
Distal [FeS]-Cluster Coordination in [NiFe]-Hydrogenase Facilitates Intermolecular Electron Transfer


Abstract
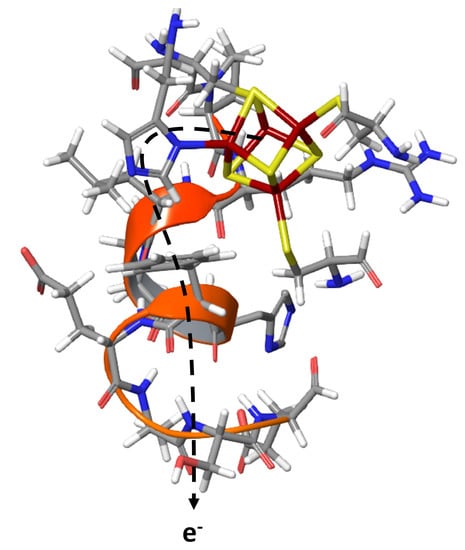
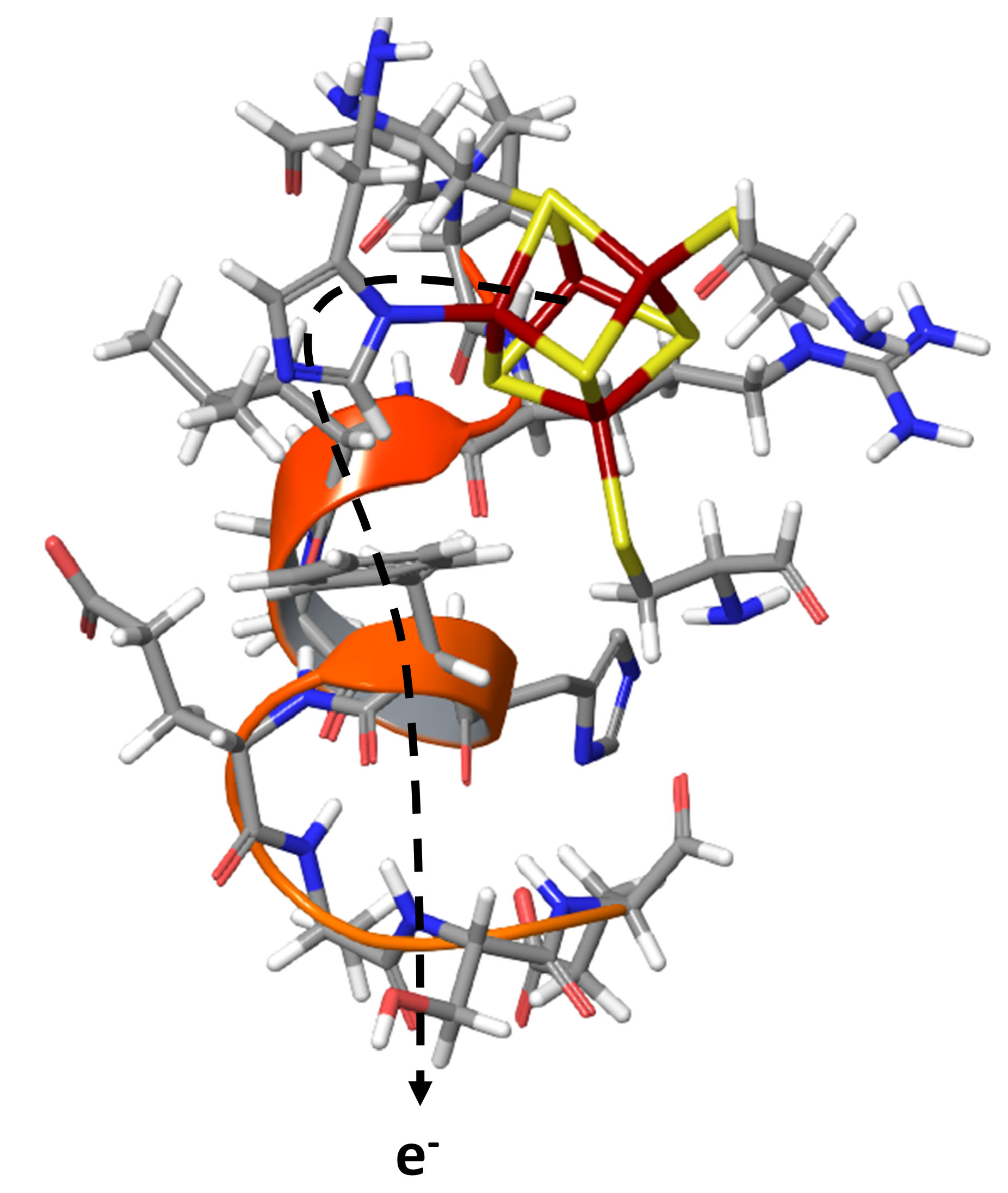
:
1. Introduction
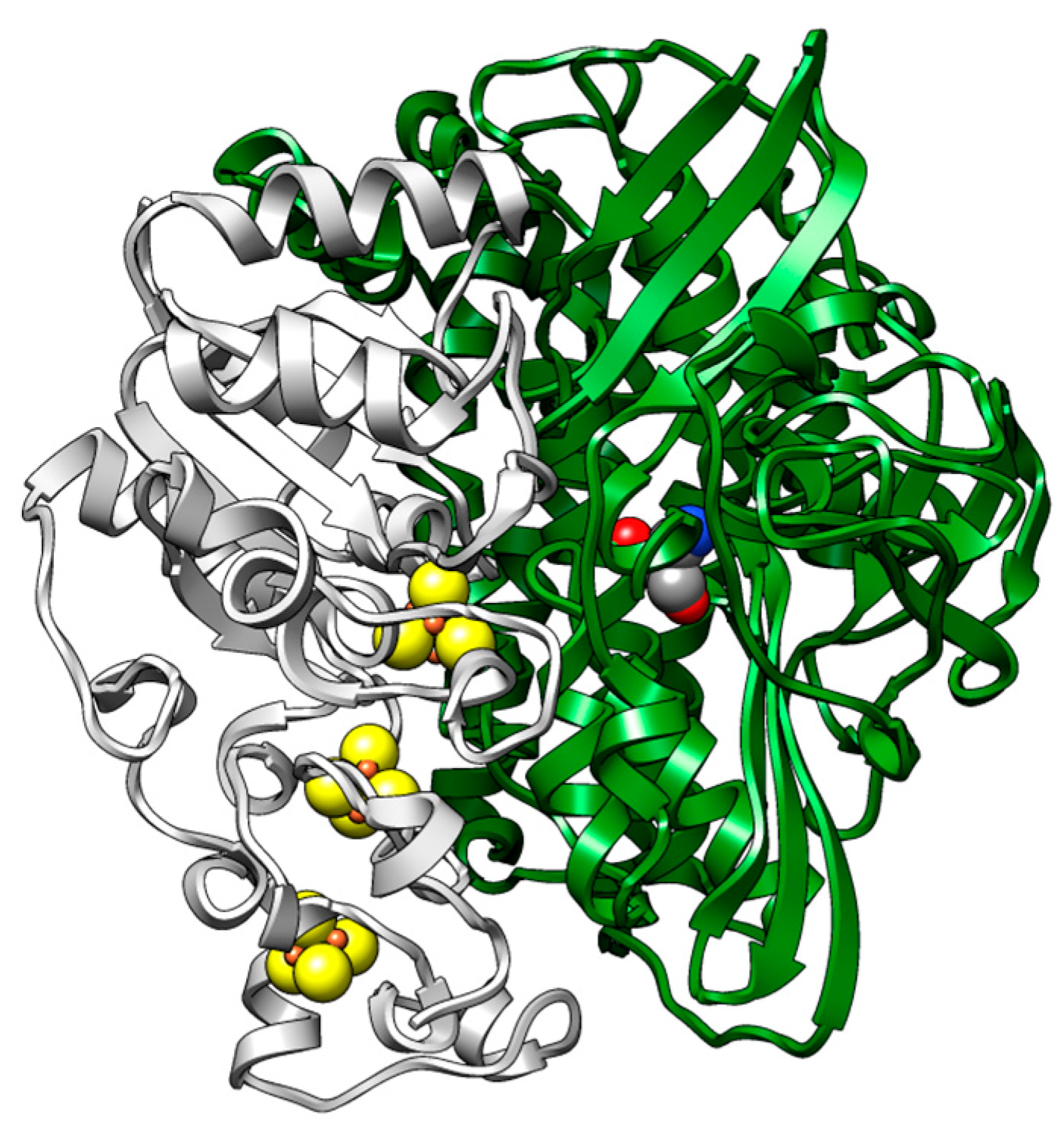
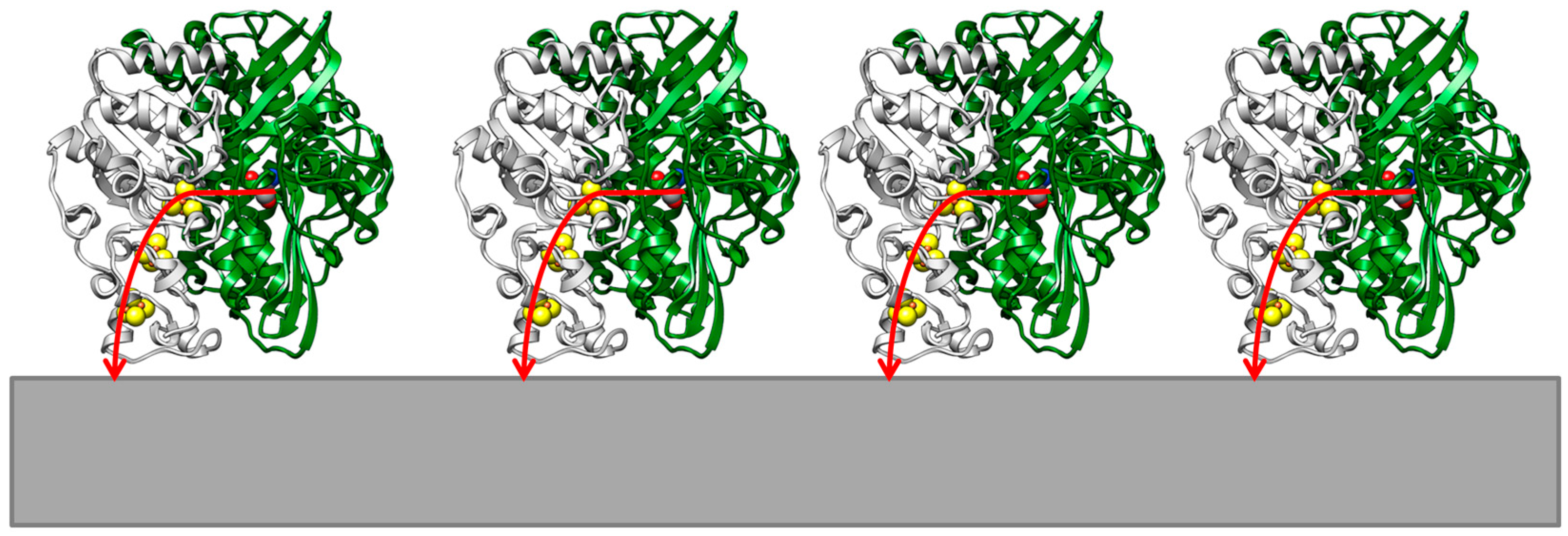
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Armstrong, R.C.; Wolfram, C.; de Jong, K.P.; Gross, R.; Lewis, N.S.; Boardman, B.; Ragauskas, A.J.; Ehrhardt-Martinez, K.; Crabtree, G.; Ramana, M.V. The frontiers of energy. Nat. Energy 2016, 1, 15020. [Google Scholar] [CrossRef]
- Jones, A.K.; Sillery, E.; Albracht, S.P.J.; Armstrong, F.A. Direct comparison of the electrocatalytic oxidation of hydrogen by an enzyme and a platinum catalyst. Chem. Commun. 2002, 2002, 866–867. [Google Scholar] [CrossRef]
- Ogata, H.; Nishikawa, K.; Lubitz, W. Hydrogens detected by subatomic resolution protein crystallography in a [NiFe]-hydrogenase. Nature 2015, 520, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, F.A.; Evans, R.M.; Hexter, S.V.; Murphy, B.J.; Roessler, M.M.; Wulff, P. Guiding