle cells deficient in osteopontin have enhanced susceptibility to calcification in vitro. Cardiovasc. Res. 2005, 66, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Debernardi, N.; Roijers, R.B.; Krams, R.; de Crom, R.; Mutsaers, P.H. A.; van der Vusse, G.J. Microcalcifications in atherosclerotic lesion of apolipoprotein E-deficient mouse. Int. J. Exp. Pathol. 2010, 91, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Towler, D.A.; Bidder, M.; Latifi, T.; Coleman, T.; Semenkovich, C.F. Diet-induced diabetes activates an osteogenic gene regulatory program in the aortas of low density lipoprotein receptor-deficient mice. J. Biol. Chem. 1998, 273, 30427–30434. [Google Scholar] [CrossRef] [PubMed]
- Browner, W.S.; Seeley, D.G.; Vogt, T.M.; Cummings, S.R. Non-trauma mortality in elderly women with low bone mineral density. Study of Osteoporotic Fractures Research Group. Lancet 1991, 338, 355–358. [Google Scholar] [CrossRef]
- Uyama, O.; Yoshimoto, Y.; Yamamoto, Y.; Kawai, A. Bone changes and carotid atherosclerosis in postmenopausal women. Stroke 1997, 28, 1730–1732. [Google Scholar] [CrossRef] [PubMed]
- Demer, L.L. Vascular calcification and osteoporosis: Inflammatory responses to oxidized lipids. Int. J. Epidemiol. 2002, 31, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, E.; Nahrendorf, M.; Figueiredo, J.L.; Swirski, F.K.; Shtatland, T.; Kohler, R.H.; Jaffer, F.A.; Aikawa, M.; Weissleder, R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation 2007, 116, 2841–2850. [Google Scholar] [CrossRef] [PubMed]
- Hjortnaes, J.; Butcher, J.; Figueiredo, J.L.; Riccio, M.; Kohler, R.H.; Kozloff, K.M.; Weissleder, R.; Aikawa, E. Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: A role for inflammation. Eur. Heart J. 2010, 31, 1975–1984. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yan, M. GW26-e5327 micro-calcification regression in ApoE−/− mice spontaneous atherosclerotic plaque by simvastatin on inhibition of endoplasmic reticulum mediated apoptosis. J. Am. Coll. Cardiol. 2015, 66. [Google Scholar] [CrossRef]
- Van Herck, J.L.; de Meyer, G.R.; Martinet, W.; van Hove, C.E.; Foubert, K.; Theunis, M.H.; Apers, S.; Bult, H.; Vrints, C.J.; Herman, A.G. Impaired fibrillin-1 function promotes features of plaque instability in apolipoprotein E-deficient mice. Circulation 2009, 120, 2478–2487. [Google Scholar] [CrossRef] [PubMed]
- Daeichin, V.; Sluimer, J.C.; van der Heiden, K.; Skachkov, I.; Kooiman, K.; Janssen, A.; Janssen, B.; Bosch, J.G.; de Jong, N.; Daemen, M.J.A.P.; et al. Live observation of atherosclerotic plaque disruption in apolipoprotein E-Deficient mouse. Ultrasound Int. Open 2015, 1, E67–E71. [Google Scholar] [CrossRef]
- Roth, L.; van Dam, D.; van der Donckt, C.; Schrijvers, D.M.; Lemmens, K.; van Brussel, I.; de Deyn, P.P.; Martinet, W.; de Meyer, G.R. Impaired gait pattern as a sensitive tool to assess hypoxic brain damage in a novel mouse model of atherosclerotic plaque rupture. Physiol. Behav. 2015, 139, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Roche-Molina, M.; Sanz-Rosa, D.; Cruz, F.M.; García-Prieto, J.; López, S.; Abia, R.; Muriana, F.J.G.; Fuster, V.; Ibáñez, B.; Bernalet, J.A. Induction of sustained hypercholesterolemia by single adeno-associated virus-mediated gene transfer of mutant hPCSK9. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Van der Heiden, K.; Hoogendoorn, A.; Daemen, M.J.; Gijsen, F.J.H. Animal models for plaque rupture: A biomechanical assessment. Thromb. Haemost. 2015, 115, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ni, M.; Ma, L.; Yang, J.; Wang, L.; Liu, F.; Dong, M.; Yang, X.; Zhang, M.; Lu, H.; et al. Targeting blood thrombogenicity precipitates atherothrombotic events in a mouse model of plaque destabilization. Sci. Rep. 2015, 5, 10225. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Wang, Y.; Zang, M.; Zang, P.F.; Ding, S.F.; Liu, C.X. Atherosclerotic plaque disruption induced by stress and lipopolysaccharide in apolipoprotein E knockout mice. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1598–H1606. [Google Scholar] [CrossRef] [PubMed]
- Steinl, D.C.; Kaufmann, B.A. Ultrasound Imaging for Risk Assessment in Atherosclerosis. Int. J. Mol. Sci. 2015, 16, 9749–9769. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis: An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Dong, Z.M.; Wagner, D.D. Leukocyte-endothelium adhesion molecules in atherosclerosis. J. Lab. Clin. Med. 1998, 132, 369–375. [Google Scholar] [CrossRef]
- Wick, G.; Grundtman, C. Inflammation and Atherosclerosis; SpringerWien: New York, NY, USA, 2012; pp. 121–122. [Google Scholar]
- Davies, M.J.; Gordon, J.L.; Gearing, A.J.; Pigott, R.; Katz, D.; Kyriakopoulos, A. The expression of the adhesion molecules ICAM-1, VCAM-1, PECAM, and E-selectin in human atherosclerosis. J. Pathol. 1993, 171, 223–229. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.D.; Allen, M.D.; McDonald, T.O.; Chait, A.; Harlan, J.M.; Fishbein, D.; McCarty, J.; Ferguson, M.; Hudkins, K.; Benjamin, C.D.; et al. Vascular cell adhesion molecule-1 is expressed in human coronary atherosclerotic plaques. Implications for the mode of progression of advanced coronary atherosclerosis. J. Clin. Investig. 1993, 92, 945–951. [Google Scholar] [CrossRef] [PubMed]
- George, S.J.; Johnson, J. Atherosclerosis: Molecular and Cellular Mechanisms; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 47–50. [Google Scholar]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- Poston, R.N.; Haskard, D.O.; Coucher, J.R.; Gall, N.P.; Johson-Tidey, R.R. Expression of intercellular adhesion molecule-1 in atherosclerotic plaques. Am. J. Pathol. 1992, 140, 665–673. [Google Scholar] [PubMed]
