
Advanced Glycation End-Products Induce Apoptosis of Vascular Smooth Muscle Cells: A Mechanism for Vascular Calcification

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
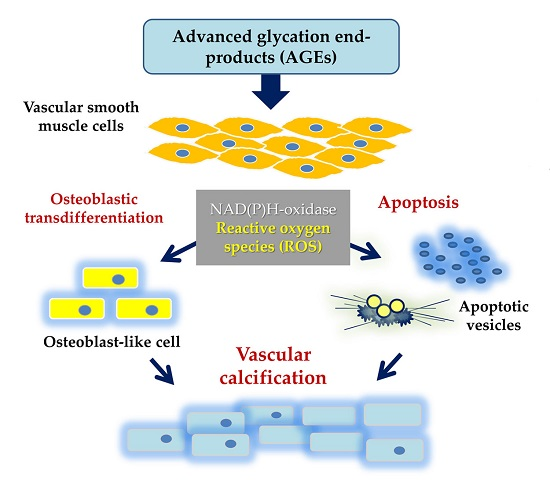
1. Introduction
2. Results
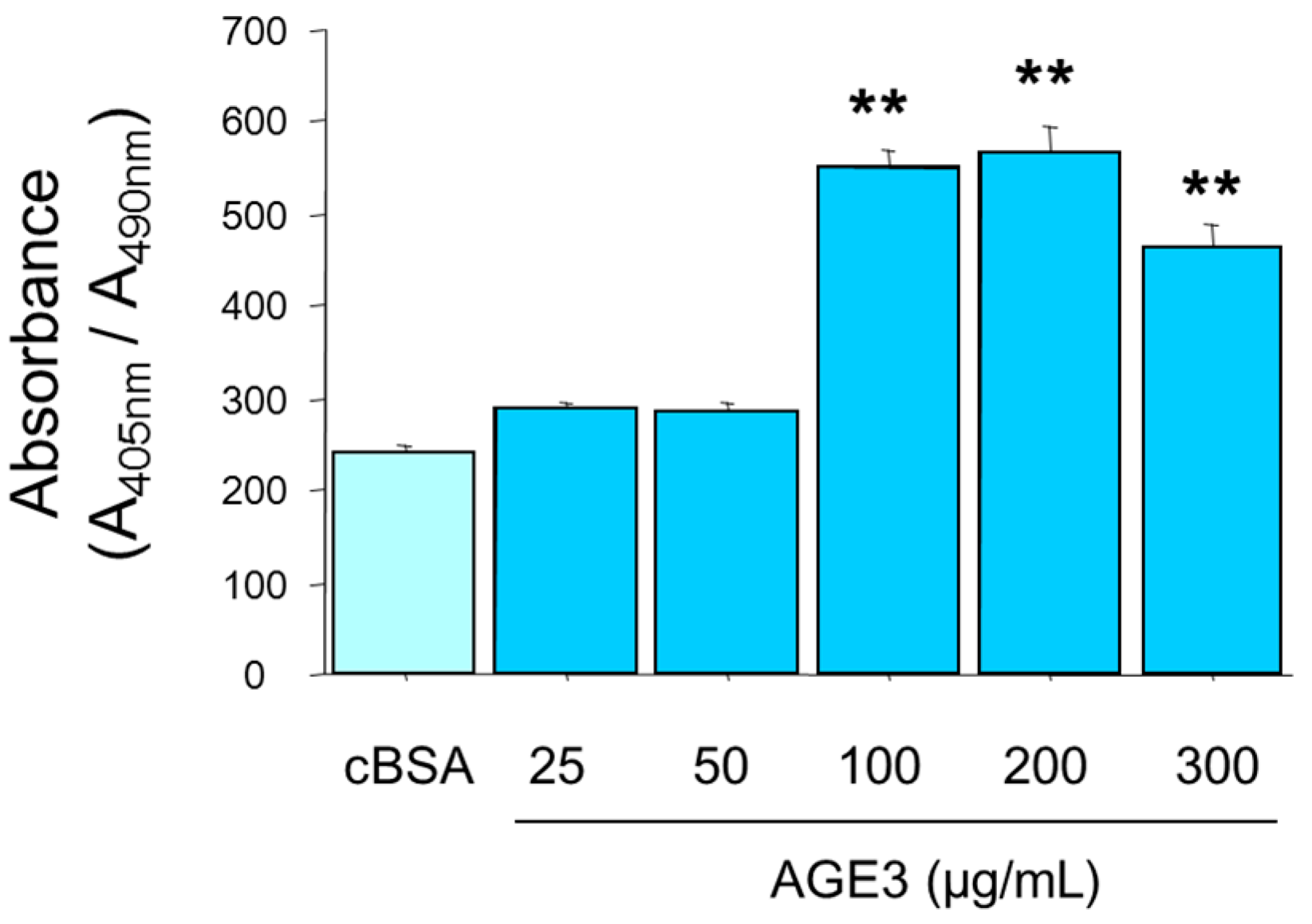
2.1. AGE3-BSA Increased Calcium Deposition in Vascular Smooth Muscle Cells (VSMCs)
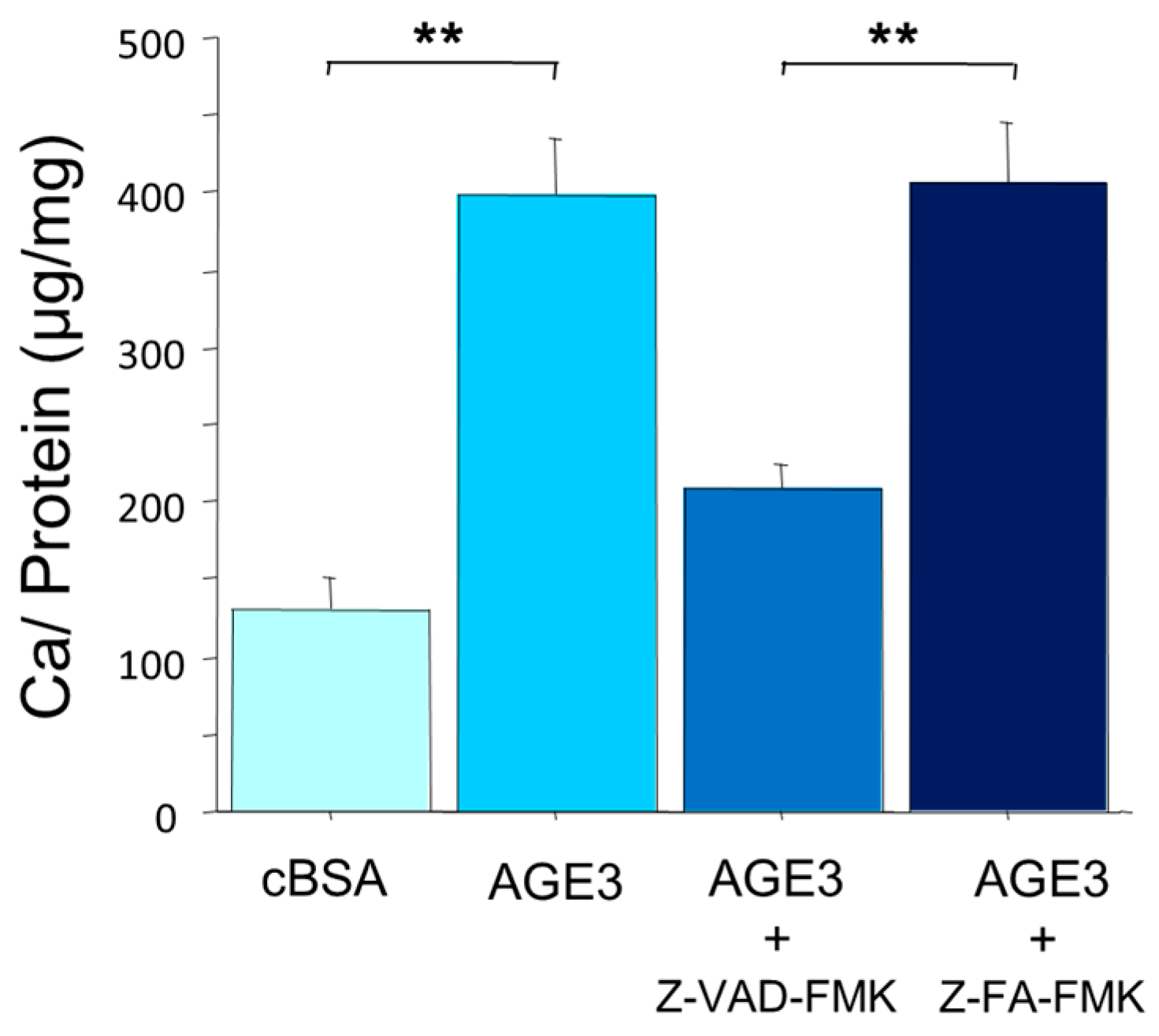
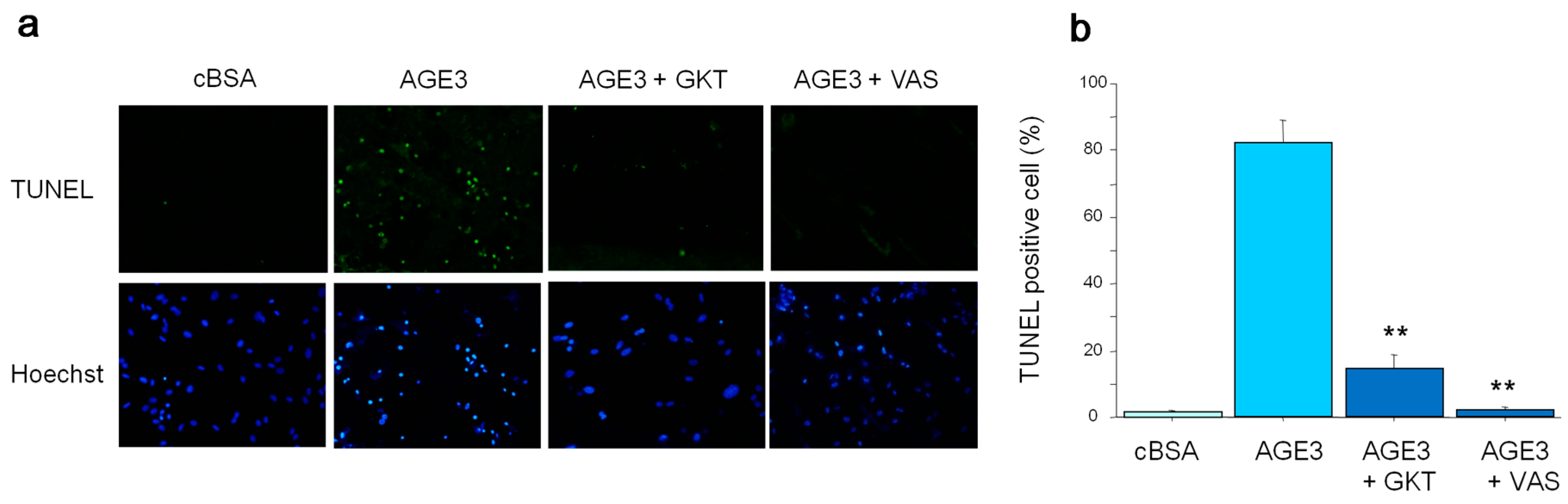
2.2. AGE3-BSA Induced Apoptosis of VSMCs
2.3. AGE3 Induced VSMC Apoptosis through NAD(P)H Oxidase Activity
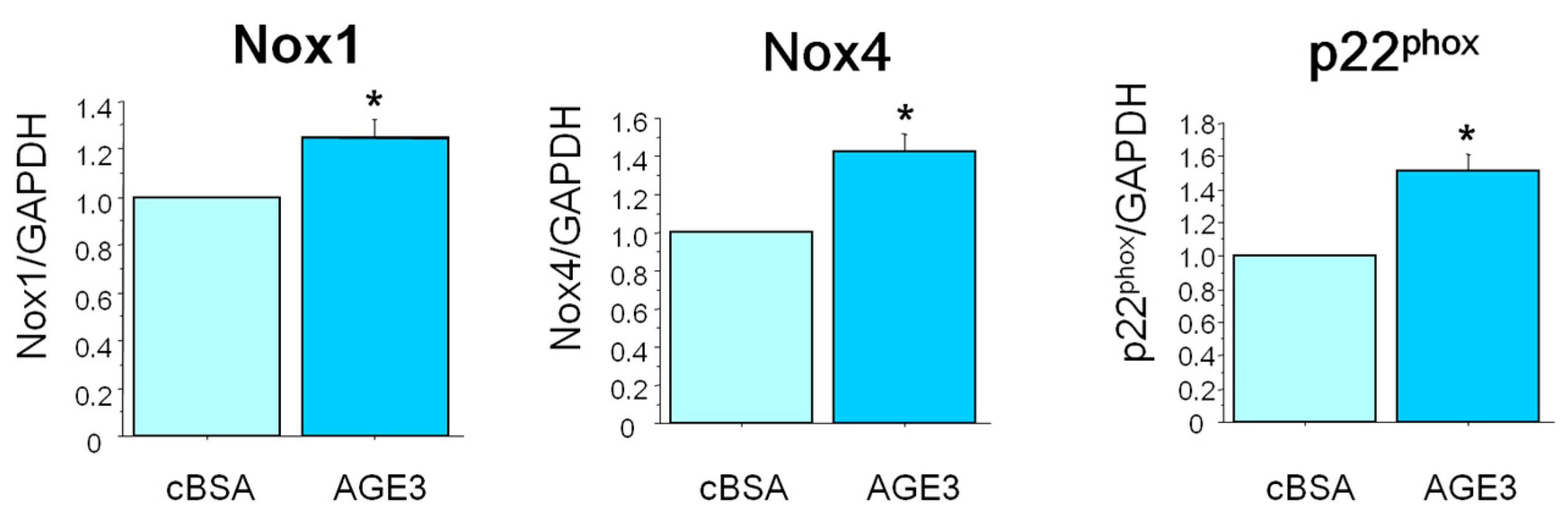
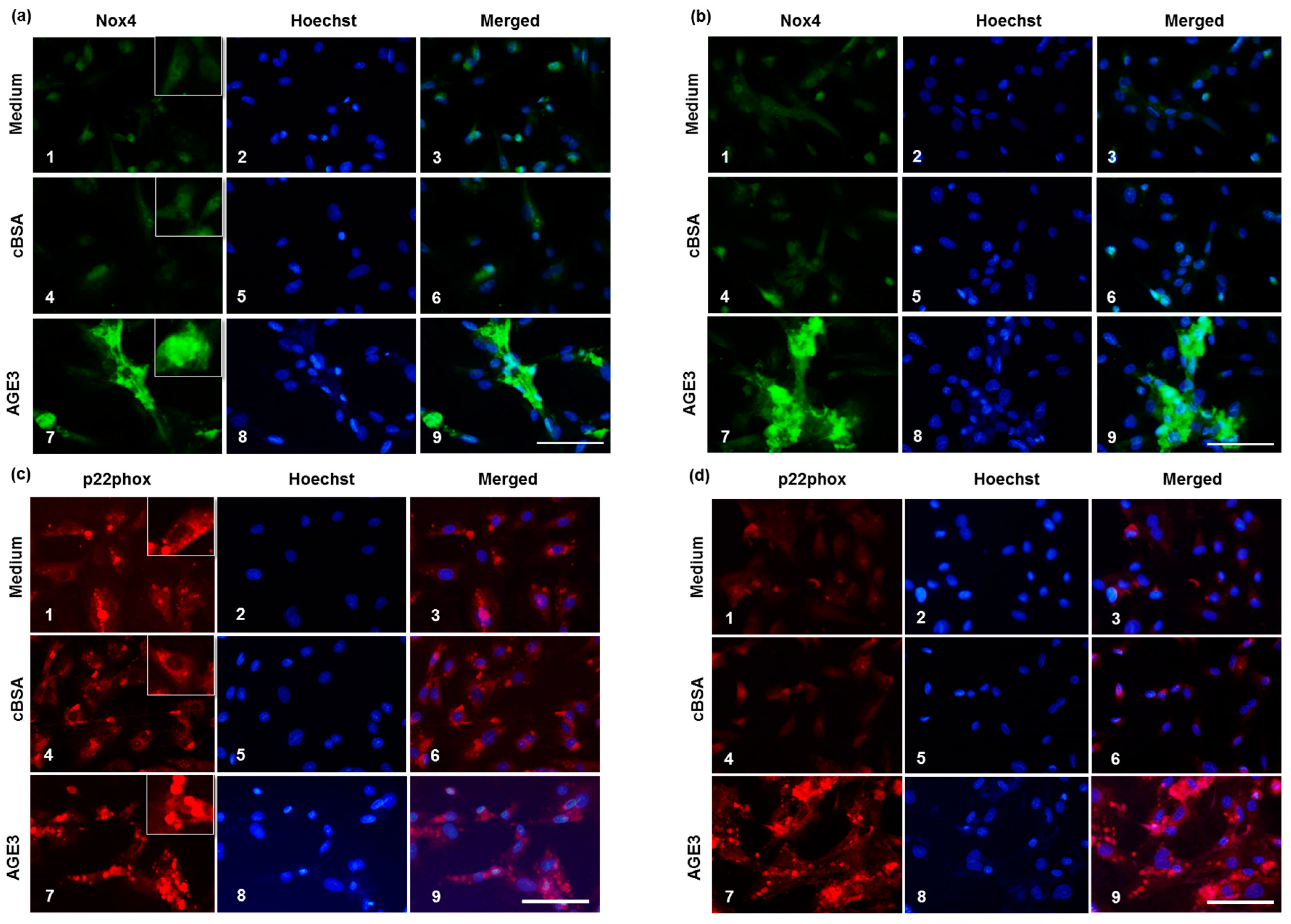
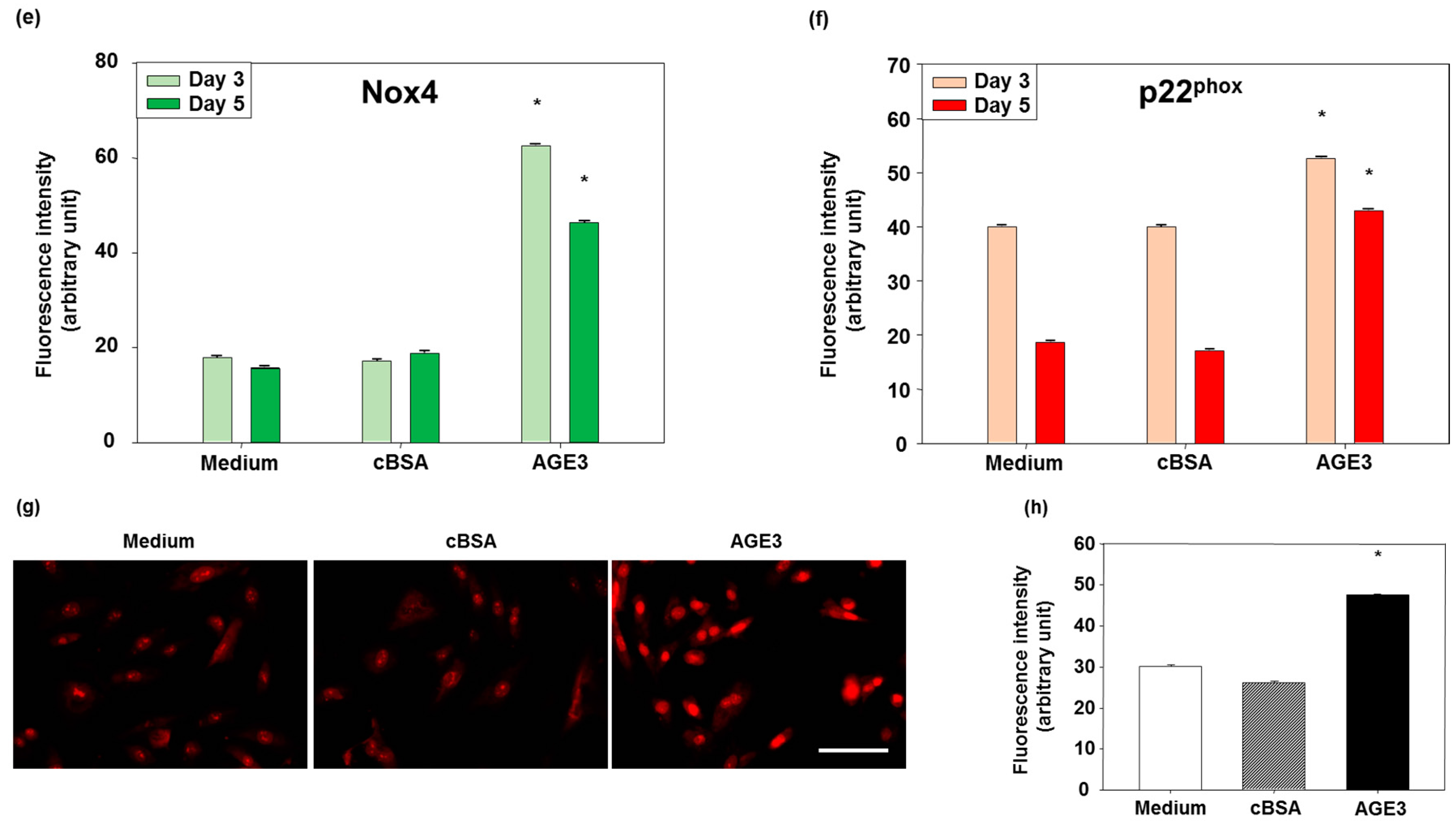
2.4. AGE3-BSA Induced Expression of NAD(P)H Oxidase Components and ROS Generation in VSMCs
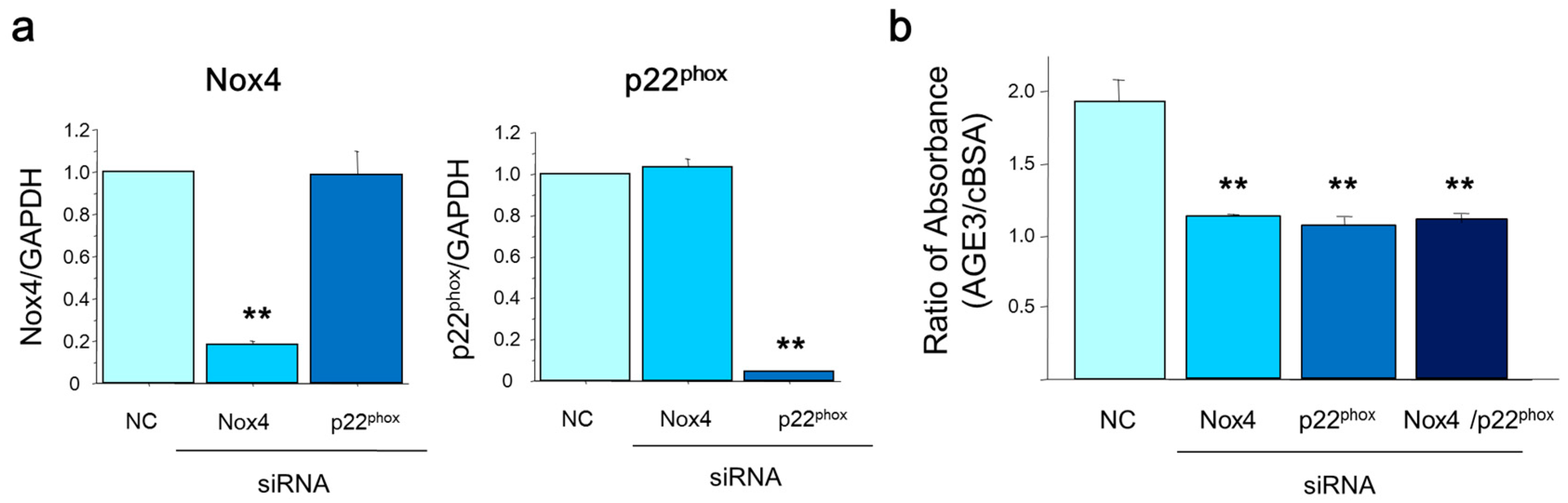
2.5. Silencing Nox4 and p22phox Suppressed AGE3-Induced Apoptosis of A7r5 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Induction of Calcification
4.3. Preparation of AGEs
4.4. Quantification of Calcium Deposition
4.5. TUNEL Assay
4.6. Apoptosis Assay Using a DNA Fragment Detection ELISA Kit
4.7. Quantification of mRNA Expression by Real-Time PCR
4.8. RNA Interference
4.9. Immunofluorescence
4.10. DHE Measurement
4.11. Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AGEs | Advanced glycation end-products |
| BSA | Bovine serum albumin |
| CKD | Chronic kidney disease |
| CV | Cardiovascular |
| ELISA | Enzyme-linked immunosorbent assay |
| NADPH | Nicotinamide adenine dinucleotide phosphate, reduced form |
| PCR | Polymerase chain reaction |
| RAGE | Receptor for AGEs |
| ROS | Reactive oxygen species |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| VSMC | Vascular smooth muscle cell |
References
- Nathan, D.M.; Cleary, P.A.; Backlund, J.Y.; Genuth, S.M.; Lachin, J.M.; Orchard, T.J.; Raskin, P.; Zinman, B.; Diabetes, C. Complications Trial/Epidemiology of Diabetes Investigation and Complications Study Research Group. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar] [PubMed]