principles of hydrogenase catalysis instigated and clarified by protein film electrochemistry. Acc. Chem. Res. 2016, 49, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Vincent, K.A.; Parkin, A.; Armstrong, F.A. Investigating and exploiting the electrocatalytic properties of hydrogenases. Chem. Rev. 2007, 107, 4366–4413. [Google Scholar] [CrossRef] [PubMed]
- Pershad, H.R.; Duff, J.L.C.; Heering, H.A.; Duin, E.C.; Albracht, S.P.J.; Armstrong, F.A. Catalytic electron transport in chromatium vinosum [NiFe]-hydrogenase: Application of voltammetry in detecting redox-active centers and establishing that hydrogen oxidation is very fast even at potentials close to the reversible H+/H2 value. Biochemistry 1999, 38, 8992–8999. [Google Scholar] [CrossRef] [PubMed]
- Lojou, E.; Luo, X.; Brugna, M.; Candoni, N.; Dementin, S.; Giudici-Orticoni, M.T. Biocatalysts for fuel cells: Efficient hydrogenase orientation for H2 oxidation at electrodes modified with carbon nanotubes. J. Biol. Inorg. Chem. 2008, 13, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Cammack, R. Hydrogenases and their activities. In Hydrogen as a Fuel; Cammack, R., Frey, M., Robson, R., Eds.; Taylor and Francis: London, UK, 2001; pp. 73–92. [Google Scholar]
- Nelsen, S.F.; Blackstock, S.C.; Kim, Y. Estimation of inner shell marcus terms for amino nitrogen-compounds by molecular-orbital calculations. J. Am. Chem. Soc. 1987, 109, 677–682. [Google Scholar] [CrossRef]
- Sigfridsson, E.; Olsson, M.H.M.; Ryde, U. Inner-sphere reorganization energy of iron-sulfur clusters studied with theoretical methods. Inorg. Chem. 2001, 40, 2509–2519. [Google Scholar] [CrossRef] [PubMed]
- Dementin, S.; Belle, V.; Bertrand, P.; Guigliarelli, B.; Adryanczyk-Perrier, G.; de Lacey, A.L.; Fernandez, V.M.; Rousset, M.; Leger, C. Changing the ligation of the distal [4Fe4S] cluster in NiFe hydrogenase impairs inter- and intramolecular electron transfers. J. Am. Chem. Soc. 2006, 128, 5209–5218. [Google Scholar] [CrossRef] [PubMed]
