
Protein Kinases and Parkinson’s Disease


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Leucine-Rich Repeat Kinase 2 (LRRK2)
3. Phosphatase and Tensin Homolog (PTEN)-Induced Putative Kinase 1 (PINK1)
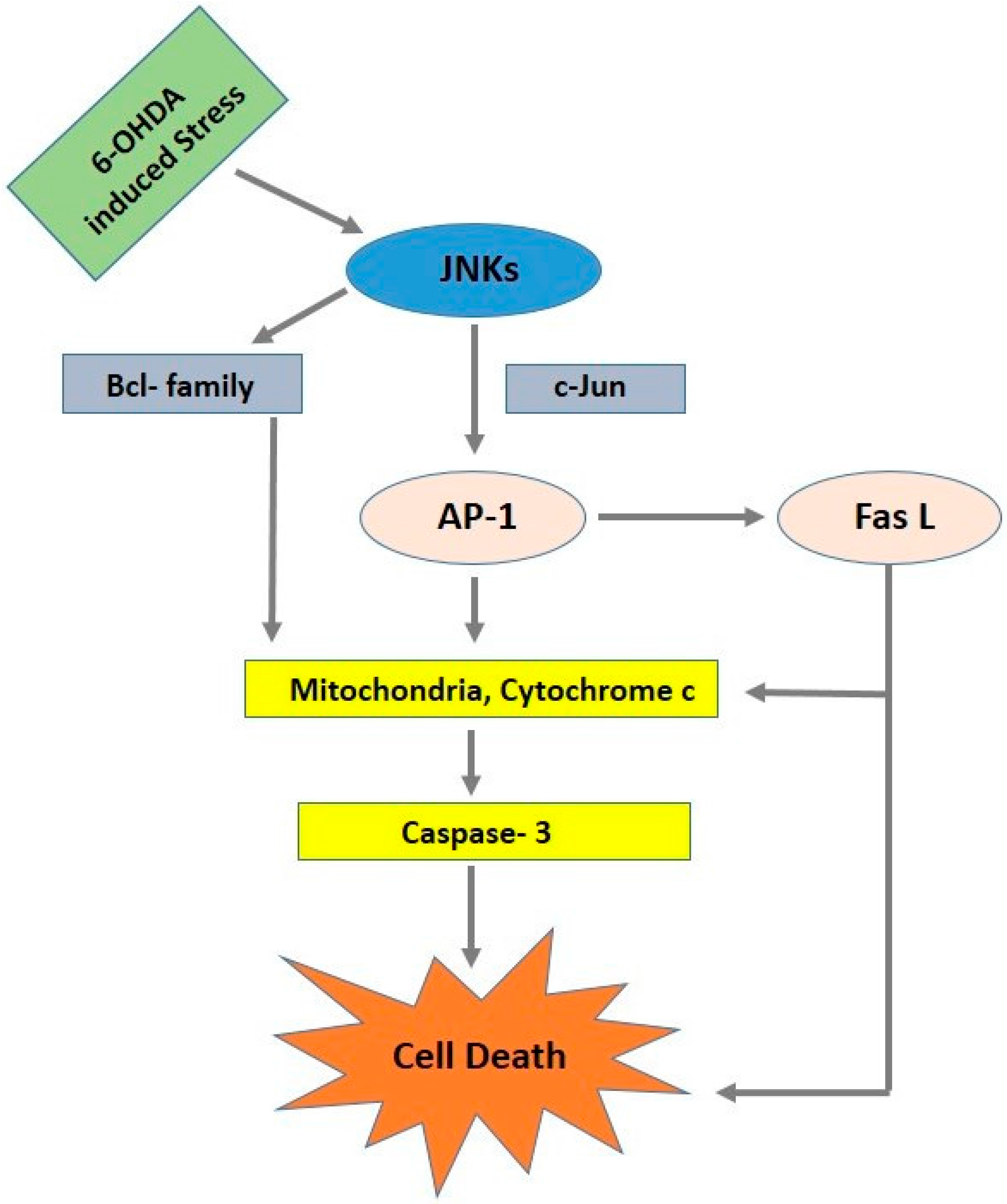
4. c-Jun N-Terminal Kinase Signaling Pathway (JNK)
5. AKT (Protein Kinase B) Signaling Pathway
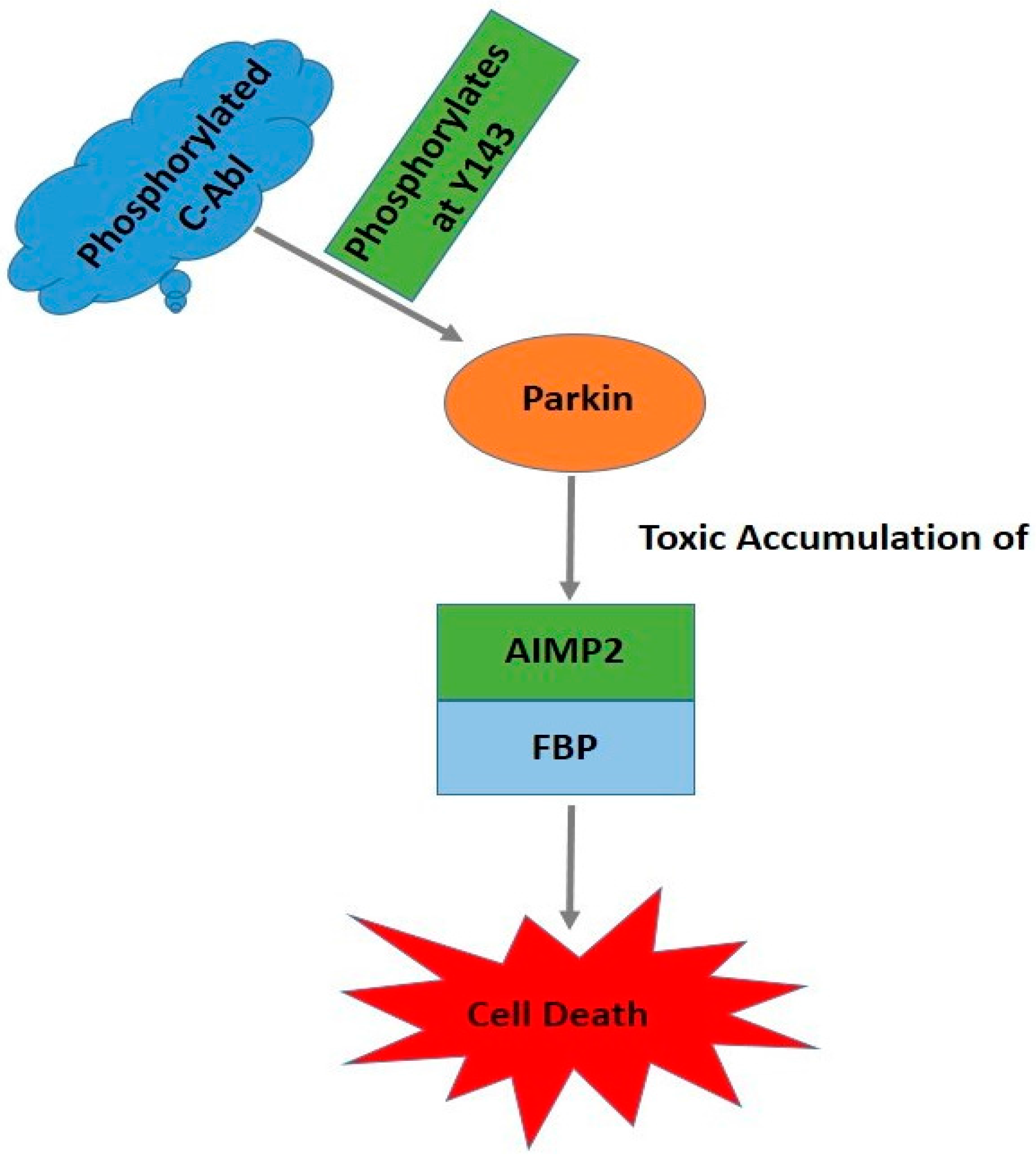
6. c-Abl
7. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Manning, G.; Plowman, G.D.; Hunter, T.; Sudarsanam, S. Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 2002, 27, 514–520. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Liu, Y.; Uno, T.; Gray, N. Creating chemical diversity to target protein kinases. Comb. Chem. High Throughput Screen. 2004, 7, 453–472. [Google Scholar] [CrossRef] [PubMed]
- Imam, S.Z.; Zhou, Q.; Yamamoto, A.; Valente, A.J.; Ali, S.F.; Bains, M.; Roberts, J.L.; Kahle, P.J.; Clark, R.A.; Li, S.L. Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: Implications for Parkinson’s disease. J. Neurosci. 2011, 31, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Pagan, F.; Hebron, M.; Valadez, E.H.; Torres-Yaghi, Y.; Huang, X.; Mills, R.R.; Wilmarth, B.M.; Howard, H.; Dunn, C.; Carlson, A.; et al. Nilotinib effects in Parkinson’s disease and dementia with lewy bodies. J. Parkinsons Dis. 2016, 6, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Moghal, S.; Rajput, A.H.; Darcy, C.; Rajput, R. Prevalence of movement-disorders in elderly community residents. Neuroepidemiology 1994, 13, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Savitt, J.M.; Dawson, V.L.; Dawson, T.M. Diagnosis and treatment of Parkinson disease: Molecules to medicine. J. Clin. Investig. 2006, 116, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Paisan-Ruiz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simon, J.; van der Brug, M.; de Munain, A.L.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.N.; Wang, Y.; Karlins, E.; Saito, L.; Mejia-Santana, H.; Harris, J.; Louis, E.D.; Cote, L.J.; Andrews, H.; Fahn, S.; et al. Frequency of LRRK2 mutations in early- and late-onset Parkinson disease. Neurology 2006, 67, 1786–1791. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R.; Bandmann, O. Parkinson’s disease: Insights from pathways. Hum. Mol. Genet. 2010, 19, R21–R27. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R.; Xiromerisiou, G.; Singleton, A. How genetics research in Parkinson’s disease is enhancing understanding of the common idiopathic forms of the disease. Curr. Opin. Neurol. 2005, 18, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Correia Guedes, L.; Ferreira, J.J.; Rosa, M.M.; Coelho, M.; Bonifati, V.; Sampaio, C. Worldwide frequency of G2019s LRRK2 mutation in Parkinson’s disease: A systematic review. Parkinsonism Relat. Disord. 2010, 16, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Dachsel, J.C.; Farrer, M.J. LRRK2 and Parkinson disease. Arch. Neurol. 2010, 67, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Kett, L.R.; Dauer, W.T. Leucine-rich repeat kinase 2 for beginners: Six key questions. Csh. Perspect. Med. 2012, 2, a009407. [Google Scholar] [CrossRef] [PubMed]