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H.A. 10-year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 2008, 359, 1577–1589. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Maeda, S.; Matsui, T.; Ueda, S.; Fukami, K.; Okuda, S. Role of advanced glycation end products (AGEs) and oxidative stress in vascular complications in diabetes. Biochim. Biophys. Acta 2012, 1820, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. European Uremic Toxin Work, G. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Wagner, Z.; Molnar, M.; Molnar, G.A.; Tamasko, M.; Laczy, B.; Wagner, L.; Csiky, B.; Heidland, A.; Nagy, J.; Wittmann, I. Serum carboxymethyllysine predicts mortality in hemodialysis patients. Am. J. Kidney Dis. 2006, 47, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Furuya, R.; Kumagai, H.; Miyata, T.; Fukasawa, H.; Isobe, S.; Kinoshita, N.; Hishida, A. High plasma pentosidine level is accompanied with cardiovascular events in hemodialysis patients. Clin. Exp. Nephrol. 2012, 16, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, Y.; Koyama, H.; Inaba, M. AGEs and cardiovascular diseases in patients with end-stage renal diseases. J. Ren. Nutr. 2012, 22, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Yano, S.; Yamaguchi, T.; Sugimoto, T. Advanced glycation end products-induced reactive oxygen species generation is partly through NF-κB activation in human aortic endothelial cells. J. Diabetes Complicat. 2013, 27, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Soro-Paavonen, A.; Watson, A.M.; Li, J.; Paavonen, K.; Koitka, A.; Calkin, A.C.; Barit, D.; Coughlan, M.T.; Drew, B.G.; Lancaster, G.I.; et al. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes 2008, 57, 2461–2469. [Google Scholar] [CrossRef] [PubMed]
- Lehto, S.; Niskanen, L.; Suhonen, M.; Ronnemaa, T.; Laakso, M. Medial artery calcification. A neglected harbinger of cardiovascular complications in non-insulin-dependent diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 978–983. [Google Scholar] [CrossRef] [PubMed]