- Bader, J.S.; Cortis, C.M.; Berne, B.J. Solvation and reorganization energies in polarizable molecular and continuum solvents. J. Chem. Phys. 1997, 106, 2372–2387. [Google Scholar] [CrossRef]
- Tan, M.L.; Dolan, E.A.; Ichiye, T. Understanding intramolecular electron transfer in ferredoxin: A molecular dynamics study. J. Phys. Chem. B 2004, 108, 20435–20441. [Google Scholar] [CrossRef]
- Petrenko, A.; Stein, M. Rates and routes of electron transfer of [NiFe]-hydrogenase in an enzymatic fuel cell. J. Phys. Chem. B 2015, 119, 13870–13882. [Google Scholar] [CrossRef] [PubMed]
- Cracknell, J.A.; Vincent, K.A.; Armstrong, F.A. Enzymes as working or inspirational electrocatalysts for fuel cells and electrolysis. Chem. Rev. 2008, 108, 2439–2461. [Google Scholar] [CrossRef] [PubMed]
- Dementin, S.; Burlat, B.; Fourmond, V.; Leroux, F.; Liebgott, P.-P.; Abou Hamdan, A.; Leger, C.; Rousset, M.; Guigliarelli, B.; Bertrand, P. Rates of intra- and intermolecular electron transfers in hydrogenase deduced from steady-state activity measurements. J. Am. Chem. Soc. 2011, 133, 10211–10221. [Google Scholar] [CrossRef] [PubMed]