- Jaleel, M.; Nichols, R.J.; Deak, M.; Campbell, D.G.; Gillardon, F.; Knebel, A.; Alessi, D.R. LRRK2 phosphorylates moesin at threonine-558: Characterization of how Parkinson’s disease mutants affect kinase activity. Biochem. J. 2007, 405, 307–317. [Google Scholar] [CrossRef] [PubMed]
- West, A.B.; Moore, D.J.; Biskup, S.; Bugayenko, A.; Smith, W.W.; Ross, C.A.; Dawson, V.L.; Dawson, T.M. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. USA 2005, 102, 16842–16847. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.L.; Wu, C.X.; Burlak, C.; Zhang, S.; Sahm, H.; Wang, M.; Zhang, Z.Y.; Vogel, K.W.; Federici, M.; Riddle, S.M.; et al. Parkinson disease-associated mutation R1441H in LRRK2 prolongs the “active state” of its GTPase domain. Proc. Natl. Acad. Sci. USA 2014, 111, 4055–4060. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.J.; Zhang, S.O.; Bustos, D.; Kleinheinz, T.; Le Pichon, C.E.; Dominguez, S.L.; Solanoy, H.O.; Drummond, J.; Zhang, X.L.; Ding, X.; et al. Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci. Transl. Med. 2012, 4, 164ra161. [Google Scholar] [CrossRef] [PubMed]
- Taymans, J.M. The GTPase function of LRRK2. Biochem. Soc. Trans. 2012, 40, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Taymans, J.M.; Vancraenenbroeck, R.; Ollikainen, P.; Beilina, A.; Lobbestael, E.; de Maeyer, M.; Baekelandt, V.; Cookson, M.R. LRRK2 kinase activity is dependent on LRRK2 GTP binding capacity but independent of LRRK2 GTP binding. PLoS ONE 2011, 6, e23207. [Google Scholar] [CrossRef] [PubMed]
- Greggio, E.; Jain, S.; Kingsbury, A.; Bandopadhyay, R.; Lewis, P.; Kaganovich, A.; van der Brug, M.P.; Beilina, A.; Blackinton, J.; Thomas, K.J.; et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 2006, 23, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Hatano, T.; Kubo, S.; Imai, S.; Maeda, M.; Ishikawa, K.; Mizuno, Y.; Hattori, N. Leucine-rich repeat kinase 2 associates with lipid rafts. Hum. Mol. Genet. 2007, 16, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Luzon-Toro, B.; de la Torre, E.R.; Delgado, A.; Perez-Tur, J.; Hilfiker, S. Mechanistic insight into the dominant mode of the Parkinson’s disease-associated G2019S LRRK2 mutation. Hum. Mol. Genet. 2007, 16, 2031–2039. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, D.; Dowman, J.; Hammond, R.; Leete, T.; Inoue, K.; Abeliovich, A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 2006, 52, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.A.; Greggio, E.; Beilina, A.; Jain, S.; Baker, A.; Cookson, M.R. The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem. Biophys. Res. Commun. 