- Okuno, S.; Ishimura, E.; Kitatani, K.; Fujino, Y.; Kohno, K.; Maeno, Y.; Maekawa, K.; Yamakawa, T.; Imanishi, Y.; Inaba, M.; et al. Presence of abdominal aortic calcification is significantly associated with all-cause and cardiovascular mortality in maintenance hemodialysis patients. Am. J. Kidney Dis. 2007, 49, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jiang, Y.; Liu, N.; Ren, L.; Zhu, Y.; An, Y.; Chen, D. Advanced glycation end-product nepsilon-carboxymethyl-lysine accelerates progression of atherosclerotic calcification in diabetes. Atherosclerosis 2012, 221, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Satheesan, S.; Figarola, J.L.; Dabbs, T.; Rahbar, S.; Ermel, R. Effects of a new advanced glycation inhibitor, LR-90, on mitigating arterial stiffening and improving arterial elasticity and compliance in a diabetic rat model: Aortic impedance analysis. Br. J. Pharmacol. 2014, 171, 3103–3114. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, K.; Yamaguchi, T.; Tanaka, K.; Notsu, M.; Ogawa, N.; Yano, S.; Sugimoto, T. Advanced glycation end products (AGEs), but not high glucose, inhibit the osteoblastic differentiation of mouse stromal ST2 cells through the suppression of osterix expression, and inhibit cell growth and increasing cell apoptosis. Calcif. Tissue Int. 2012, 91, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Yamaguchi, T.; Kanazawa, I.; Sugimoto, T. Effects of high glucose and advanced glycation end products on the expressions of sclerostin and RANKL as well as apoptosis in osteocyte-like MLO-Y4-A2 cells. Biochem. Biophys. Res. Commun. 2015, 461, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Yu, T.; Yan, Q.C.; Wang, W.; Meng, N.; Li, X.J.; Luo, Y.H. AGEs promote oxidative stress and induce apoptosis in retinal pigmented epithelium cells RAGE-dependently. J. Mol. Neurosci. 