- Wood, K.M.; Cadogan, M.D.; Ramshaw, A.L.; Parus, D.V. The distribution of adhesion molecules in human atherosclerosis. Histopathology 1993, 22, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Raines, E.W.; Plump, A.S.; Breslow, J.L.; Ross, R. Upregulation of VCAM-1 and ICAM-1 at Atherosclerosis-Prone Sites on the Endothelium in the ApoE-Deficient Mouse. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Henninger, D.D.; Panes, J.; Eppihimer, M.; Russel, J.; Gerritsen, M.; Anderson, D.C.; Granger, D.N. Cytokine-induced VCAM-1, and ICAM-1 expression in different organs of the mouse. J. Immunol. 1997, 158, 1825–1832. [Google Scholar] [PubMed]
- Johnson-Tidey, R.R.; McGregor, J.L.; Taylor, P.R.; Poston, R.N. Increase in the adhesion molecule P-selectin in endothelium overlying atherosclerotic plaques. Coexpression with intercellular adhesion molecule-1. Am. J. Pathol. 1994, 144, 952–961. [Google Scholar] [PubMed]
- Dong, Z.M.; Brown, A.A.; Wagner, D.D. Prominent role of P-selectin in the development of advanced atherosclerosis in ApoE-deficient mice. Circulation 2000, 101, 2290–2295. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.L.; Huo, Y.; Jung, U.; Ghosh, S.; Manka, D.R.; Sarembock, I.J.; Ley, K. Direct demonstration of P-selectin- and VCAM-1-dependent mono-nuclear cell rolling in early atherosclerotic lesions of apolipoprotein E-deficient mice. Circ. Res. 1999, 84, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Burger, P.C.; Wagner, D.D. Platelet P-selectin facilitates atherosclerotic lesion development. Platelet P-selectin facilitates atherosclerotic lesion development. Blood 2003, 101, 2661–2666. [Google Scholar] [CrossRef] [PubMed]
- Manka, D.; Forlow, S.B.; Sanders, J.M.; Hurwitz, D.; Bennett, D.K.; Green, S.A.; Ley, K.; Sarembock, I.J. Critical role of platelet P-selectin in the response to arterial injury in apolipoprotein-E–deficient mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1124–1129. [Google Scholar] [CrossRef] [PubMed]
- Narula, J.; Garg, P.; Achenbach, S.; Motoyama, S.; Virmani, R.; Strauss, H.W. Arithmetic of vulnerable plaques for noninvasive imaging. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, S2–S10. [Google Scholar] [CrossRef] [PubMed]
- Jaffer, F.A.; Libby, P.; Weissleder, R. Molecular and cellular imaging of atherosclerosis: Emerging applications. J. Am. Coll. Cardiol. 2006, 47, 1328–1338. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.H.; Shah, P.K.; Faure, E.; Equils, O.; Thomas, L.; Fishbein, M.C.; Luthringer, D.; Xu, X.P.; Rajavashisth, T.B.; Yano, J.; et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation 2001, 104, 3103–3108. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Lòpez, W.; Xia, W.; Varghese, B.; Low, P.S. Imaging of atherosclerosis in apoliprotein E knockout mice: Targeting of a folate-conjugated radiopharmaceutical to activated macrophages. J. Nucl. Med. 2010, 51, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Beck, K.; Rancic, Z.; Müller, C.; Fischer, C.R.; Betzel, T.; Kaufmann, P.A.; Schibli, R.; Krämer, S.D.; Ametamey, S.M. Imaging atherosclerotic plaque inflammation via folate receptor targeting using a novel 18F-folate radiotracer. Mol. Imaging 2014, 13, 1–11. [Google Scholar] [PubMed]
- Chen, Y.X.; Nakashima, Y.; Tanaka, K.; Shiraishi, S.; Nakagawa, K.; Sueishi, K. Immunohistochemical expression of vascular endothelial growth factor/vascular permeability factor in atherosclerotic intimas of human coronary arteries. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Celletti, F.L.; Hilfiker, P.R.; Ghafouri, P.; Dake, M.D. Effect of human recombinant vascular endothelial growth factor165 on progression of atherosclerotic plaque. J. Am. Coll. Cardiol. 2001, 37, 2126–2130. [Google Scholar] [CrossRef]
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Finn, A.V.; Gold, H.K.; Tulenko, T.N.; Wrenn, S.P.; Narula, J. Atherosclerotic plaque progression and vulnerability to rupture: Angiogenesis as a source of intraplaque hemorrhage. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2054–2061. [Google Scholar] [CrossRef] [PubMed]
- Holm, P.W.; Slart, R.H.; Zeebregts, C.J.; Hillebrands, J.L.; Tio, R.A. Atherosclerotic plaque development and instability: A dual role for VEGF. Ann. Med. 2009, 41, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Olson, F.J.; Strömbert, S.; Hjelmgren, O.; Kjelldahl, J.; Fagerberg, B.; Bergström, G.M. Increased vascularization of shoulder regions of carotid atherosclerotic plaques from patients with diabetes. J. Vasc. Surg. 2011, 54, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Johansson, F.; Kramer, F.; Barnhart, S.; Kanter, J.E.; Vaisar, T.; Merrill, R.D.; Merrill, R.D.; Geng, L.; Oka, K.; Chan, L.; et al. Type 1 diabetes promotes disruption of advanced atherosclerotic lesions in LDL receptor-deficient mice. Proc. Natl. Acad. Sci. USA 2008, 105, 2082–2087. [Google Scholar] [CrossRef] [PubMed]
- Parathath, S.; Mick, S.L.; Feig, J.E.; Joaquin, V.; Grauer, L.; Habiel, D.M.; Gassmann, M.; Gardner, L.B.; Fisher, E.A. Hypoxia is present in murine atherosclerotic plaques and has multiple effects on macrophage lipid metabolism. Circ. Res. 2011, 109, 1141–1152. [Google Scholar] [PubMed]
- Zamir, M.; Silver, M.D. Vasculature in the walls of human coronary arteries. Arch. Pathol. Lab. Med. 1985, 109, 659–662. [Google Scholar] [PubMed]
- Kamat, B.R.; Galli, S.J.; Barger, A.C.; Lainey, L.L.; Silverman, K.J. Neovascularization and coronary atherosclerotic plaque: Cinematographic localization and quantitative histologic analysis. Hum. Pathol. 1987, 18, 1036–1042. [Google Scholar] [PubMed]
- Zhang, Y.; Cliff, W.J.; Schoefl, G.I.; Higgins, G. Immunohistochemical study of intimal microvessels in coronary atherosclerosis. Am. J. Pathol. 1993, 143, 164–172. [Google Scholar] [PubMed]
- Sueishi, K.; Yonemitsu, Y.; Nakagawa, K.; Kaneda, Y.; Kumamoto, M.; Nakashima, Y. Atherosclerosis and angiogenesis: Its pathophysiological significance in humans as well as in an animal model induced by the gene transfer of vascular endothelial growth factor. Ann. N. Y. Acad. Sci. 1997, 811, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Moulton, K.S.; Heller, E.; Konerding, M.A.; Flynn, E.; Palinski, W.; Folkman, J. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E–deficient mice. Circulation 1999, 99, 1726–1732. [Google Scholar] [CrossRef] [PubMed]