2007, 357, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Li, X.T.; Tan, Y.C.; Poulose, S.; Olanow, C.W.; Huang, X.Y.; Yue, Z.Y. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson’s disease R1441C/G mutants. J. Neurochem. 2007, 103, 238–247. [Google Scholar] [PubMed]
- Sakaguchi-Nakashima, A.; Meir, J.Y.; Jin, Y.; Matsumoto, K.; Hisamoto, N. LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr. Biol. 2007, 17, 592–598. [Google Scholar] [PubMed]
- Lee, B.D.; Shin, J.H.; VanKampen, J.; Petrucelli, L.; West, A.B.; Ko, H.S.; Lee, Y.I.; Maguire-Zeiss, K.A.; Bowers, W.J.; Federoff, H.J.; et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat. Med. 2010, 16, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Kawajiri, S.; Saiki, S.; Sato, S.; Hattori, N. Genetic mutations and functions of PINK1. Trend Pharmacol. Sci. 2011, 32, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Abou-Sleiman, P.M.; Muqit, M.M.; Wood, N.W. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat. Rev. Neurosci. 2006, 7, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Woodroof, H.I.; Pogson, J.H.; Begley, M.; Cantley, L.C.; Deak, M.; Campbell, D.G.; van Aalten, D.M.; Whitworth, A.J.; Alessi, D.R.; Muqit, M.M. Discovery of catalytically active orthologues of the Parkinson’s disease kinase PINK1: Analysis of substrate specificity and impact of mutations. Open Biol. 2011, 1, 110012. [Google Scholar] [CrossRef] [PubMed]
- Moretti, R.; Pansiot, J.; Bettati, D.; Strazielle, N.; Ghersi-Egea, J.F.; Damante, G.; Fleiss, B.; Titomanlio, L.; Gressens, P. Blood-brain barrier dysfunction in disorders of the developing brain. Front. Neurosci. 2015, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Petit, A.; Kawarai, T.; Paitel, E.; Sanjo, N.; Maj, M.; Scheid, M.; Chen, F.S.; Gu, Y.J.; Hasegawa, H.; Salehi-Rad, S.; et al. Wild-type pink1 prevents basal and induced neuronal apoptosis, a protective effect abrogated by Parkinson disease-related mutations. J. Biol. Chem. 2005, 280, 34025–34032. [Google Scholar] [CrossRef] [PubMed]
- Dagda, R.K.; Cherra, S.J.; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009, 284, 13843–13855. [Google Scholar] [CrossRef] [PubMed]
- Okatsu, K.; Oka, T.; Iguchi, M.; Imamura, K.; Kosako, H.; Tani, N.; Kimura, M.; Go, E.; Koyano, F.; Funayama, M.; et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 2012, 3, 1016. [Google Scholar] [CrossRef] [PubMed]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Muqit, M.M.K.; Stanyer, L.; Healy, D.G.; Abou-Sleiman, P.M.; Hargreaves, I.; Heales, S.; Ganguly, M.; Parsons, L.; Lees, A.J.; et al. Pink1 protein in normal human brain and Parkinson’s disease. Brain 2006, 129, 1720–1731. [Google Scholar] [CrossRef] [PubMed]