- Raleiras, P.; Kellers, P.; Lindblad, P.; Styring, S.; Magnuson, A. Isolation and characterization of the small subunit of the uptake hydrogenase from the cyanobacterium Nostoc punctiforme. J. Biol. Chem. 2013, 288, 18345–18352. [Google Scholar] [CrossRef] [PubMed]
- Schröder, O.; Bleijlevens, B.; de Jongh, T.E.; Chen, Z.; Li, T.; Fischer, J.; Förster, J.; Friedrich, C.G.; Bagley, K.A.; Albracht, S.P.J.; et al. Characterization of a cyanobacterial-like uptake [NiFe]-hydrogenase: Epr and Ftir spectroscopic studies of the enzyme from acidithiobacillus ferrooxidans. J. Biol. Inorg. Chem. 2007, 12, 212–233. [Google Scholar] [CrossRef] [PubMed]
- Raleiras, P.; Khanna, N.; Miranda, H.; Meszaros, L.S.; Krassen, H.; Ho, F.; Battchikova, N.; Aro, E.-M.; Magnuson, A.; Lindblad, P.; et al. Turning around the electron flow in an uptake hydrogenase. EPR spectroscopy and in vivo activity of a designed mutant in Hupsl from Nostoc punctiforme. Energy Environ. Sci. 2016, 9, 581–594. [Google Scholar] [CrossRef]