- Moulton, K.S.; Vakili, K.; Zurakowski, D.; Soliman, M.; Butterfield, C.; Sylvin, E.; Lo, K.M.; Gillies, S.; Javaherian, K.; Folkman, J. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc. Natl. Acad. Sci. USA 2003, 8, 4736–4741. [Google Scholar] [CrossRef] [PubMed]
- Tekabe, Y.; Li, Q.; Luma, J.; Weisenberger, D.; Sedlar, M.; Harja, E.; Narula, J.; Johnson, L.L. Noninvasive monitoring the biology of atherosclerotic plaque development with radiolabeled annexin V and matrix metalloproteinase inhibitor in spontaneous atherosclerotic mice. J. Nucl. Cardiol. 2010, 17, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Waldeck, J.; Häger, F.; Höltke, C.; Lanckohr, C.; von Wallbrunn, A.; Torsello, G.; Heindel, W.; Theilmeier, G.; Schäfers, M.; Bremer, C. Fluorescence reflectance imaging of macrophage-rich atherosclerotic plaques using an αvβ3 integrin-targeted fluorochrome. J. Nucl. Med. 2008, 49, 1845–1851. [Google Scholar] [CrossRef] [PubMed]
- Winter, P.M.; Caruthers, S.D.; Allen, J.S.; Cai, K.; Williams, T.A.; Lanza, G.M.; Wickline, S.A. Molecular imaging of angiogenic therapy in peripheral vascular disease with αvβ3-integrin-targeted nanoparticles. Magn. Reson. Med. 2010, 64, 369–376. [Google Scholar] [PubMed]
- Hoshiga, M.; Alpers, C.E.; Smith, L.L.; Giachelli, C.M.; Schwartz, S.M. αvβ3 integrin expression in normal and atherosclerotic artery. Circ. Res. 1995, 77, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Antonov, A.S.; Kolodgie, F.D.; Munn, D.H.; Gerrity, R.G. Regulation of macrophage foam cell formation by αvβ3 integrin—Potential role in human atherosclerosis. Am. J. Pathol. 2004, 165, 247–258. [Google Scholar] [CrossRef]
- Beer, A.J.; Schwaiger, M. Imaging of integrin αvβ3 expression. Cancer Metastasis Rev. 2008, 27, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Dufourcq, P.; Louis, H.; Moreau, C.; Daret, D.; Boisseau, M.R.; Lamaziere, J.M.D.; Bonnet, J. Vitronectin expression and interaction with receptors in smooth muscle cells from human atheromatous plaque. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Macrophage apoptosis in atherosclerosis: Consequences on plaque progression and the role of endoplasmic reticulum stress. Antioxid. Redox Signal. 2009, 11, 2333–2339. [Google Scholar] [CrossRef] [PubMed]
- Seimon, T.; Tabas, I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J. Lipid Res. 2009, 50, S382–S387. [Google Scholar] [CrossRef] [PubMed]
- Van Engeland, M.; Nieland, L.J.; Ramaekers, F.C.; Schutte, B.; Reutelingsperger, C.P. Annexin V-affinity assay: A review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry 1998, 31, 1–9. [Google Scholar] [CrossRef]
- Koopman, G.; Reutelingsperger, C.P.; Kuijten, G.A.; Keehnen, R.M.; Pals, S.T.; van Oers, M.H. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 1994, 84, 1415–1420. [Google Scholar] [PubMed]
- Laufer, E.M.; Reutelingsperger, C.P.; Narula, J.; Hofstra, L. Annexin A5: An imaging biomarker of cardiovascular risk. Basic Res. Cardiol. 2008, 103, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Isobe, S.; Tsimikas, S.; Zhou, J.; Fujimoto, S.; Sarai, M.; Branks, M.J.; Fujimoto, A.; Hofstra, L.; Reutelingsperger, C.P.; Murohara, T.; et al. Noninvasive imaging of atherosclerotic lesions in apolipoprotein E-deficient and low-density-lipoprotein receptor- deficient mice with annexin A5. J. Nucl. Med. 2006, 47, 1497–1505. [Google Scholar] [PubMed]
- Van Tilborg, G.A.; Vucic, E.; Strijkers, G.J.; Cormode, D.P.; Mani, V.; Skajaa, T.; Reutelingsperger, C.P.; Fayad, Z.A.; Mulder, W.J.; Nicolay, K. Annexin A5-functionalized bimodal nanoparticles for MRI and fluorescence imaging of atherosclerotic plaques. Bioconjug. Chem. 2010, 21, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Laufer, E.M.; Winkens, H.M.; Corsten, M.F.; Reutelingsperger, C.P.; Narula, J.; Hofstra, L. PET and SPECT imaging of apoptosis in vulnerable atherosclerotic plaques with radiolabeled Annexin A5. Q. J. Nucl. Med. Mol. Imaging 2009, 53, 26–34. [Google Scholar] [PubMed]
- Kietselaer, B.L.; Reutelingsperger, C.P.; Heidendal, G.A.; Daemen, M.J.; Mess, W.H.; Hofstra, L.; Narula, J. Noninvasive detection of plaque instability with use of radiolabeled annexin A5 in patients with carotid-artery atherosclerosis. N. Engl. J. Med. 2004, 350, 1472–1473. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Shen, S.; Wang, J.; Wang, H.; Li, M.; Liu, Y.; Hou, F.; Liao, Y.; Bin, J. Detection of high-risk atherosclerotic plaques with ultrasound molecular imaging of glycoprotein IIb/IIIa receptor on activated platelets. Theranostics 2015, 5, 418–430. [Google Scholar] [CrossRef] [PubMed]
- Metzger, K.; Vogel, S.; Chatterjee, M.; Borst, O.; Seizer, P.; Schönberger, T.; Geisler, T.; Lang, F.; Langer, H.; Rheinlaender, J.; et al. High-frequency ultrasound-guided disruption of glycoprotein VI-targeted microbubbles targets atheroprogressison in mice. Biomaterials 2015, 36, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Haage, P.; Wiethoff, A.J.; Gunther, R.W.; Bucker, A.; Tacke, J.; Spuentrup, E. Molecular magnetic resonance imaging of deep vein thrombosis using a fibrin-targeted contrast agent: A feasibility study. Investig. Radiol. 2009, 44, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Vymazal, J.; Spuentrup, E.; Cardenas-Molina, G.; Wiethoff, A.J.; Hartmann, M.G.; Caravan, P.; Parsons, E.C., Jr. Thrombus imaging with fibrin-specific gadolinium-based MR contrast agent EP-2104R: Results of a phase II clinical study of feasibility. Investig. Radiol. 2009, 44, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Jaffer, F.A.; Tung, C.H.; Wykrzykowska, J.J.; Ho, N.H.; Houng, A.K.; Reed, G.L.; Weissleder, R. Molecular imaging of factor XIIIa activity in thrombosis using a novel, near-infrared fluorescent contrast agent that covalently links to thrombi. Circulation 2004, 110, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Miserus, R.J.; Herias, M.V.; Prinzen, L.; Lobbes, M.B.; van Suylen, R.J.; Dirksen, A.; Hackeng, T.M.; Heemskerk, J.W.; van Engelshoven, J.M.; Daemen, M.J.; et al. Molecular MRI of early thrombus formation using a bimodal α2-antiplasmin-based contrast agent. JACC Cardiovasc. Imaging 2009, 2, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; Mancini, M.; Gargiulo, S.; Gramanzini, M.; Claudio, P.P.; Brunetti, A.; Salvatore, M. Ultrasound biomicroscopy in small animal research: Applications in molecular and preclinical imaging. J. Biomed. Biotechnol. 2012, 2012, 519238. [Google Scholar] [CrossRef] [PubMed]