- Samaranch, L.; Lorenzo-Betancor, O.; Arbelo, J.M.; Ferrer, I.; Lorenzo, E.; Irigoyen, J.; Pastor, M.A.; Marrero, C.; Isla, C.; Herrera-Henriquez, J.; et al. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 2010, 133, 1128–1142. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Casado, M.E.; Lima, E.; Garcia, J.A.; Doerrier, C.; Aranda, P.; Sayed, R.K.; Guerra-Librero, A.; Escames, G.; Lopez, L.C.; Acuna-Castroviejo, D. Melatonin rescues zebrafish embryos from the parkinsonian phenotype restoring the parkin/PINK1/DJ-1/MUL1 network. J. Pineal Res. 2016, 61, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: A 10-year update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [PubMed]
- Derijard, B.; Hibi, M.; Wu, I.H.; Barrett, T.; Su, B.; Deng, T.; Karin, M.; Davis, R.J. JNK1: A protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 1994, 76, 1025–1037. [Google Scholar] [CrossRef]
- Maroney, A.C.; Finn, J.P.; Bozyczko-Coyne, D.; O’Kane, T.M.; Neff, N.T.; Tolkovsky, A.M.; Park, D.S.; Yan, C.Y.I.; Troy, C.M.; Greene, L.A. CEP-1347 (KT7515), an inhibitor of JNK activation, rescues sympathetic neurons and neuronally differentiated PC12 cells from death evoked by three distinct insults. J. Neurochem. 1999, 73, 1901–1912. [Google Scholar] [PubMed]
- Trotter, L.; Panton, W.; Hajimohamadreza, I.; Petalidis, L.; Ward, R.; Fleming, Y.; Armstrong, C.G.; Cohen, P.; Karran, E.H.; Kinloch, R.A. Mitogen-activated protein kinase kinase 7 is activated during low potassium-induced apoptosis in rat cerebellar granule neurons. Neurosci. Lett. 2002, 320, 29–32. [Google Scholar] [CrossRef]
- Troy, C.M.; Rabacchi, S.A.; Xu, Z.; Maroney, A.C.; Connors, T.J.; Shelanski, M.L.; Greene, L.A. β-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J. Neurochem. 2001, 77, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Xu, Z.; Kukekov, N.V.; Gire, S.; Greene, L.A. Proapoptotic Nix activates the JNK pathway by interacting with POSH and mediates death in a Parkinson disease model. J. Biol. Chem. 2007, 282, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Greene, L.A. Activation of the apoptotic JNK pathway through the Rac1-binding scaffold protein POSH. Method. Enzymol. 2006, 406, 479–489. [Google Scholar]
- Xu, Z.; Kukekov, N.V.; Greene, L.A. POSH acts as a scaffold for a multiprotein complex that mediates JNK activation in apoptosis. EMBO J. 2003, 22, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Kukekov, N.V.; Greene, L.A. Regulation of apoptotic c-Jun N-terminal kinase signaling by a stabilization-based feed-forward loop. Mol. Cell. Biol. 2005, 25, 9949–9959. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Maroney, A.C.; Dobrzanski, P.; Kukekov, N.V.; Greene, L.A. The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol. Cell. Biol. 2001, 21, 4713–4724. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Sproul, A.; Wang, W.; Kukekov, N.; Greene, L.A. Siah1 interacts with the scaffold protein POSH to promote JNK activation and apoptosis. J. Biol. Chem. 2006, 281, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.D.; Kuan, C.Y.; Whitmarsh, A.J.; Rincon, M.; Zheng, T.S.; Davis, R.J.; Rakic, P.; Flavell, R.A. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the JNK3 gene. Nature 1997, 389, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.S.; Yoon, S.Y.; Oh, T.H.; Choi, E.J.; O’Malley, K.L.; Oh, Y.J. Two distinct mechanisms are involved in 6-hydroxydopamine- and MPP+-induced dopaminergic neuronal cell death: Role of caspases, ROS, and JNK. J. Neurosci. Res. 1999, 57, 86–94. [Google Scholar] [CrossRef]
- Saporito, M.S.; Thomas, B.A.; Scott, R.W. MPTP activates c-Jun NH2-terminal kinase (JNK) and its upstream regulatory kinase MKK4 in nigrostriatal neurons in vivo. J. Neurochem. 2000, 75, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Xing, B.; Liu, M.; Bing, G. Neuroprotection with pioglitazone against LPS insult on dopaminergic neurons may be associated with its inhibition of NF-κB and JNK activation and suppression of COX-2 activity. J. Neuroimmunol. 2007, 192, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Hunot, S.; Vila, M.; Teismann, P.; Davis, R.J.; Hirsch, E.C.; Przedborski, S.; Rakic, P.; Flavell, R.A. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.W.; Howard, S.; LoGrasso, P.V. Blocking c-Jun N-terminal kinase (JNK) translocation to the mitochondria prevents 6-hydroxydopamine-induced toxicity in vitro and in vivo. J. Biol. Chem. 2013, 288, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.W.; Pachori, A.; Howard, S.; Ganno, M.; Hansen, D., Jr.; Kamenecka, T.; Song, X.; Duckett, D.; Chen, W.; Ling, Y.Y.; et al. Small molecule c-Jun-N-terminal kinase (JNK) inhibitors protect dopaminergic neurons in a model of Parkinson’s disease. ACS Chem. Neurosci. 2011, 2, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Saporito, M.S.; Brown, E.M.; Miller, M.S.; Carswell, S. CEP-1347/KT-7515, an inhibitor of c-jun N-terminal kinase activation, attenuates the 1-methyl-4-phenyl tetrahydropyridine-mediated loss of nigrostriatal dopaminergic neurons in vivo. J. Pharmacol. Exp. Ther. 1999, 288, 421–427. [Google Scholar] [PubMed]