2015, 56, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.D.; Xu, C.; Feng, L.; Wang, F. The augmentation of O-GlcNAcylation reduces glyoxal-induced cell injury by attenuating oxidative stress in human retinal microvascular endothelial cells. Int. J. Mol. Med. 2015, 36, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yin, L.; Xu, Z.; An, F.M.; Liu, A.R.; Wang, Y.; Yao, W.B.; Gao, X.D. Inhibiting receptor for advanced glycation end product (AGE) and oxidative stress involved in the protective effect mediated by glucagon-like peptide-1 receptor on AGE induced neuronal apoptosis. Neurosci. Lett. 2016, 612, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Tada, Y.; Yano, S.; Yamaguchi, T.; Okazaki, K.; Ogawa, N.; Morita, M.; Sugimoto, T. Advanced glycation end products-induced vascular calcification is mediated by oxidative stress: Functional roles of NAD(P)H-oxidase. Horm. Metab. Res. 2013, 45, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Parhami, F.; Morrow, A.D.; Balucan, J.; Leitinger, N.; Watson, A.D.; Tintut, Y.; Berliner, J.A.; Demer, L.L. Lipid oxidation products have opposite effects on calcifying vascular cell and bone cell differentiation. A possible explanation for the paradox of arterial calcification in osteoporotic patients. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 680–687. [Google Scholar] [CrossRef] [PubMed]
- San Martin, A.; Du, P.; Dikalova, A.; Lassegue, B.; Aleman, M.; Gongora, M.C.; Brown, K.; Joseph, G.; Harrison, D.G.; Taylor, W.R.; et al. Reactive oxygen species-selective regulation of aortic inflammatory gene expression in Type 2 diabetes. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2073–H2082. [Google Scholar] [CrossRef] [PubMed]
- Hata, I.; Kaji, M.; Hirano, S.; Shigematsu, Y.; Tsukahara, H.; Mayumi, M. Urinary oxidative stress markers in young patients with type 1 diabetes. Pediatr. Int. 2006, 48, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Santilli, F.; D’Ardes, D.; Davi, G. Oxidative stress in chronic vascular disease: From prediction to prevention. Vasc. Pharmacol. 2015, 74, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell transformation by the superoxide-generating oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar] [PubMed]