- Mancini, M.; Greco, A.; Salvatore, G.; Liuzzi, R.; di Maro, G.; Vergara, E.; Chiappetta, G.; Pasquinelli, R.; Brunetti, A.; Salvatore, M. Imaging of thyroid tumor angiogenesis with microbubbles targeted to vascular endothelial growth factor receptor type 2 in mice. BMC Med. Imaging 2013, 13, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Bekeredjian, R.; Chen, S.; Frenkel, P.A.; Grayburn, P.A.; Shohet, R.V. Ultrasound-targeted microbubble destruction can repeatedly direct highly specific plasmid expression to the heart. Circulation 2003, 108, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Bekeredjian, R.; Chen, S.; Grayburn, P.A.; Shohet, R.V. Augmentation of cardiac protein delivery using ultrasound targeted microbubble destruction. Ultrasound Med. Biol. 2005, 31, 687–691. [Google Scholar] [CrossRef] [PubMed]
- Inaba, Y.; Linder, J.R. Molecular Imaging of disease with targeted contrast ultrasound imaging. Transl. Res. 2012, 159, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Khanicheh, E.; Mitterhuber, M.; Xu, L.; Haeuselmann, S.P.; Kuster, G.M.; Kaufmann, B.A. Noninvasive ultrasound molecular imaging of the effect of statins on endothelial inflammatory phenotype in early atherosclerosis. PLoS ONE 2013, 8, e58761. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Davidson, B.P.; Yue, Q.; Belcik, T.; Xie, A.; Inaba, Y.; McCarty, O.J.; Tormoen, G.W.; Zhao, Y.; Ruggeri, Z.M.; et al. Molecular imaging of inflammation and platelet adhesion in advanced atherosclerosis effects of antioxidant therapy with NADPH oxidase inhibition. Circ. Cardiovasc. Imaging 2013, 6, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hagemeyer, C.E.; Hohmann, J.D.; Leitner, E.; Armstrong, P.C.; Jia, F.; Olschewski, M.; Needles, A.; Peter, K.; Ahrens, I. Novel single-chain antibody-targeted microbubbles for molecular ultrasound imaging of thrombosis: Validation of a unique noninvasive method for rapid and sensitive detection of thrombi and monitoring of success or failure of thrombolysis in mice. Circulation 2012, 125, 3117–3126. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, F.; Fokong, S.; Koczera, P.; Lederle, W.; Lammers, T. Ultrasound microbubbles for molecular diagnosis, therapy, and theranostics. J. Nucl. Med. 2012, 53, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.F.L.; Kaspar, M.; Staub, D. Contrast-enhanced ultrasound: Clinical applications in patients with atherosclerosis. Int. J. Cardiovasc. Imaging 2016, 32, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, B.A.; Sanders, J.M.; Davis, C.; Xie, A.; Aldred, P.; Sarembock, I.J.; Linder, J.R. Molecular imaging of inflammation in atherosclerosis with targeted ultrasound detection of vascular cell adhesion molecule-1. Circulation 2007, 116, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Leong-Poi, H.; Bin, L.; Yang, L.; Liao, Y.; Liu, Y.; Cai, J.; Xie, J.; Liu, Y. Efficacy of contrast-enhanced US and magnetic microbubbles targeted to vascular cell adhesion molecule–1 for molecular imaging of atherosclerosis. Radiology 2011, 2, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Khanicheh, E.; Qi, Y; Xie, A.; Mitterhuber, M.; Xu, L.; Mochizuki, M.; Daali, Y.; Jaquet, V.; Krause, K.H.; Ruggeri, Z.M.; et al. Molecular imaging reveals rapid reduction of endothelial activation in early atherosclerosis with apocynin independent of antioxidative properties. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2187–2192. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, B.A.; Carr, C.L.; Belcik, J.T.; Xie, A.; Yue, Q.; Chadderdon, S.; Caplan, E.S.; Khangura, J.; Bullens, S.; Bunting, S.; et al. Molecular imaging of the initial inflammatory response in atherosclerosis. Implications for early detection of disease. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 54–59. [Google Scholar] [CrossRef] [PubMed]
- McCarty, O.J.T.; Conley, R.B.; Shentu, W.; Tormoen, J.W.; Zha, D.; Xie, A.; Qi, Y.; Zhao, Y.; Carr, C.; Belchic, T.; et al. Molecular imaging of activated von willebrand factor to detect high-risk atherosclerotic phenotype. ACC Cardiovasc. Imaging 2010, 3, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Shim, C.Y.; Liu, Y.N.; Atkinson, T.; Xie, A.; Foster, T.; Davidson, B.P.; Treible, M.; Qi, Y.; López, J.A.; Munday, A.; et al. molecular imaging of platelet–endothelial interactions and endothelial von willebrand factor in early and mid-stage atherosclerosis. Circ. Cardiovasc. Imaging 2015, 8, e002765. [Google Scholar] [CrossRef] [PubMed]
- Massberg, S.; Gawaz, M.; Gruner, S.; Schulte, V.; Konrad, I.; Zohlnhofer, D.; Heinzmann, U.; Nieswandt, B. A crucial role of glycoprotein VI for platelet recruitment to the injured arterial wall in vivo. J. Exp. Med. 2003, 197, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Massberg, S.; Konrad, I.; Bultmann, A.; Schulz, C.; Munch, G.; Peluso, M.; Lorenz, M.; Schneider, S.; Besta, F.; Müller, I.; et al. Soluble glycoprotein VI dimer inhibits platelet adhesion and aggregation to the injured vessel wall in vivo. FASEB J. 2004, 18, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P. Arterial thrombosis-insidious, unpredictable and deadly. Nat. Med. 2011, 17, 1423–1436. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.L.; Mascelli, M.A.; Neblock, D.S.; Weisman, H.F.; Coller, B.S.; Jordan, R.E. Analysis of GPIIb/IIIa receptor number by quantification of 7E3 binding to human platelets. Blood 1996, 88, 907–914. [Google Scholar] [PubMed]