- Wang, W.; Shi, L.; Xie, Y.; Ma, C.; Li, W.; Su, X.; Huang, S.; Chen, R.; Zhu, Z.; Mao, Z.; et al. Sp600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Neurosci. Res. 2004, 48, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; O’Connell, J.R.; McArdle, P.F.; Wade, J.B.; Dorff, S.E.; Shah, S.J.; Shi, X.L.; Pan, L.; Rampersaud, E.; Shen, H.Q.; et al. Whole-genome association study identifies STK39 as a hypertension susceptibility gene. Proc. Natl. Acad. Sci. USA 2009, 106, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Castro-Caldas, M.; Carvalho, A.N.; Rodrigues, E.; Henderson, C.J.; Wolf, C.R.; Rodrigues, C.M.; Gama, M.J. Tauroursodeoxycholic acid prevents MPTP-induced dopaminergic cell death in a mouse model of Parkinson’s disease. Mol. Neurobiol. 2012, 46, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Im, J.Y.; Woo, J.M.; Grosso, H.; Kim, Y.S.; Cristovao, A.C.; Sonsalla, P.K.; Schuster, D.S.; Jalbut, M.M.; Fernandez, J.R.; et al. Neuroprotective and anti-inflammatory properties of a coffee component in the MPTP model of Parkinson’s disease. Neurotherapeutics 2013, 10, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Zhai, A.; Zhu, X.; Wang, X.; Chen, R.; Wang, H. Secalonic acid A protects dopaminergic neurons from 1-methyl-4-phenylpyridinium MPP+-induced cell death via the mitochondrial apoptotic pathway. Eur. J. Pharmacol. 2013, 713, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Pei, D.S.; Wang, X.T.; Liu, Y.; Sun, Y.F.; Guan, Q.H.; Wang, W.; Yan, J.Z.; Zong, Y.Y.; Xu, T.L.; Zhang, G.Y. Neuroprotection against ischaemic brain injury by a GluR6-9c peptide containing the TAT protein transduction sequence. Brain 2006, 129, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Faris, M.; Latinis, K.M.; Kempiak, S.J.; Koretzky, G.A.; Nel, A. Stress-induced FAS ligand expression in T cells is mediated through a MEK kinase 1-regulated response element in the FAS ligand promoter. Mol. Cell. Biol. 1998, 18, 5414–5424. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Ying, C.Y.; Yuan, Z.M.; Song, B.; Li, D.; Liu, Y.L.; Lai, B.Q.; Li, W.M.; Chen, R.Z.; Ching, Y.P.; et al. dp5/HRK is a c-Jun target gene and required for apoptosis induced by potassium deprivation in cerebellar granule neurons. J. Biol. Chem. 2007, 282, 30901–30909. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, J.; Neame, S.J.; Paquet, L.; Bernard, O.; Ham, J. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron 2001, 29, 629–643. [Google Scholar] [CrossRef]
- Pan, J.; Wang, G.; Yang, H.Q.; Hong, Z.; Xiao, Q.; Ren, J.; Zhou, H.Y.; Bai, L.; Chen, S.D. K252a prevents nigral dopaminergic cell death induced by 6-hydroxydopamine through inhibition of both mixed-lineage kinase 3/c-Jun NH2-terminal kinase 3 (JNK3) and apoptosis-inducing kinase 1/JNK3 signaling pathways. Mol. Pharmacol. 2007, 72, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Xiao, Q.; Sheng, C.Y.; Hong, Z.; Yang, H.Q.; Wang, G.; Ding, J.Q.; Chen, S.D. Blockade of the translocation and activation of c-Jun N-terminal kinase 3 (JNK3) attenuates dopaminergic neuronal damage in mouse model of Parkinson’s disease. Neurochem. Int. 2009, 54, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Fayard, E.; Xue, G.D.; Parcellier, A.; Bozulic, L.; Hemmings, B.A. Protein kinase B (PKB/Akt), a key mediator of the PI3K signaling pathway. Curr. Top. Microbiol. Immunol. 2010, 346, 31–56. [Google Scholar] [PubMed]