- Lassegue, B.; Sorescu, D.; Szocs, K.; Yin, Q.; Akers, M.; Zhang, Y.; Grant, S.L.; Lambeth, J.D.; Griendling, K.K. Novel gp91phox homologues in vascular smooth muscle cells: Nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ. Res. 2001, 88, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Fukui, T.; Lassegue, B.; Kai, H.; Alexander, R.W.; Griendling, K.K. Cytochrome B-558 α-subunit cloning and expression in rat aortic smooth muscle cells. Biochim. Biophys. Acta 1995, 1231, 215–219. [Google Scholar] [CrossRef]
- Bleeke, T.; Zhang, H.; Madamanchi, N.; Patterson, C.; Faber, J.E. Catecholamine-induced vascular wall growth is dependent on generation of reactive oxygen species. Circ. Res. 2004, 94, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Ellmark, S.H.; Dusting, G.J.; Fui, M.N.; Guzzo-Pernell, N.; Drummond, G.R. The contribution of Nox4 to NADPH oxidase activity in mouse vascular smooth muscle. Cardiovasc. Res. 2005, 65, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A.; Brandes, R.P.; Bassus, S.; Kronemann, N.; Kirchmaier, C.M.; Busse, R.; Schini-Kerth, V.B. Oxidative stress and expression of p22phox are involved in the up-regulation of tissue factor in vascular smooth muscle cells in response to activated platelets. FASEB J. 2000, 14, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Khatri, J.J.; Johnson, C.; Magid, R.; Lessner, S.M.; Laude, K.M.; Dikalov, S.I.; Harrison, D.G.; Sung, H.J.; Rong, Y.; Galis, Z.S. Vascular oxidant stress enhances progression and angiogenesis of experimental atheroma. Circulation 2004, 109, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Brewer, A.C.; Schroder, K.; Santos, C.X.; Grieve, D.J.; Wang, M.; Anilkumar, N.; Yu, B.; Dong, X.; Walker, S.J.; et al. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18121–18126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, Y.H.; Quach, T.; Saito, R.; Pham, J.; Sharma, K. Metabolomics reveals a key role for fumarate in mediating the effects of NADPH Oxidase 4 in diabetic kidney disease. J. Am. Soc. Nephrol. 2016, 27, 466–481. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Lee, M.S.; Son, S.M.; Kim, I.J.; Lee, W.S.; Rhim, B.Y.; Hong, K.W.; Kim, C.D. Vascular NADH oxidase is involved in impaired endothelium-dependent vasodilation in OLETF rats, a model of type 2 diabetes. Diabetes 2002, 51, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Serrander, L.; Cartier, L.; Bedard, K.; Banfi, B.; Lardy, B.; Plastre, O.; Sienkiewicz, A.; Forro, L.; Schlegel, W.; Krause, K.H. Nox4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 2007, 406, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Son, B.K.; Kozaki, K.; Iijima, K.; Eto, M.; Kojima, T.; Ota, H.; Senda, Y.; Maemura, K.; Nakano, T.; Akishita, M.; et al. Statins protect human aortic smooth muscle cells from inorganic phosphate-induced calcification by restoring Gas6-Axl survival pathway. Circ. Res. 2006, 98, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Iyemere, V.P.; Proudfoot, D.; Weissberg, P.L.; Shanahan, C.M. Vascular smooth muscle cell phenotypic plasticity and the regulation of vascular calcification. J. Intern. Med. 2006, 260, 192–210. [Google Scholar] [CrossRef] [PubMed]
- Adeva-Andany, M.M.; Fernandez-Fernandez, C.; Sanchez-Bello, R.; Donapetry-Garcia, C.; Martinez-Rodriguez, J. The role of carbonic anhydrase in the pathogenesis of vascular calcification in humans. Atherosclerosis 2015, 241, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Harper, E.; Forde, H.; Davenport, C.; Rochfort, K.D.; Smith, D.; Cummins, P.M. Vascular calcification in type-2 diabetes and cardiovascular disease: Integrative roles for OPG, RANKL and TRAIL. Vasc. Pharmacol. 2016, 82, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Tanikawa, T.; Okada, Y.; Tanikawa, R.; Tanaka, Y. Advanced glycation end products induce calcification of vascular smooth muscle cells through RAGE/p38 MAPK. J. Vasc. Res. 2009, 46, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Wu, S.; Wang, Q.; Shi, Y.; Liu, G.; Zhi, J.; Wang, F. Saikosaponin-D reduces HO-induced PC12 cell apoptosis by removing ROS and blocking MAPK-dependent oxidative damage. Cell. Mol. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pai, A.; Leaf, E.M.; El-Abbadi, M.; Giachelli, C.M. Elastin degradation and vascular smooth muscle cell phenotype change precede cell loss and arterial medial calcification in a uremic mouse model of chronic kidney disease. Am. J. Pathol. 2011, 178, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Son, B.K.; Akishita, M.; Iijima, K.; Ogawa, S.; Arai, T.; Ishii, H.; Maemura, K.; Aburatani, H.; Eto, M.; Ouchi, Y. Thrombomodulin, a novel molecule regulating inorganic phosphate-induced vascular smooth muscle cell calcification. J. Mol. Cell Cardiol. 2013, 56, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Suzuki, M.; Hattori, S.; Kasai, K. Vascular smooth muscle cell activation by glycated albumin (Amadori adducts). Hypertension 2002, 39, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Casella, S.; Bielli, A.; Mauriello, A.; Orlandi, A. Molecular pathways regulating macrovascular pathology and vascular smooth muscle cells phenotype in type 2 diabetes. Int. J. Mol. Sci. 2015, 16, 24353–24368. [Google Scholar] [CrossRef] [PubMed]
- Maltais, J.S.; Simard, E.; Froehlich, U.; Denault, J.B.; Gendron, L.; Grandbois, M. iRAGE as a novel carboxymethylated peptide that prevents advanced glycation end product-induced apoptosis and endoplasmic reticulum stress in vascular smooth muscle cells. Pharmacol. Res. 2016, 104, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Simard, E.; Sollradl, T.; Maltais, J.S.; Boucher, J.; D’Orleans-Juste, P.; Grandbois, M. Receptor for advanced glycation end-products signaling interferes with the vascular smooth muscle cell contractile phenotype and function. PLoS ONE 2015, 10, e0128881. [Google Scholar]
- Chawla, V.; Simionescu, A.; Langan, E.M., 3rd; LaBerge, M. Influence of clinically relevant mechanical forces on vascular smooth muscle cells under chronic high glucose: An in vitro dynamic disease model. Ann. Vasc. Surg. 2016, 34, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Shioi, A.; Nishizawa, Y.; Jono, S.; Koyama, H.; Hosoi, M.; Morii, H. β-Glycerophosphate accelerates calcification in cultured bovine vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 2003–2009. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, N.; Yamaguchi, T.; Yano, S.; Yamauchi, M.; Yamamoto, M.; Sugimoto, T. The combination of high glucose and advanced glycation end-products (AGEs) inhibits the mineralization of osteoblastic MC3T3-E1 cells through glucose-induced increase in the receptor for AGEs. Horm. Metab. Res. 2007, 39, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Yamaguchi, T.; Kanazawa, I.; Ogawa, N.; Hayashi, K.; Yamauchi, M.; Sugimoto, T. The uraemic toxin phenylacetic acid inhibits osteoblastic proliferation and differentiation: An implication for the pathogenesis of low turnover bone in chronic renal failure. Nephrol. Dial. Transplant. 2007, 22, 3160–3165. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.M.; Michikawa, M.; Kim, S.U.; Nagai, A. Lysophosphatidylcholine increases the neurotoxicity of Alzheimer’s amyloid β1–42 peptide: Role of oligomer formation. Neuroscience 2015, 292, 159–169. [Google Scholar] [CrossRef] [PubMed]







© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koike, S.; Yano, S.; Tanaka, S.; Sheikh, A.M.; Nagai, A.; Sugimoto, T. Advanced Glycation End-Products Induce Apoptosis of Vascular Smooth Muscle Cells: A Mechanism for Vascular Calcification. Int. J. Mol. Sci. 2016, 17, 1567. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091567
Koike S, Yano S, Tanaka S, Sheikh AM, Nagai A, Sugimoto T. Advanced Glycation End-Products Induce Apoptosis of Vascular Smooth Muscle Cells: A Mechanism for Vascular Calcification. International Journal of Molecular Sciences. 2016; 17(9):1567. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091567
Chicago/Turabian StyleKoike, Sayo, Shozo Yano, Sayuri Tanaka, Abdullah M. Sheikh, Atsushi Nagai, and Toshitsugu Sugimoto. 2016. "Advanced Glycation End-Products Induce Apoptosis of Vascular Smooth Muscle Cells: A Mechanism for Vascular Calcification" International Journal of Molecular Sciences 17, no. 9: 1567. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091567