- Aukrust, P.; Halvorsen, B.; Ueland, T.; Michelsen, A.E.; Skjelland, M.; Gullestad, L.; Yndestad, A.; Otterdal, K. Activated platelets and atherosclerosis. Expert Rev. Cardiovasc. Ther. 2010, 8, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Burtea, C.; Laurent, S.; Murariu, O.; Rattat, D.; Toubeau, G.; Verbruggen, A.; Vansthertem, D.; Vander Elst, L.; Muller, R.N. Molecular imaging of αvβ3 integrin expression in atherosclerotic plaques with a mimetic of RGD peptide grafted to Gd-DTPA. Cardiovasc. Res. 2008, 78, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Burtea, C.; Laurent, S.; Lancelot, E.; Ballet, S.; Murariu, O.; Rousseaux, O.; Port, M.; Vander Elst, L.; Corot, C.; Muller, R.N. Peptidic targeting of phosphatidylserine for the MRI detection of apoptosis in atherosclerotic plaques. Mol. Pharmcol. 2009, 6, 1903–1919. [Google Scholar] [CrossRef] [PubMed]
- Burtea, C.; Ballet, S.; Laurent, S.; Rousseaux, O.; Dencausse, A.; Gonzalez, W.; Port, M.; Corot, C.; Vander Elst, L.; Muller, R.N. Development of a magnetic resonance imaging protocol for the characterization of atherosclerotic plaque by using vascular cell adhesion molecule-1 and apoptosis-targeted ultrasmall superparamagnetic iron oxide derivatives. Arterioscler. Thromb. Vasc. Biol. 2012, 32, e36–e48. [Google Scholar] [CrossRef] [PubMed]
- Michalska, M.; Machtoub, L.; Manthey, H.D.; Bauer, E.; Herold, V.; Krohne, G.; Lykowsky, G.; Hildenbrand, M.; Kampf, T.; Jakob, P.; et al. Visualization of vascular inflammation in the atherosclerotic mouse by ultrasmall superparamagnetic iron oxide vascular cell adhesion molecule-1-specific nanoparticles. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2350–2357. [Google Scholar] [CrossRef] [PubMed]
- Makowski, M.R.; Forbes, S.C.; Blume, U.; Warley, A.; Jansen, C.H.; Schuster, A.; Wiethoff, A.J.; Botnar, R.M. In vivo assessment of intraplaque and endothelial fibrin in ApoE−/− mice by molecular MRI. Atherosclerosis 2012, 222, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Balu, N.; Li, W.; Chen, Y.; Shi, X.; Kummitha, C.M.; Yu, X.; Yuan, C.; Lu, Z.R. Molecular MRI of atherosclerotic plaque progression in an ApoE−/− mouse model with a CLT1 peptide targeted macrocyclic Gd(III) chelate. Am. J. Nucl. Med. Mol. Imaging 2013, 3, 446–455. [Google Scholar] [PubMed]
- Segers, F.M.; den Adel, B.; Bot, I.; van der Graaf, L.M.; van der Veer, E.P.; Gonzalez, W.; Raynal, I.; de Winther, M.; Wodzig, W.K.; Poelmann, R.E.; et al. Scavenger receptor-AI-targeted iron oxide nanoparticles for in vivo MRI detection of atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Parolini, C.; Busnelli, M.; Ganzetti, G.S.; Dellera, F.; Manzini, S.; Scanziani, E.; Johnson, J.L.; Sirtori, C.R.; Chiesa, G. Magnetic resonance imaging visualization of vulnerable atherosclerotic plaques at the brachiocephalic artery of apolipoprotein E knockout mice by the blood pool contrast agent B22956/1. Mol. Imaging 2014, 13. [Google Scholar] [CrossRef]
- Tarin, C.; Carril, M.; Martin-Ventura, J.L.; Markuerkiaga, I.; Padro, D.; Llamas-Granda, P.; Moreno, J.A.; García, I.; Genicio, N.; Plaza-Garcia, S.; et al. Targeted gold-coated iron oxide nanoparticles for CD163 detection in atherosclerosis by MRI. Sci. Rep. 2015, 5, 17135. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Liu, D.F.; Cui, Y.; Harris, S.S.; Chen, Y.C.; Li, K.C.; Ju, S.H.; Teng, G.J. In vivo MRI detection of carotid atherosclerotic lesions and kidney inflammation in ApoE-deficient mice by using LOX-1 targeted iron nanoparticles. Nanomedicine 2014, 10, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, J.; Yang, B.; Qiao, H.; Gao, L.; Su, T.; Ma, S.; Zhang, X.; Li, X.; Liu, G.; et al. In vivo MR and fluorescence dual-modality imaging of atherosclerosis characteristics in mice using profilin-1 targeted magnetic nanoparticles. Theranostics 2016, 6, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Nahrendorf, M.; Zhang, H.; Hembrador, S.; Panizzi, P.; Sosnovik, D.E.; Aikawa, E.; Libby, P.; Swirski, F.K.; Weissleder, R. Nanoparticle PET-CT imaging of macrophages in inflammatory atherosclerosis. Circulation 2008, 117, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.W.; Baek, H.; Mahackian, L.M.; Kusunose, J.; Hamzah, J.; Ruoslahti, E.; Ferrara, K.W. 64Cu-labeled LyP-1-dendrimer for PET-CT imaging of atherosclerotic plaque. Bioconjug. Chem. 2014, 25, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Patel, A.R.; Klibanov, A.L.; Kramer, C.M.; Ruiz, M.; Kang, B.Y.; Metha, J.L.; Beller, J.L.; Glover, D.K.; Meyer, C.H. Molecular imaging of atherosclerotic plaques targeted to oxidized LDL receptor LOX-1 by SPECT/CT and magnetic resonance. Circ. Cardiovasc. Imaging 2010, 3, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Foss, C.A.; Bedja, D.; Mease, R.C.; Wang, H.; Kass, D.A.; Chatterjee, S.; Pomper, M.G. Molecular imaging of inflammation in the ApoE−/− mouse model of atherosclerosis with IodoDPA. Biochem. Biophys. Res. Commun. 