- Song, G.; Ouyang, G.L.; Bao, S.D. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Timmons, S.; Coakley, M.F.; Moloney, A.M.; O’Neill, C. Akt signal transduction dysfunction in Parkinson’s disease. Neurosci. Lett. 2009, 467, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Malagelada, C.; Jin, Z.H.; Greene, L.A. RTP801 is induced in Parkinson’s disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J. Neurosci. 2008, 28, 14363–14371. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.A.; Levy, O.; Malagelada, C. Akt as a victim, villain and potential hero in Parkinson’s disease pathophysiology and treatment. Cell. Mol. Neurobiol. 2011, 31, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Ries, V.; Henchcliffe, C.; Kareva, T.; Rzhetskaya, M.; Bland, R.; During, M.J.; Kholodilov, N.; Burke, R.E. Oncoprotein Akt/PKB induces trophic effects in murine models of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 18757–18762. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.H.; Khursigara, G.; Sun, X.; Franke, T.F.; Chao, M.V. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol. Cell. Biol. 2001, 21, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.H.; Yano, H.; Cho, H.; Meyer, D.; Monks, B.; Margolis, B.; Birnbaum, M.J.; Chao, M.V. Akt1 regulates a JNK scaffold during excitotoxic apoptosis. Neuron 2002, 35, 697–709. [Google Scholar] [CrossRef]
- Moresco, E.M.Y.; Koleske, A.J. Regulation of neuronal morphogenesis and synaptic function by Abl family kinases. Curr. Opin. Neurobiol. 2003, 13, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Hantschel, O.; Superti-Furga, G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat. Rev. Mol. Cell Biol. 2004, 5, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Sirvent, A.; Benistant, C.; Roche, S. Oncogenic signaling by tyrosine kinases of the SRC family in advanced colorectal cancer. Am. J. Cancer Res. 2012, 2, 357–371. [Google Scholar] [PubMed]
- Ko, H.S.; Lee, Y.; Shin, J.H.; Karuppagounder, S.S.; Gadad, B.S.; Koleske, A.J.; Pletnikova, O.; Troncoso, J.C.; Dawson, V.L.; Dawson, T.M. Phosphorylation by the c-Abl protein tyrosine kinase inhibits Parkin’s ubiquitination and protective function. Proc. Natl. Acad. Sci. USA 2010, 107, 16691–16696. [Google Scholar] [CrossRef] [PubMed]
- Hebron, M.L.; Lonskaya, I.; Moussa, C.E. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of α-synuclein in Parkinson’s disease models. Hum. Mol. Genet. 2013, 22, 3315–3328. [Google Scholar] [CrossRef] [PubMed]