- Turbomole Program Package for Ab Initio Electronic Structure Calculations, version 6.6; Turbomole GmbH: Karlruhe, Germany, 2014.
- Furche, F.; Ahlrichs, R.; Hättig, C.; Klopper, W.; Sierka, M.; Weigend, F. Turbomole. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 91–100. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Stephens, P.; Devlin, F.; Chabalowski, C.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple ζ valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Stein, M.; Kaur-Ghumaan, S. Microbial hydrogen splitting in the presence of oxygen. Biochem. Soc. Trans. 2013, 41, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Cammack, R.; Patil, D.S.; Hatchikian, E.C.; Fernández, V.M. Nickel and iron-sulphur centres in Desulfovibrio gigas hydrogenase: ESR spectra, redox properties and interactions. Biochim. Biophys. Acta 1987, 912, 98–109. [Google Scholar] [CrossRef]
- Casado-Pascual, J.; Morillo, M.; Goychuk, I.; Hänggi, P. The role of different reorganization energies within the Zusman theory of electron transfer. J. Chem. Phys. 2003, 118, 291–303. [Google Scholar] [CrossRef]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. 1993, 2, 799–805. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
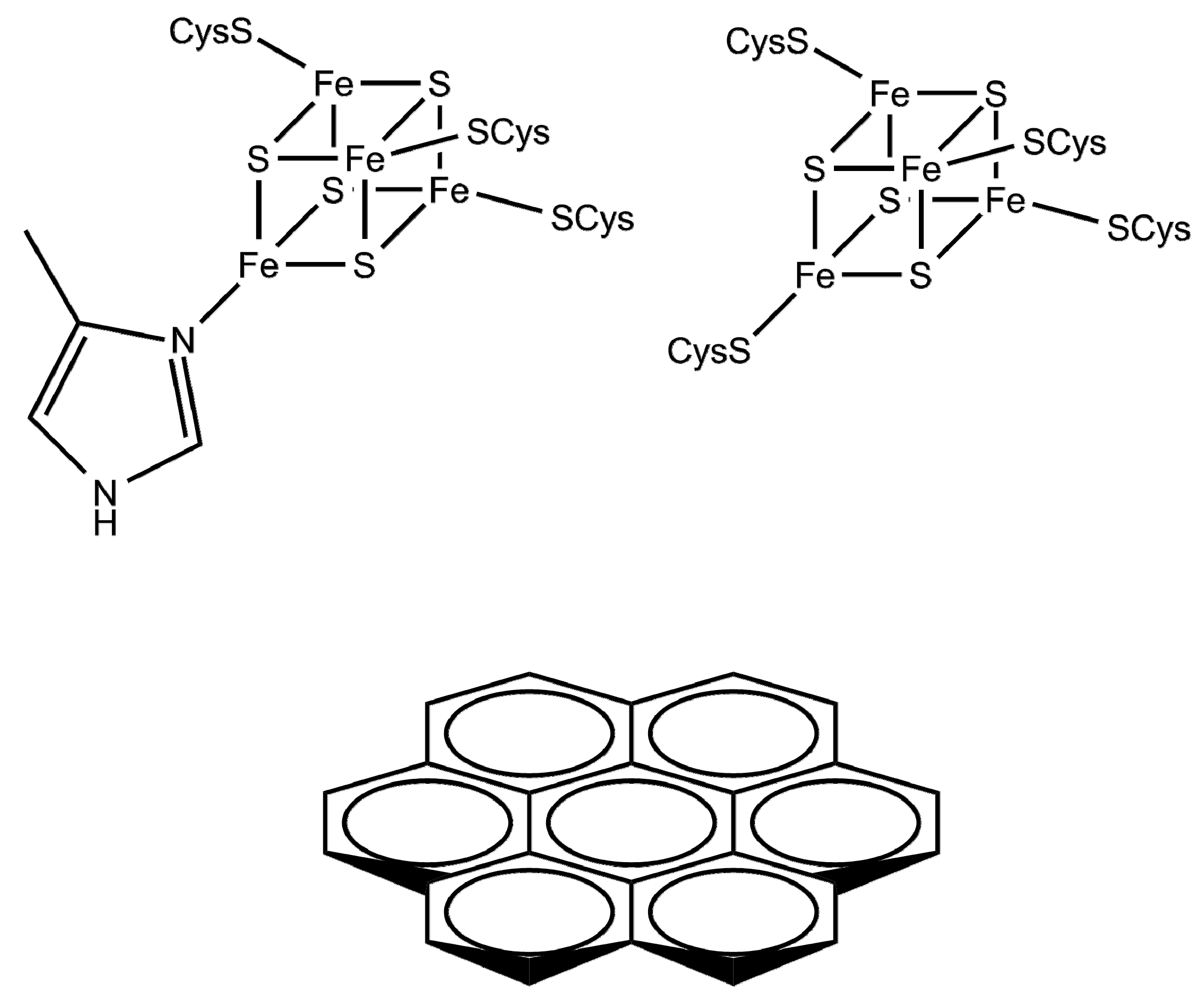{kind=link}
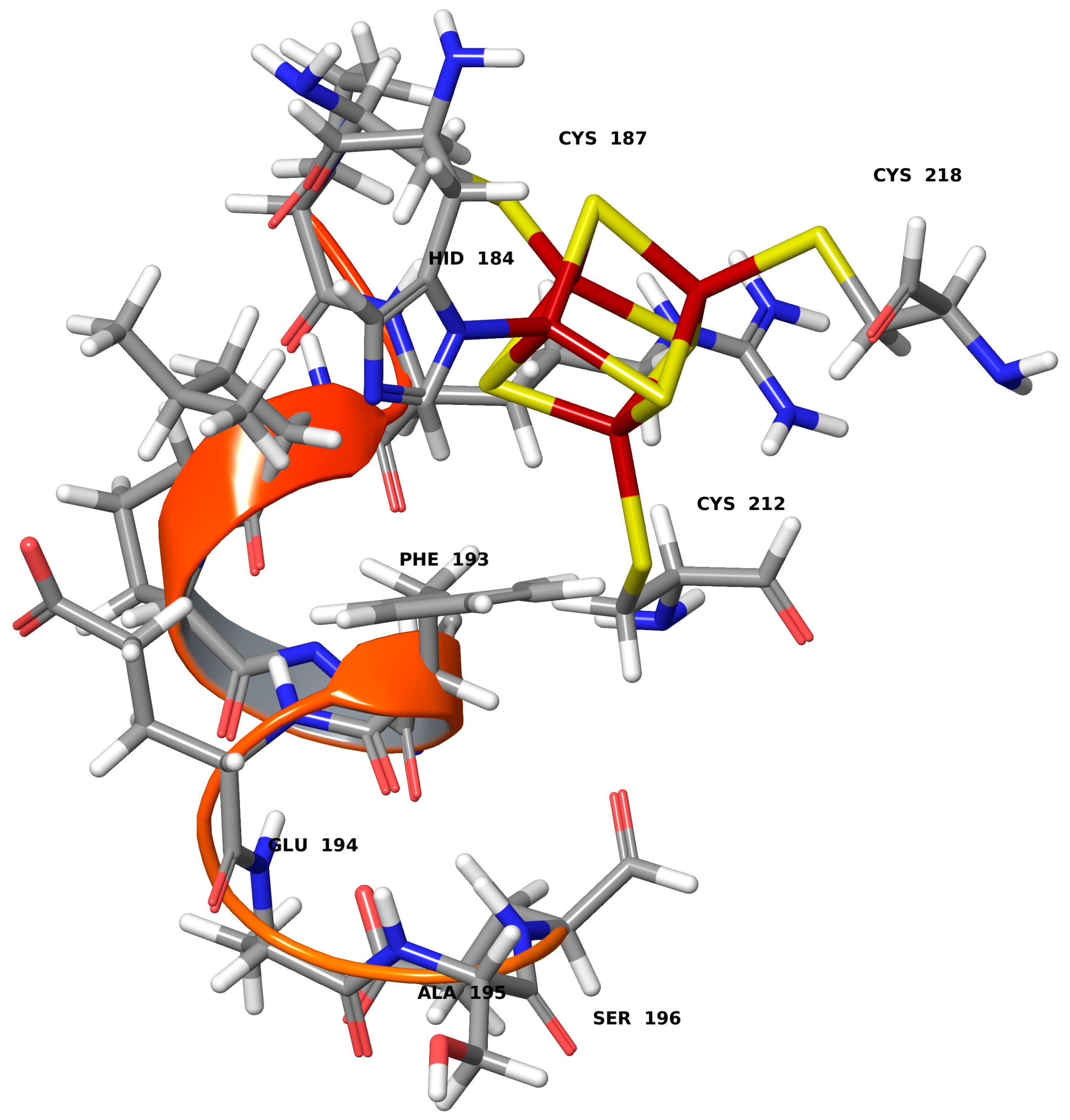{kind=link}
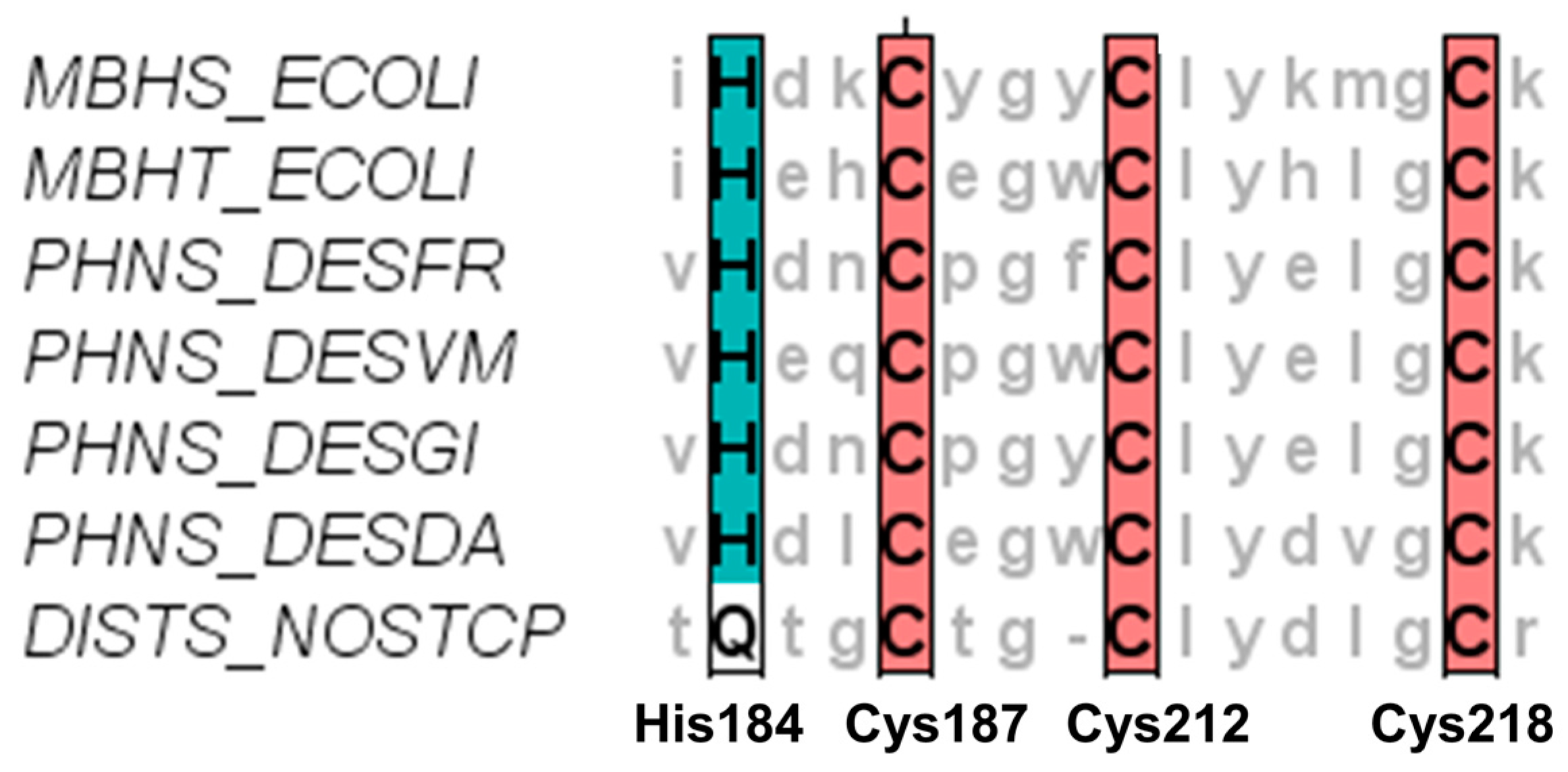{kind=link}
| Distal FeS-Cluster Model | Redox State | λi (eV) |
|---|---|---|
| ([Fe4S4](Cys)3(His))−2/−1 | ox | 0.18 |
| red | 0.32 | |
| ([Fe4S4](Cys)4)−3/−2 | ox | 0.19 [10] |
| red | 0.30 [10] |
| Donor/Acceptor for ET | Cluster Composition | Redox State | λS (eV) |
|---|---|---|---|
| Donor [FeS]d-Cluster Model | ([Fe4S4](Cys)3(His))−2/−1 | ox/red | 0.38/0.18 |
| ([Fe4S4](Cys)4)−3/−2 | ox/red | 0.30/0.19 | |
| Acceptor Graphite Model | Coronene C24H12−1/0 | ox/red | 0.09/0.07 |
| Distal FeS-Cluster Model | Z (Å) | S | VDA (cm−1) | kET (s−1) |
|---|---|---|---|---|
| [Fe4S4](Cys)3(His) | 12.3 | 5.3 × 10−6 | 4.4 × 10−2 | 5.1 × 103 |
| [Fe4S4](Cys)4 | 12.3 | 1.1 × 10−7 | 1.5 × 10−3 | 6.0 |
| Distal FeS-Cluster Model | Z (Å) | S | VDA (cm−1) | kET (s−1) | Experiment [16] (s−1) |
|---|---|---|---|---|---|
| [Fe4S4](Cys)3(His) | 15.3 | 8.2 × 10-6 | 2.9 × 10-2 | 2.2 × 103 | 1–3.5 × 103 |
| [Fe4S4](Cys)4 | 15.3 | 2.9 × 10-6 | 1.1 × 10-2 | 3.6 × 102 | >40 |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrenko, A.; Stein, M. Distal [FeS]-Cluster Coordination in [NiFe]-Hydrogenase Facilitates Intermolecular Electron Transfer. Int. J. Mol. Sci. 2017, 18, 100. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18010100
Petrenko A, Stein M. Distal [FeS]-Cluster Coordination in [NiFe]-Hydrogenase Facilitates Intermolecular Electron Transfer. International Journal of Molecular Sciences. 2017; 18(1):100. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18010100
Chicago/Turabian StylePetrenko, Alexander, and Matthias Stein. 2017. "Distal [FeS]-Cluster Coordination in [NiFe]-Hydrogenase Facilitates Intermolecular Electron Transfer" International Journal of Molecular Sciences 18, no. 1: 100. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18010100