2015, 461, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Nahrendorf, M.; Keliher, E.; Panizzi, P.; Zhang, H.; Hembrador, S.; Figueiredo, J.L.; Aikawa, E.; Kelly, K.; Libby, P.; Weissleder, R. 18F-4V for PET-CT imaging of VCAM-1 expression in atherosclerosis. J. Am. Coll. Cardiol. Cardiovasc. Imag. 2009, 2, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Bala, G.; Blykers, A.; Xavier, C.; Descamps, B.; Broisat, A.; Ghezzi, C.; Fagret, D.; van Camp, G.; Caveliers, V.; Vanhove, C.; et al. Targeting of vascular cell adhesion molecule-1 by 18F-lbelled nanobodies for PET/CT imaging of inflammed atherosclerotic plaques. Eur. Hearth J. Cardiovasc. Imaging 2016, 17, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Broisat, A.; Hernot, S.; Toczek, J.; de Vos, J.; Riou, L.M.; Martin, S.; Ahmadi, M.; Thielens, N.; Wernery, U.; Caveliers, V.; et al. Nanobodies targeting mouse/human VCAM1 for the nuclear imaging of atherosclerotic lesions. Circ. Res. 2012, 110, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Dimastromatteo, J.; Broisat, A.; Perret, P.; Ahmadi, M.; Boturyn, D.; Dumy, P.; Fagret, D.; Riou, L.M.; Ghezzi, C. In vivo molecular imaging of atherosclerotic lesions in ApoE−/− mice using VCAM-1-specific, 99mTc-labeled peptidic sequences. J. Nucl. Med. 2013, 54, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bauer, W.; Israel, I.; Kreissl, M.C.; Weirather, J.; Richter, D.; Bauer, E.; Herold, V.; Jakob, P.; Buck, A.; et al. Targeting P-selectin by gallium-68-labeled fucoidan positron emission tomography for noninvasive characterization of vulnerable plaques: Correlation with in vivo 17.6T MRI. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.; Hasegawa, K.; Wada, Y.; Hirase, T.; Node, K.; Watanabe, Y. Detection of early stage atherosclerotic plaques using PET and CT fusion imaging targeting P-selectin in low density lipoprotein receptor-deficient mice. Biochem. Biophys. Res. Commun. 2013, 433, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, I.; Saraste, A.; Weidl, E.; Poethko, T.; Weber, A.W.; Nekolla, S.G.; Leppänen, P.; Ylä-Herttuala, S.; Hölzlwimmer, G.; Walch, A.; et al. Evaluation of αvβ3 integrin-targeted positron emission tomography tracer 18F-galacto-RGD for imaging of vascular inflammation in atherosclerotic mice. Circ. Cardiovasc. Imaging 2009, 2, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Gorodny, N.; Gomez, L.F.; Gangadharmath, U.B.; Mu, F.; Chen, G.; Walsh, J.C.; Szardenings, K.; Berman, D.S.; Kolb, H.C.; et al. Atherosclerotic plaque uptake of a novel integrin tracer 18F-Flotegatide in a mouse model of atherosclerosis. J. Nucl. Cardiol. 2014, 21, 553–562. [Google Scholar] [CrossRef] [PubMed]
- De Saint-Hubert, M.; Bauwens, M.; Deckers, N.; Drummen, M.; Douma, K.; Granton, P.; Hendrikx, G.; Kusters, D.; Bucerius, J.; Reutelingsperger, C.P.; et al. In vivo molecular imaging of apoptosis and necrosis in atherosclerotic plaques using microSPECT-CT and microPET-CT imaging. Mol. Imaging Biol. 2014, 16, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Bigalke, B.; Phinikaridou, A.; Andia, M.E.; Cooper, M.S.; Schuster, A.; Schönberger, T.; Griessinger, C.M.; Wurster, T.; Onthank, D.; Ungerer, M.; et al. Positron emission tomography/computed tomographic and magnetic resonance imaging in a murine model of progressive atherosclerosis using 64Cu-labeled glycoprotein VI-Fc. Circ. Cardiovasc. Imaging 2013, 6, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, T.; Berndorff, D.; Heinrich, T.; Hucko, T.; Stepina, E.; Hauff, P.; Dinkelborg, L.M.; Atrott, K.; Giovannoni, L.; Neri, D.; et al. Targeted ED-B fibronectin SPECT in vivo imaging in experimental atherosclerosis. Q. J. Nucl. Med. Mol. Imaging 2015, 59, 228–237. [Google Scholar] [PubMed]
- Bhavane, R.; Badea, C.; Ghaghada, K.B.; Clark, D.; Vela, D.; Moturu, A.; Annapragada, A.; Johnson, G.A.; Willerson, J.T.; Annapragada, A. Dual-energy computed tomography imaging of atherosclerotic plaques in a mouse model using a liposomal-iodine nanoparticle contrast agent. Circ. Cardiovasc. Imaging 2013, 6, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Peng, C.; Zhao, B.; Ye, K.; Yuan, F.; Peng, Z.; Yang, X.; Huang, L.; Jiang, M.; Zhao, Q.; et al. Noninvasive detection of macrophages in atherosclerotic lesions by computed tomography enhanced with PEGylated gold nanoparticles. Int. J. Nanomed. 2014, 9, 5575–5590. [Google Scholar]
- Ntziachristos, V.; Tung, C.H.; Bremer, C.; Weissleder, R. Fluorescence molecular tomography resolves protease activity in vivo. Nat. Med. 2002, 8, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Nahrendorf, M.; Waterman, P.; Thurber, G.; Groves, K.; Rajopadhye, M.; Panizzi, P.; Marinelli, B.; Aikawa, E.; Pittet, M.J.; Swirski, F.K.; et al. Hybrid in vivo FMT-CT imaging of protease activity in atherosclerosis with customized nanosensors. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- Ale, A.; Ermolayev, V.; Herzog, E.; Cohrs, C.; de Angelis, M.H.; Ntziachristos, V. FMT-XCT: In vivo animal studies with hybrid fluorescence molecular tomography-X-ray computed tomography. Nat. Methods 2012, 9, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Maafi, F.; Berti, R.; Pouliot, P.; Rhéaume, E.; Tardif, J.C.; Lesage, F. Hybrid FMT-MRI applied to in vivo atherosclerosis imaging. Biomed. Opt. Express 2014, 5, 1664–1676. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, J.; Aikawa, M.; Tung, C.H.; Aikawa, E.; Kim, D.E.; Ntziachristos, V.; Weissleder, R.; Libby, P. Inflammation in atherosclerosis visualizing matrix metalloproteinase action in macrophages in vivo. Circulation 2006, 114, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Larmann, J.; Frenzel, T.; Schmitz, M.; Hahnenkamp, A.; Demmer, P.; Immenschuh, S.; Tietge, U.J.; Bremer, C.; Theilmeier, G. In vivo fluorescence-mediated tomography imaging demonstrates atorvastatin-mediated reduction of lesion macrophages in ApoE−/− mice. Anesthesiology 2013, 119, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Duivenvoorden, R.; Tang, J.; Cormode, D.P.; Mieszawska, A.J.; Izquierdo-Garcia, D.; Ozcan, C.; Otten, M.J.; Zaidi, N.; Lobatto, M.E.; van Rijs, S.M.; et al. A statin-loaded reconstituted high-density lipoprotein nanoparticle in-hibits atherosclerotic plaque inflammation. Nat. Commun. 2014, 5, 3065–3090. [Google Scholar] [PubMed]