- Karuppagounder, S.S.; Brahmachari, S.; Lee, Y.; Dawson, V.L.; Dawson, T.M.; Ko, H.S. The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson’s disease. Sci. Rep. 2014, 4, 4874. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.S.; Zhang, Y.X.; Wei, Z.F.; Li, H.; Zhou, H.X.; Zhang, Z.Y.; Zhang, Z.F. JNK inhibitor protects dopaminergic neurons by reducing COX-2 expression in the MPTP mouse model of subacute Parkinson’s disease. J. Neurol. Sci. 2009, 285, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Gilligan, P.J. Inhibitors of leucine-rich repeat kinase 2 (LRRK2): Progress and promise for the treatment of Parkinson’s disease. Curr. Top. Med. Chem. 2015, 15, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Daher, J.P.L.; Abdelmotilib, H.A.; Hu, X.Z.; Volpicelli-Daley, L.A.; Moehle, M.S.; Fraser, K.B.; Needle, E.; Chen, Y.; Steyn, S.J.; Galatsis, P.; et al. Leucine-rich repeat kinase 2 (LRRK2) pharmacological inhibition abates α-synuclein gene-induced neurodegeneration. J. Biol. Chem. 2015, 290, 19433–19444. [Google Scholar] [CrossRef] [PubMed]
- Li, T.X.; Yang, D.J.; Zhong, S.J.; Thomas, J.M.; Xue, F.T.; Liu, J.N.; Kong, L.B.; Voulalas, P.; Hassan, H.E.; Park, J.S.; et al. Novel LRRK2 GTP-binding inhibitors reduced degeneration in Parkinson’s disease cell and mouse models. Hum. Mol. Genet. 2014, 23, 6212–6222. [Google Scholar] [CrossRef] [PubMed]
- Imam, S.Z.; Trickler, W.; Kimura, S.; Binienda, Z.K.; Paule, M.G.; Slikker, W., Jr.; Li, S.; Clark, R.A.; Ali, S.F. Neuroprotective efficacy of a new brain-penetrating c-Abl inhibitor in a murine Parkinson’s disease model. PLoS ONE 2013, 8, e65129. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, A.; Yamamura, Y.; Kasahara, J.; Morigaki, R.; Kaji, R.; Goto, S. A novel tyrosine kinase inhibitor AMN107 (nilotinib) normalizes striatal motor behaviors in a mouse model of Parkinson’s disease. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehdi, S.J.; Rosas-Hernandez, H.; Cuevas, E.; Lantz, S.M.; Barger, S.W.; Sarkar, S.; Paule, M.G.; Ali, S.F.; Imam, S.Z. Protein Kinases and Parkinson’s Disease. Int. J. Mol. Sci. 2016, 17, 1585. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091585
Mehdi SJ, Rosas-Hernandez H, Cuevas E, Lantz SM, Barger SW, Sarkar S, Paule MG, Ali SF, Imam SZ. Protein Kinases and Parkinson’s Disease. International Journal of Molecular Sciences. 2016; 17(9):1585. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091585
Chicago/Turabian StyleMehdi, Syed Jafar, Hector Rosas-Hernandez, Elvis Cuevas, Susan M. Lantz, Steven W. Barger, Sumit Sarkar, Merle G. Paule, Syed F. Ali, and Syed Z. Imam. 2016. "Protein Kinases and Parkinson’s Disease" International Journal of Molecular Sciences 17, no. 9: 1585. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091585