- Lin, S.A.; Patel, M.; Suresch, D.; Connolly, B.; Bao, B.; Groves, K.; Rajopadhye, M.; Peterson, J.D.; Klimas, M.; Sur, C.; Bednar, B. Quantitative longitudinal imaging of vascular inflammation and treatment by ezetimibe in ApoE mice by FMT using new optical imaging biomarkers of cathepsin activity and αvβ3 Integrin. Int. J. Mol. Imaging 2012, 2012, 189254. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Jiang, I.; Sheng, Z.; Zhang, Y.; An, Y.; Yan, F.; Ma, G.; Liu, N.; Teng, G.; Cheng, Z. Analysis of in situ and ex vivo αvβ3 integrin expression during experimental carotid atherogenesis. Int. J. Nanomed. 2012, 7, 641–649. [Google Scholar]
- Oraevsky, A.A.; Karabutov, A.A. Optoacoustic tomography. In Biomedical Photonics Handbook; Vo-Dinh, T., Ed.; CRC Press/Francis and Taylor Group: Boca Raton, FL, USA, 2003; pp. 1–34. [Google Scholar]
- Xu, M.H.; Wang, L.H.V. Photoacoustic imaging in biomedicine. Rev. Sci. Instrum. 2006, 77, 041101. [Google Scholar] [CrossRef]
- Kim, C.; Favazza, C.; Wang, L.V. In vivo photoacoustic tomography of chemicals: High-resolution functional and molecular optical imaging at new depths. Chem. Rev. 2010, 110, 2756–2782. [Google Scholar] [CrossRef] [PubMed]
- Razansky, D.; Deliolanis, N.C.; Vinegoni, C.; Ntziachristos, V. Deep tissue optical and optoacoustic molecular imaging technologies for pre-clinical research and drug discovery. Curr. Pharm Biotechnol. 2012, 13, 504–522. [Google Scholar] [CrossRef] [PubMed]
- Luke, G.P.; Yeager, D.; Emelianov, S.Y. Biomedical applications of photoacoustic imaging with exogenous contrast agents. Ann. Biomed. Eng. 2012, 40, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, R.; Sahin, O.; Emelianov, S. Ultrasound-guided photoacoustic imaging: Current state and future development. IEEE Trans. Ultrason. Ferroelectr. Freq. Control 2014, 61, 450–466. [Google Scholar] [CrossRef] [PubMed]
- Jeon, M.; Song, W.; Huynh, E.; Kim, J.; Kim, J.; Helfield, B.L.; Leung, B.Y.; Goertz, D.E.; Zheng, G.; Oh, J.; et al. Methylene blue microbubbles as a model dual-modality contrast agent for ultrasound and activatable photoacoustic imaging. J. Biomed. Opt. 2014, 19, 16005. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chen, X. Gold nanoparticles for photoacoustic imaging. Nanomedicine 2015, 10, 299–320. [Google Scholar] [CrossRef] [PubMed]
- Taruttis, A.; Ntziachristos, V. Advances in real-time multispectral optoacoustic imaging and its applications. Nat. Photonics 2015, 9, 219–227. [Google Scholar] [CrossRef]
- Kim, K.; Huang, S.W.; Ashkenazi, S.; O’Donnell, M.; Agarwal, A.; Kotov, N.A.; Denny, M.F.; Kaplan, M.J. Photoacoustic imaging of early inflammatory response using gold nanorods. Appl. Phys. Lett. 2007, 90, 223901. [Google Scholar] [CrossRef]
- Wang, B.; Yantsen, E.; Larson, T.; Karpiouk, A.B.; Sethuraman, S.; Su, J.L.; Sokolov, K.; Emelianov, S.Y. Plasmonic intravascular photoacoustic imaging for detection of macrophages in atherosclerotic plaques. Nano Lett. 2008, 9, 2212–2217. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.; Carson, A.; Agarwal, A.; Kotov, N.A.; Kim, K. Detection and monitoring of the multiple in ammatory responses by photoacoustic molecular imaging using selectively targeted gold nanorods. Biomed. Opt. Express 2011, 2, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Rouleau, L.; Berti, R.; Ng, V.W.K.; Matteau-Pelletier, C.; Lam, T.; Saboural, P.; Kakkar, A.K.; Lesage, F.; Rhéaume, E.; Tardif, J.C. VCAM-1-targeting gold nanoshell probe for photoacoustic imaging of atherosclerotic plaque in mice. Contrast Media Mol. Imaging 2013, 8, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhang, Y.; Li, Z.; Li, C.; Wang, Q. A novel photoacoustic nanoprobe of ICG@PEG-Ag2S for atherosclerosis targeting and imaging in vivo. Nanoscale 2016, 8, 12531–12539. [Google Scholar] [CrossRef] [PubMed]
- Simsekyilmaz, S.; Cabrera-Fuentes, H.A.; Meiler, S.; Kostin, S.; Baumer, Y.; Liehn, E.A.; Weber, C.; Boisvert, W.A.; Preissner, K.T.; Zernecke, A. Role of extracellular RNA in atherosclerotic plaque formation in mice. Circulation 2014, 129, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Simsekyilmaz, S.; Cabrera-Fuentes, H.A.; Meiler, S.; Kostin, S.; Baumer, Y.; Liehn, E.A.; Weber, C.; Boisvert, W.A.; Preissner, K.T.; Zernecke, A. Response to letter regarding article “Role of extracellular RNA in atherosclerotic plaque formation in mice”. Circulation 2014, 130, e144–e145. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C.W. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed]










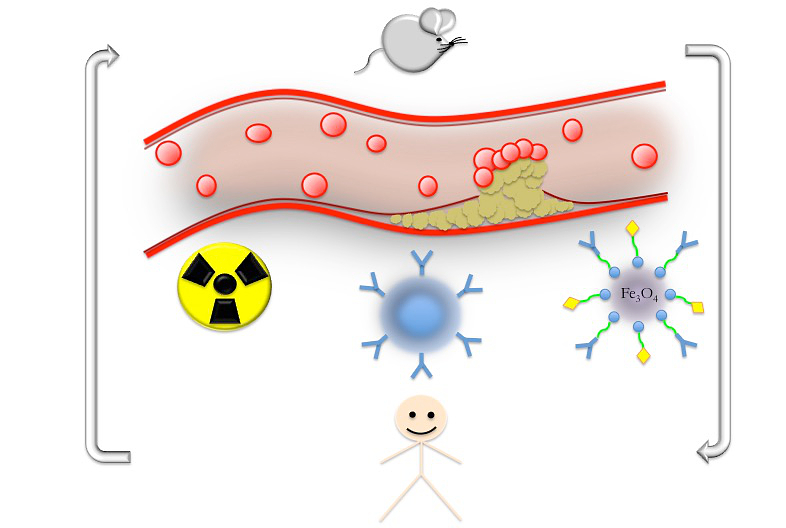{kind=link}
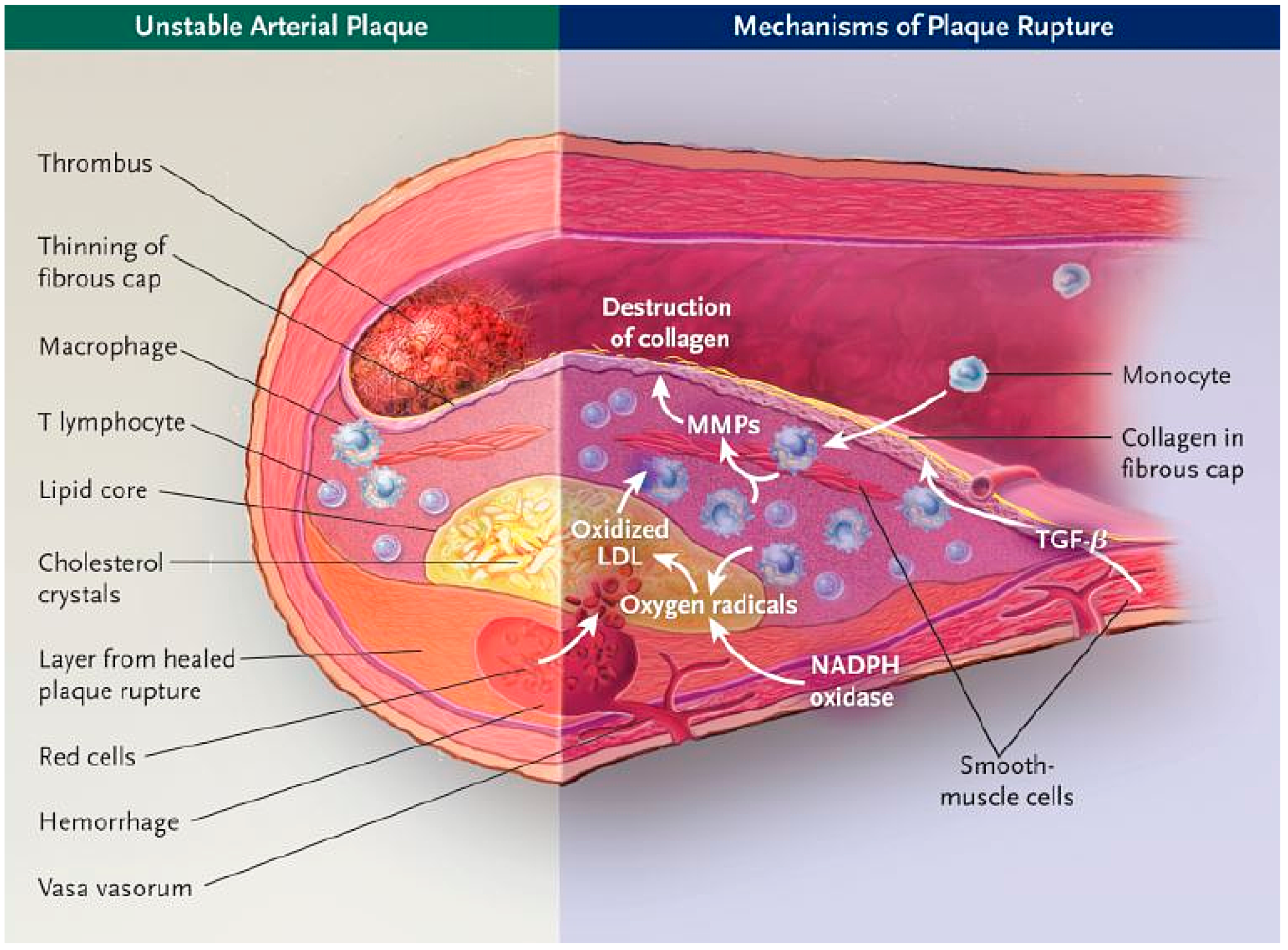{kind=link}
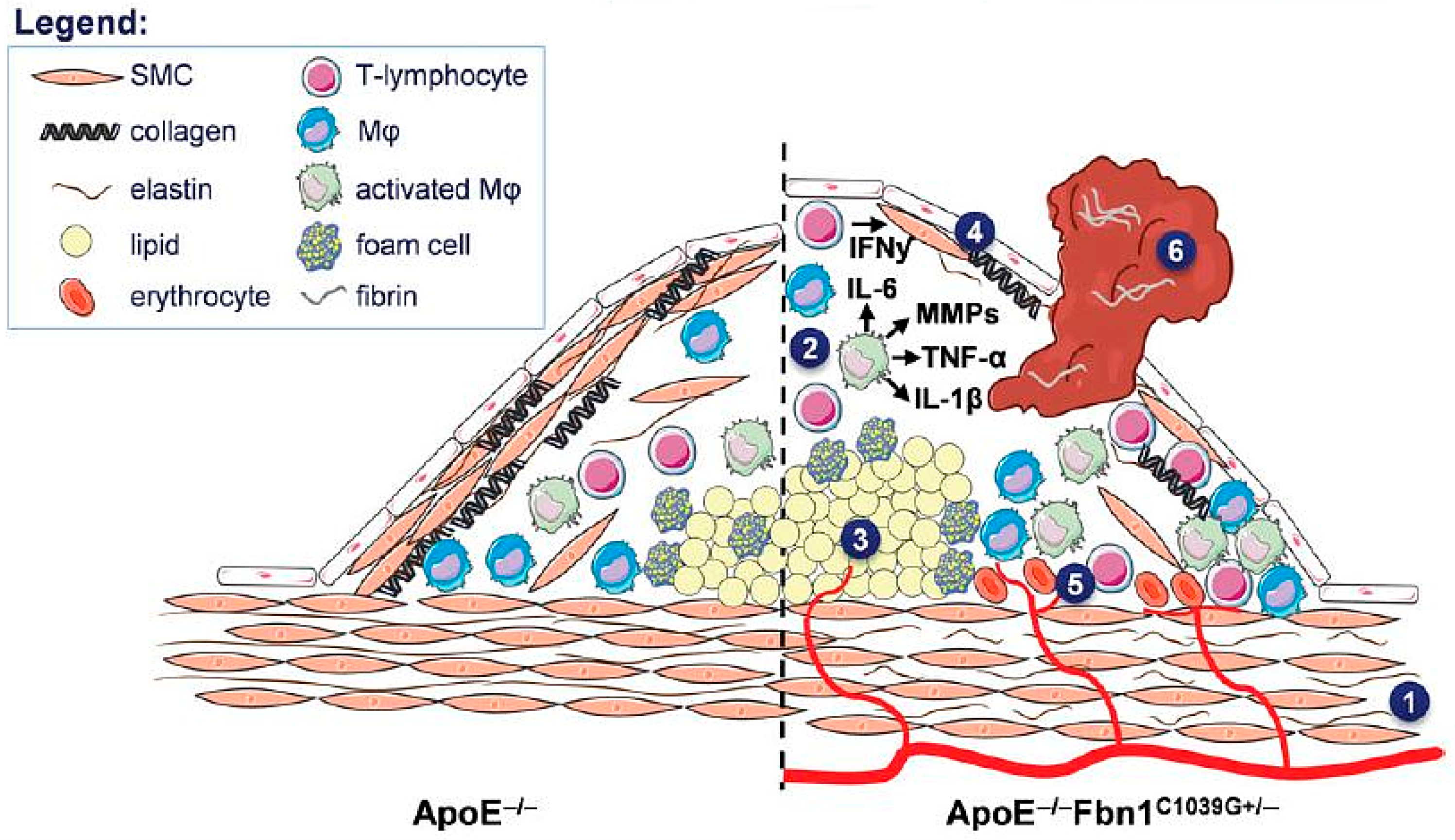{kind=link}
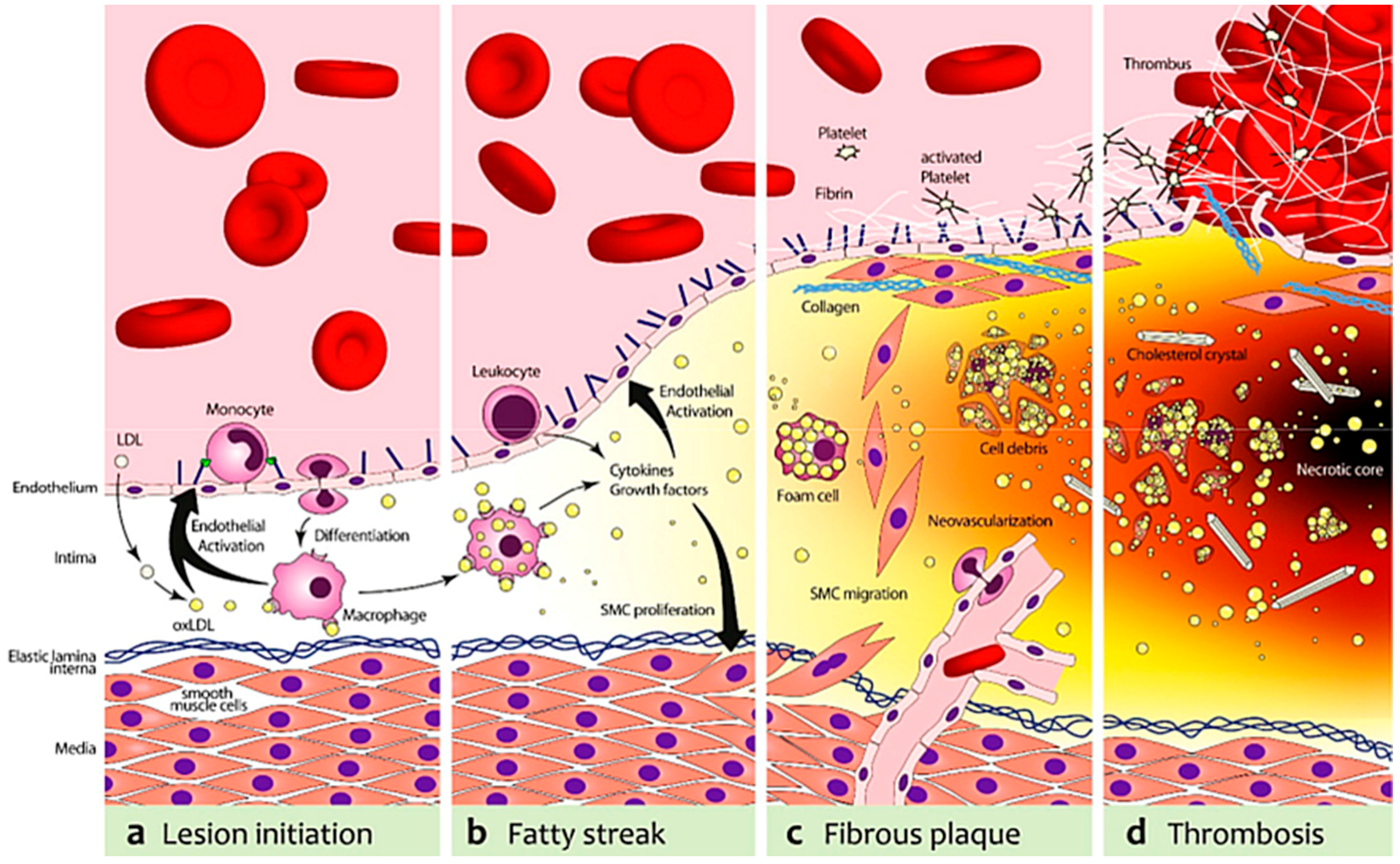{kind=link}
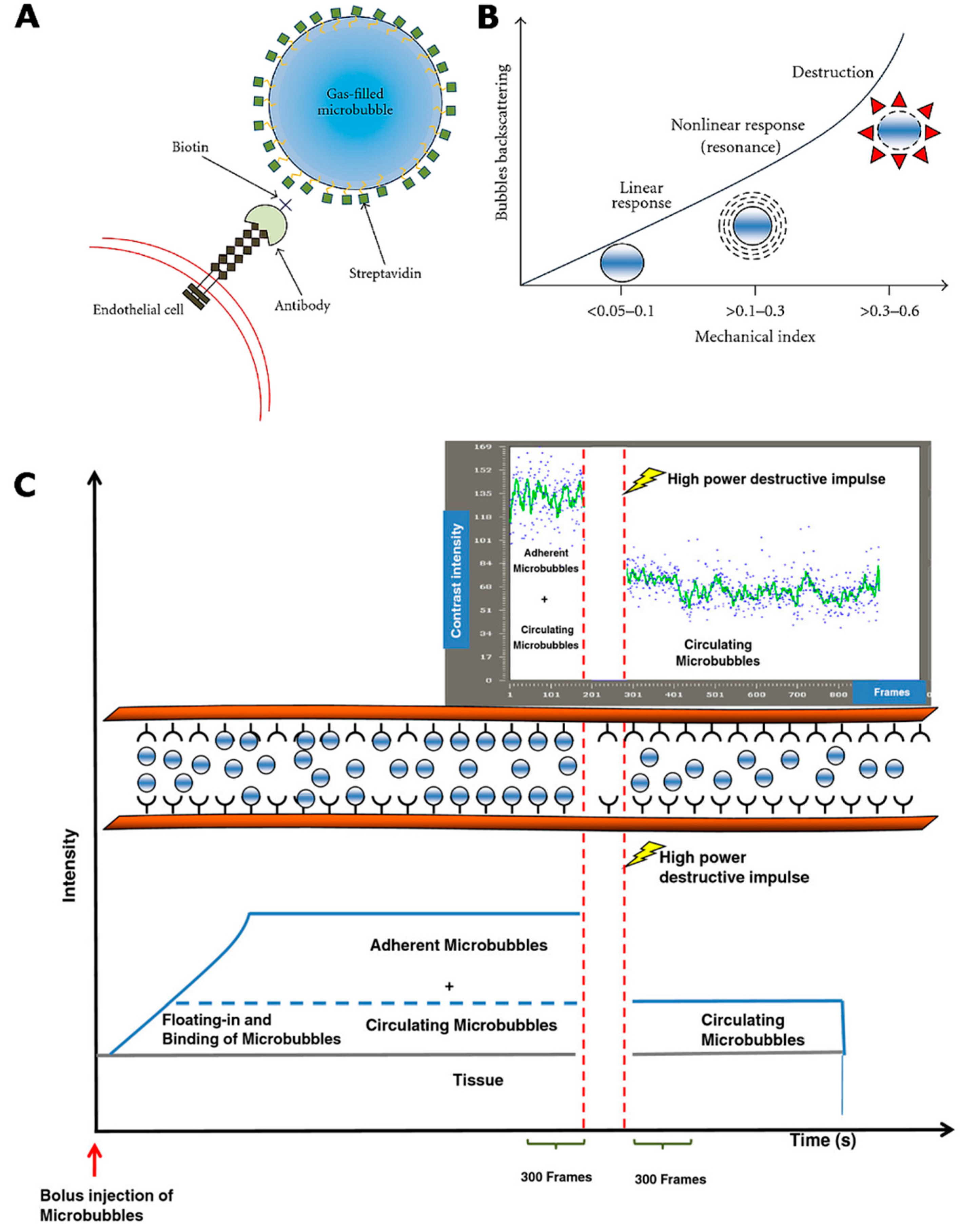{kind=link}
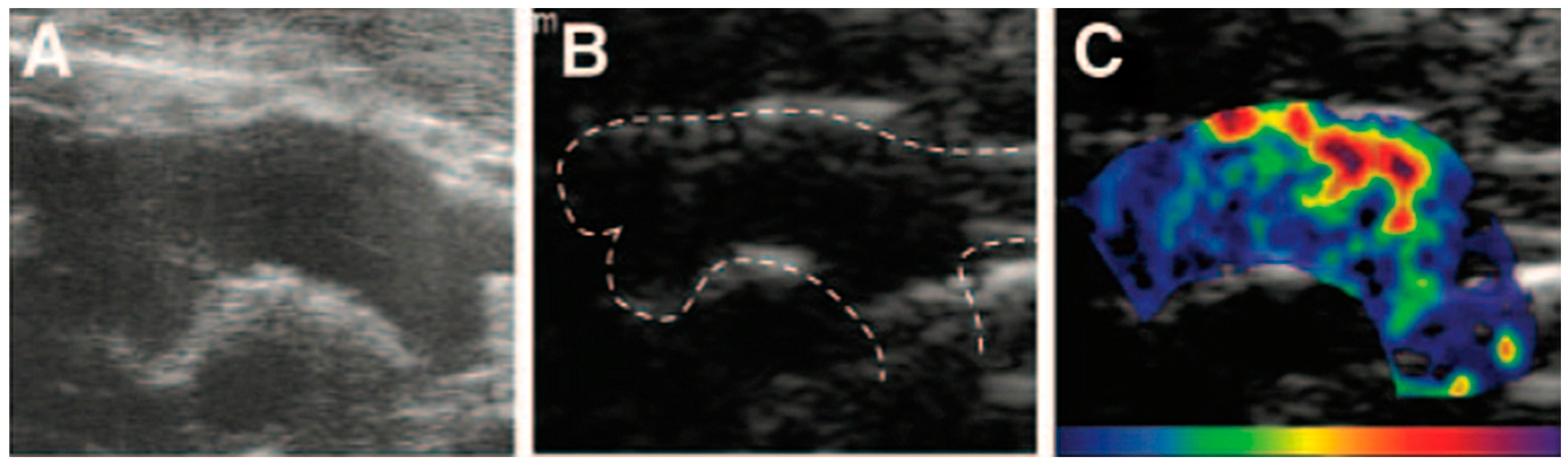{kind=link}
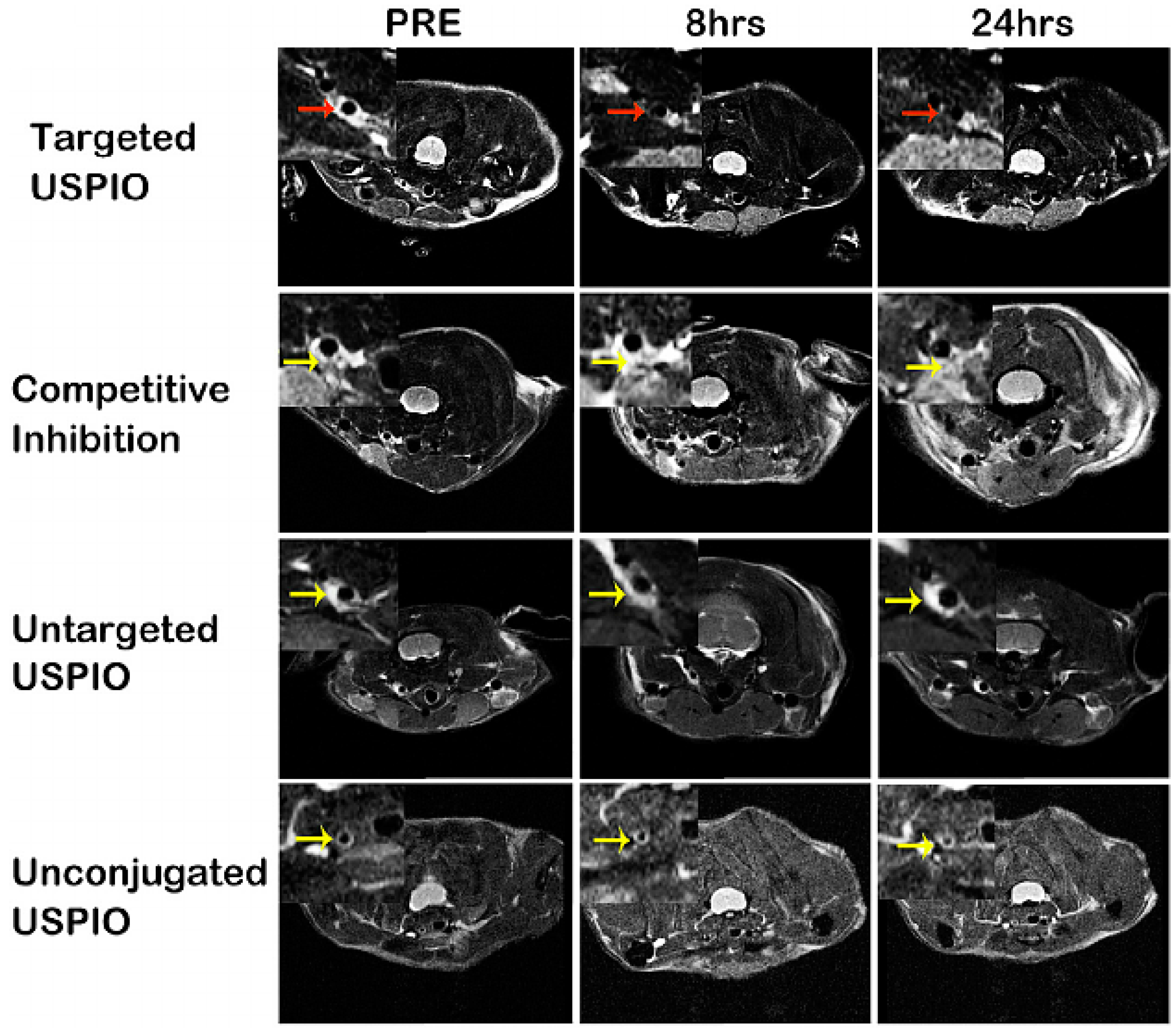{kind=link}
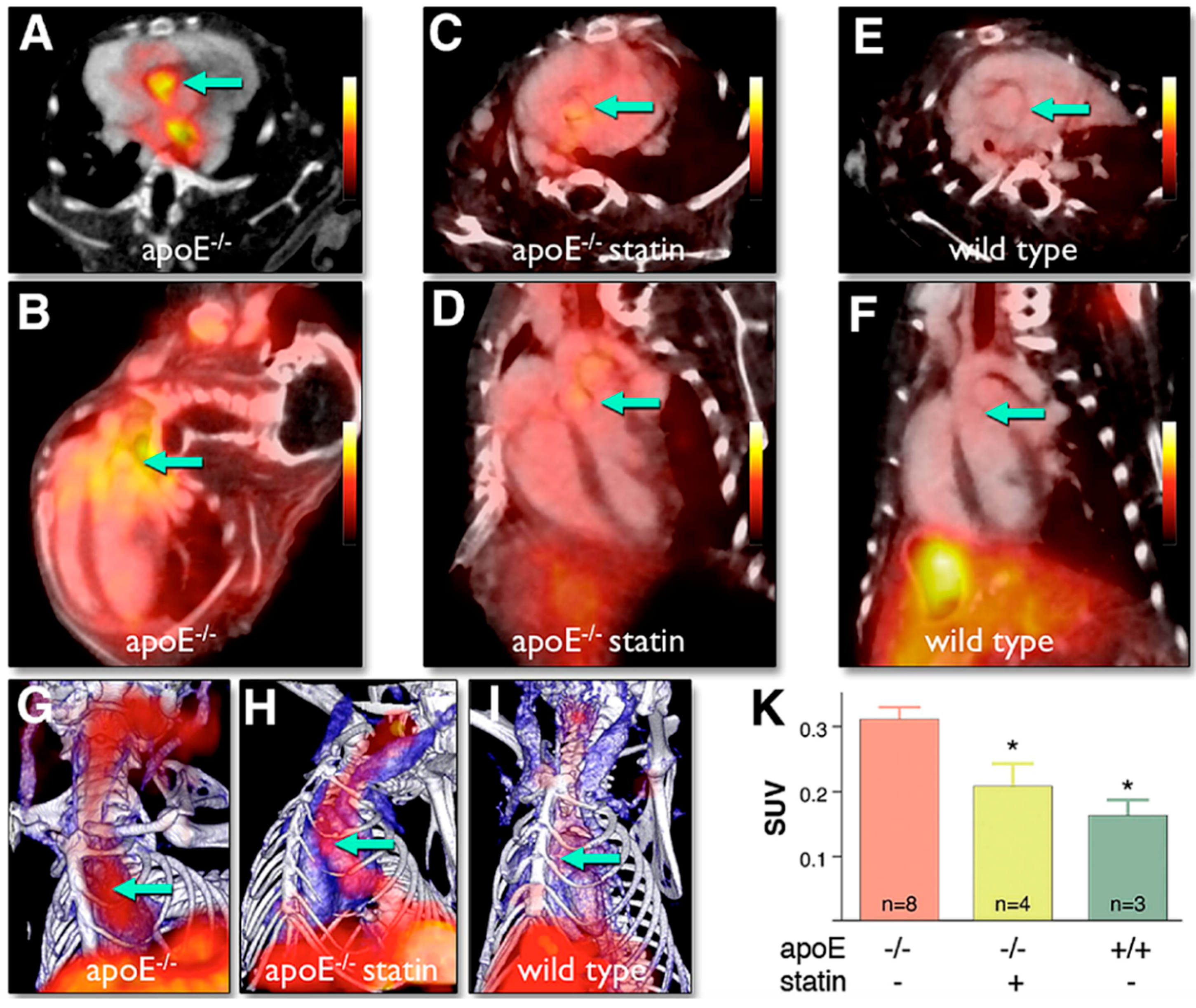{kind=link}
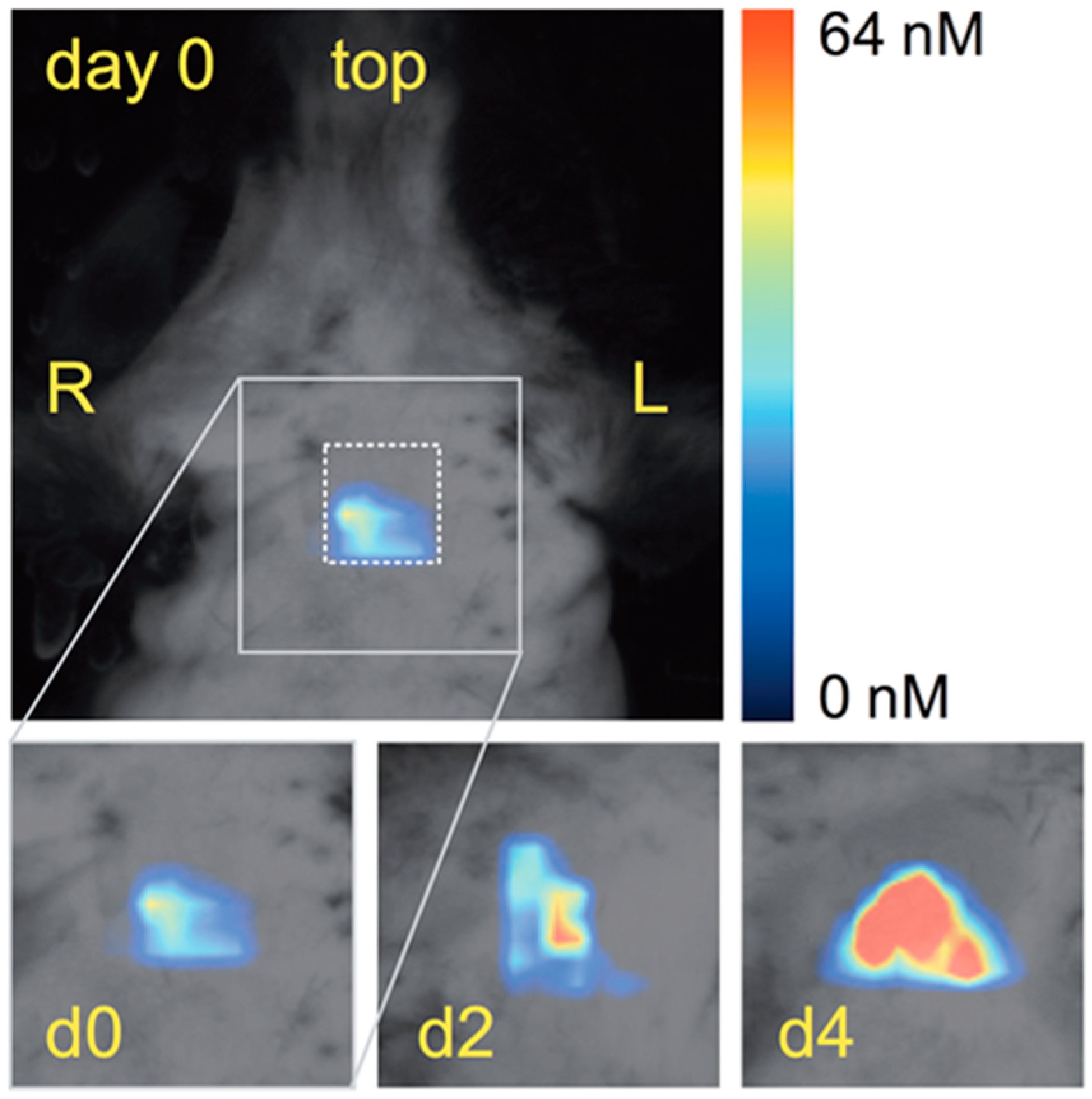{kind=link}
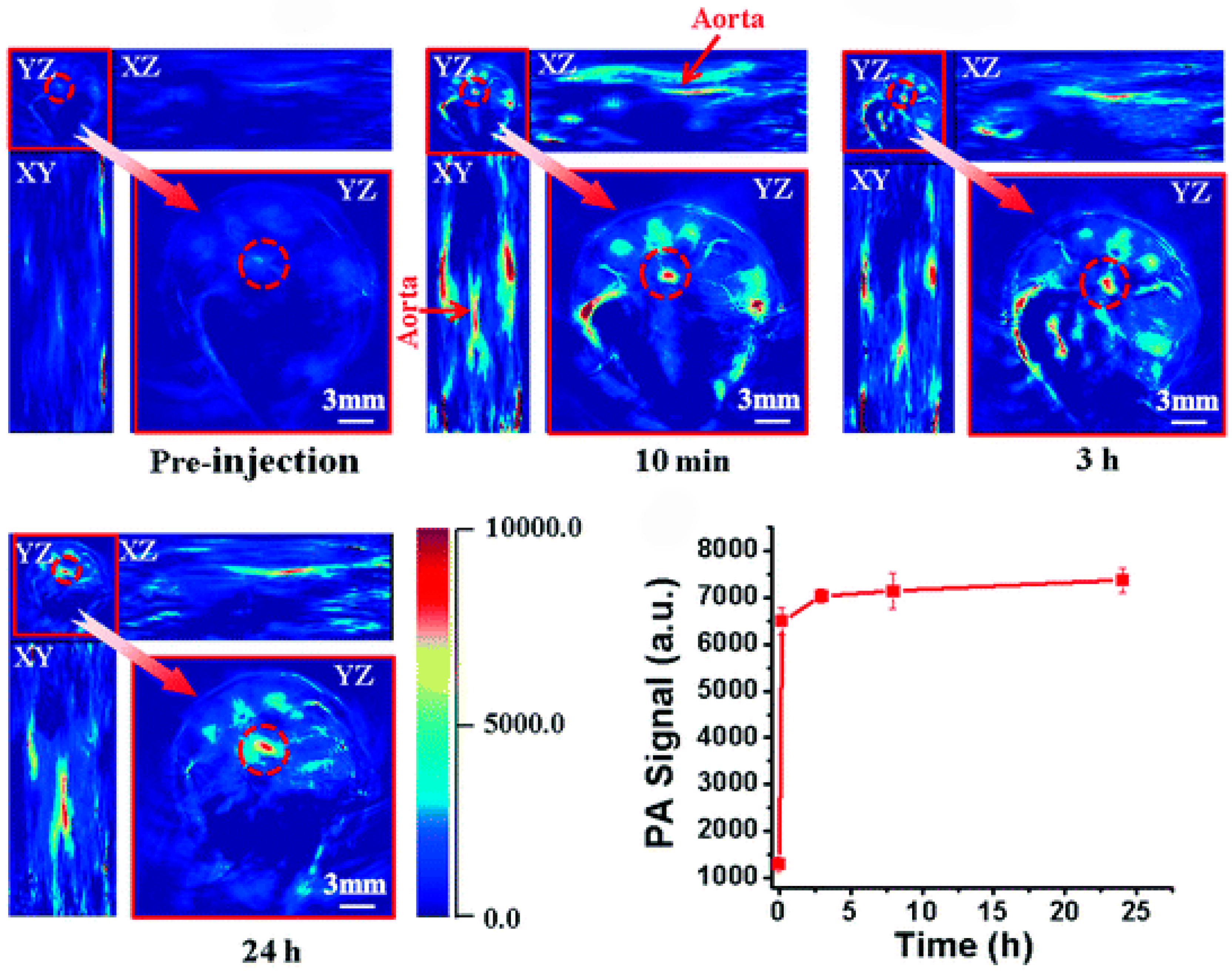{kind=link}
| Model | Mouse Strain | Stage of Lesion Formed (I–VI) | Lesion Characteristics | Area of Lesion Characterization | Modified Diet Required | Commercial Availability of Strain |
|---|---|---|---|---|---|---|
| Diet-induced | C57Bl/6J | I | Predominantly lipid-laden foam cells | Aortic root, aorta | Yes | Jackson ,Taconic, Charles River |
| Genetically modified | LDL−/− [64] | I–IV | Progression from predominantly lipid-laden foam cells to lesions with necrotic core and fibrous caps | Aortic root, aorta | Yes | Jackson |
| Genetically modified | ApoE−/− [56,57] | I–V | Progression from predominantly lipid-laden foam cells to lesions with necrotic core and fibrous caps | Aortic root, aorta; rare vulnerable plaques in innominate artery | No; fat diet accellerates atherogenesis | Jackson |
| Genetically modified | ApoE−/− Fbn1(C1039G)+/− [142] | I–VI | Development of vulnerable plaques prone to rupture; acute cardiovascular events (stroke, miocardial infarction) | Aortic root, aorta, innominate artery | No; fat diet accellerates atherogenesis | Jackson for ApoE−/− and Fbn1(C1039G+/−) breeders |
| Genetically modified | Apo E*3-Leiden [73] | I–IV | Progression from predominantly lipid-laden foam cells to lesions with necrotic core and fibrous caps | Aortic root, aorta, common carotid arteries | No; fat diet accellerates atherogenesis | Jackson |
| Collar-induced | ApoE−/− | I–VI | Development of vulnerable plaques for biomechanical alterations | Common carotid arteries | No; fat diet accellerates atherogenesis | Jackson |
| Tandem stenosis | ApoE−/− | I–VI | Development of vulnerable plaques for biomechanical alterations | Common carotid arteries | No; fat diet accellerates atherogenesis | Jackson |
| Collar + Ad-p53 + phenylephrine- induced | ApoE−/− | I–VI | Development of vulnerable plaques for biomechanical alterations, apoptosis of SMCs and hypertension | Common carotid arteries | No; fat diet accellerates atherogenesis | Jackson |
| Ligation plus cuff | ApoE−/− | I–VI | Development of vulnerable plaques for biomechanical alterations | Common carotid arteries | No; fat diet accellerates atherogenesis | Jackson |
| Partial ligation of carotid and renal arteries | ApoE−/− | I–VI | Development of vulnerable plaques for biomechanical alterations and hypertension | Common carotid arteries, renal arteries | No; fat diet accellerates atherogenesis | Jackson |
| Angiotensin II | ApoE−/− | I–VI | Development of vulnerable plaques with neovascularization, hemorragies and inflammation | Aortic root, aorta, innominate artery | No; fat diet accellerates atherogenesis | Jackson |
| Adenovirus-induced gene mutation | ApoE−/− | I–VI | PCSK9DY mutation, prothrombin overespression; fibrous cap disruption, hemorrhagies and thrombosis | Aortic root, aorta, innominate artery | No; fat diet accellerates atherogenesis | Jackson |
| Imaging Modality | Spatial Resolution | Sensitivity (mol/L) | Contrast Agent | Probe Concentration | Advantages | Limits |
|---|---|---|---|---|---|---|
| Ultrasound | 50–500 μm | Not well characterized yet | Microbubbles | µM to nM | Real-time Low cost High temporal resolution (0.1–100 s) No ionizing radiation | Operator-dependent |
| Magnetic Resonance | 10–100 μm | 10−3–10−5 | Gadolinium-based contrast agents Iron oxide and other superparamagnetic nanoparticles (USPIO, SPIO) | mM to nM | High tissue contrast and functional parameters No ionizing radiation | High cost Operator-dependent |
| Nuclear imaging | PET 1–2 mm | Positron or gamma ray emitting radionuclides (18F, 64Cu, 99mTc tracers) | pM | Molecular and functional parameters High sensitivity | Ionizing radiation Limited spatial resolution (mm) High-medium cost | |
| 10−11–10−12 | ||||||
| SPECT 0.5–2 mm | 10−10–10−11 | |||||
| X-ray computed tomography | 30–400 μm | 10−2–10−3 | Iodinated particles Gold nanorods | mM to nM | Fast acquisition time High temporal resolution (1–300 s) Provides molecular and structural information | Ionizing radiation Low soft tissue contrast resolution Medium cost |
| Fluorescence tomographic imaging | 1–2 mm | 10−10–10−11 | NIR Fluorophores | nM to pM | High sensitivity No ionizing radiation Low cost | Limited depth of penetration (1–20 mm) Limited spatial resolution (mm) |
| Photoacoustic imaging | <100 μm | <10−12 | NIR Fluorophores | nM to pM | High sensitivity No ionizing radiation High depth of penetration (<5 cm) Low cost | Data post-processing and acquisition procedures still being optimized |
| Molecular Target | Biological Events | Imaging Techniques | Imaging Probes |
|---|---|---|---|
| VCAM1-R; ICAM1-R; P-selectin | Vascular inflammation | UBM, MRI, PET, SPECT, PAI | Targeted microbubbles, targeted USPIO, 18F-, 99mTc-labeled VCAM1 antibodies, NIR Fluorophores |
| Phosphatidylserine | Apoptosis, vulnerable plaque, atherothrombosis | MRI, SPECT, FMT | Targeted USPIO, 99mTc-labeled annexin 5 or other tracers, NIR dyes conjugated with annexin 5 |
| αvβ3 | Neoangiogenesis | MRI, PET, FMT | Gadolinium-labeled RGD probes, 18F-labeled RGD or other tracers, NIR dyes conjugated with RGD or other probes |
| GPVI-R | Platelet adhesion, atherothrombosis | UBM, PET | Targeted microbubbles, 64Cu-labeled GPVI fragment |
| GP IIb/IIIa-R | Platelet adhesion, atherothrombosis | UBM | Targeted microbubbles |
| Fibrin-fibronectin complex | Atherothrombosis | MRI, SPECT | Gadolinium-labeled CLT1 peptide or other agents, 99mTc-labeled antibodies |
| Von Willebrand factor | Atherothrombosis | MRI, SPECT | Targeted microbubbles, |
| LOX-1 | Macrophagic lipid uptake | MRI, SPECT | Targeted USPIO, 99mTc-labeled antibodies |
| TSPO | Activated macrophages | SPECT | [125I]iodo-DPA-713 |
| Cathepsins and metalloproteinases | Macrophagic proteinases activity | FMT | NIR dyes |
| Macrophages infiltration | Macrophage-rich, rupture-prone plaques | CT, MRI, PET, FMT, PAI | Liposomal-iodine formulations, PEGylated gold nanoparticles, gold-coated iron oxide nanoparticles targeted for CD163 receptor antibody, trimodality 64Cu- iron oxide-NIR dye nanoparticle targeted for CD68, 18F-LyP-1 targeted for p32, NIR Fluorophores |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gargiulo, S.; Gramanzini, M.; Mancini, M. Molecular Imaging of Vulnerable Atherosclerotic Plaques in Animal Models. Int. J. Mol. Sci. 2016, 17, 1511. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091511
Gargiulo S, Gramanzini M, Mancini M. Molecular Imaging of Vulnerable Atherosclerotic Plaques in Animal Models. International Journal of Molecular Sciences. 2016; 17(9):1511. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091511
Chicago/Turabian StyleGargiulo, Sara, Matteo Gramanzini, and Marcello Mancini. 2016. "Molecular Imaging of Vulnerable Atherosclerotic Plaques in Animal Models" International Journal of Molecular Sciences 17, no. 9: 1511. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091511