
Pancreatic Neuroendocrine Neoplasms: Basic Biology, Current Treatment Strategies and Prospects for the Future

Abstract
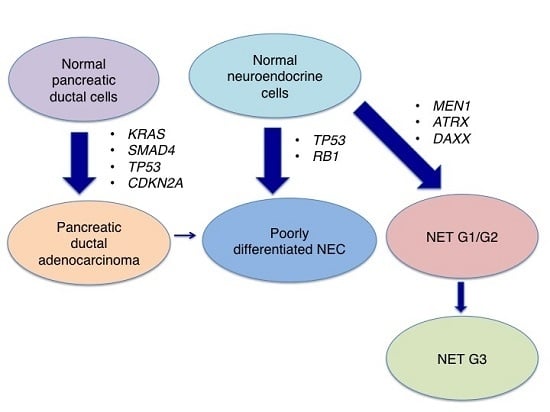
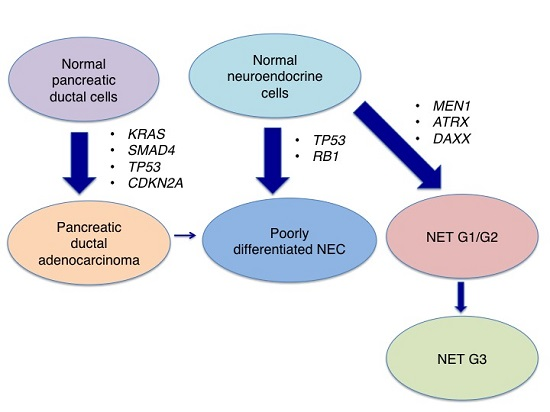
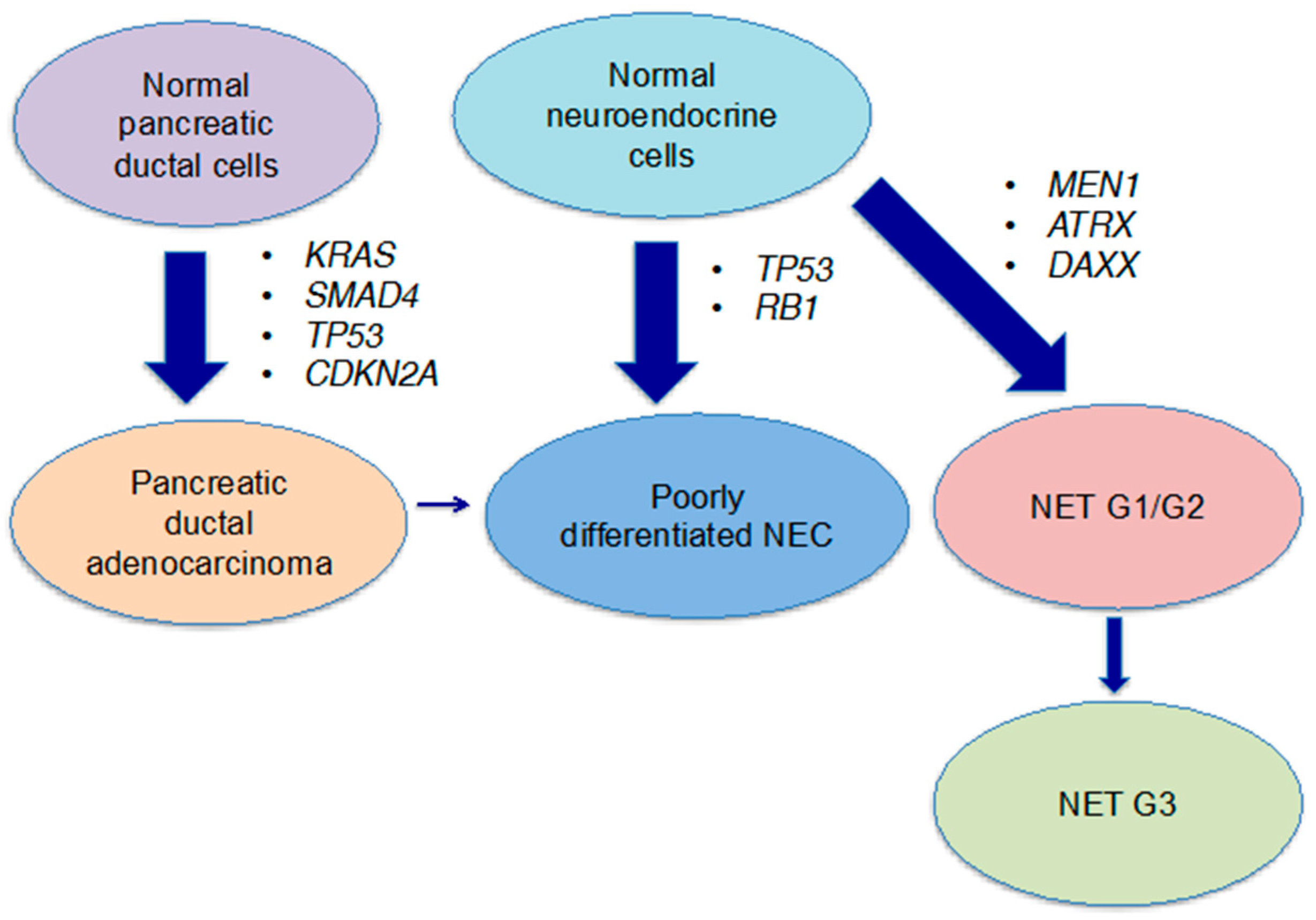
:
1. Introduction
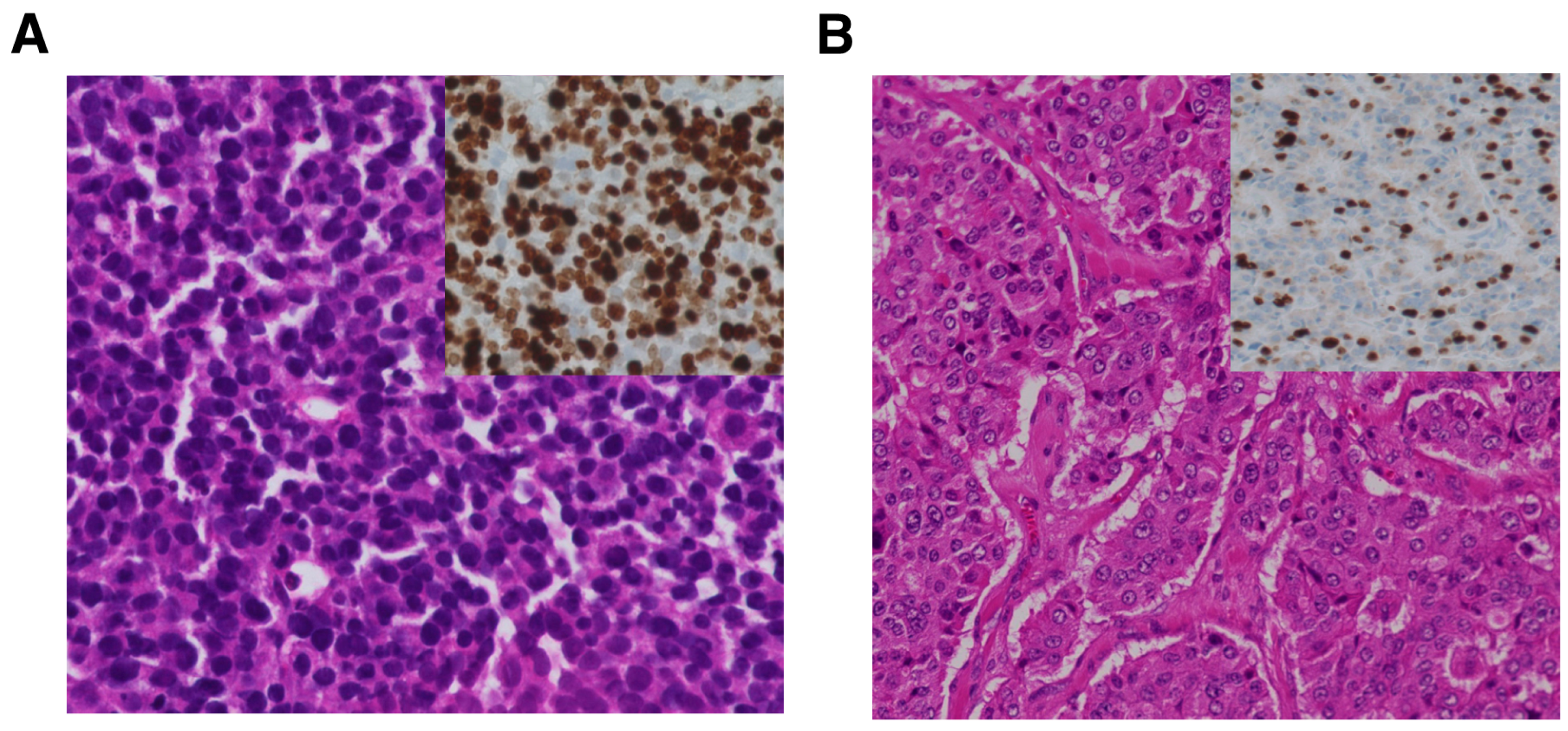
2. Histologic Classification
3. Genetic Characterisics
4. Current Treatment Strategies
4.1. Well-Differentiated pNENs (NET G1/G2)
4.2. Poorly Differentiated NEC
5. Conclusions and Future Directions
Author Contributions
Conflicts of Interest
References
- Yao, J.C.; Hassan, M.; Phan, A.; Dagohoy, C.; Leary, C.; Mares, J.E.; Abdalla, E.K.; Fleming, J.B.; Vauthey, J.N.; Rashid, A.; et al. One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J. Clin. Oncol. 2008, 26, 3063–3072. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Phan, A.T.; Yao, J.C. New strategies for advanced neuroendocrine tumors in the era of targeted therapy. Clin. Cancer Res. 2012, 18, 1830–1836. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Eisner, M.P.; Leary, C.; Dagohoy, C.; Phan, A.; Rashid, A.; Hassan, M.; Evans, D.B. Population-based study of islet cell carcinoma. Ann. Surg. Oncol. 2007, 14, 3492–3500. [Google Scholar] [CrossRef] [PubMed]
- Halfdanarson, T.R.; Rabe, K.G.; Rubin, J.; Petersen, G.M. Pancreatic neuroendocrine tumors (PNETs): Incidence, prognosis and recent trend toward improved survival. Ann. Oncol. 2008, 19, 1727–1733. [Google Scholar] [CrossRef] [PubMed]
- Bosman, F.T.; Carneiro, F.; Hruban, R.H.; Theise, N.D. WHO Classification of Tumours of the Digestive System, 4th ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2010. [Google Scholar]
- Yamaguchi, T.; Machida, N.; Morizane, C.; Kasuga, A.; Takahashi, H.; Sudo, K.; Nishina, T.; Tobimatsu, K.; Ishido, K.; Furuse, J.; et al. Multicenter retrospective analysis of systemic chemotherapy for advanced neuroendocrine carcinoma of the digestive system. Cancer Sci. 2014, 105, 1176–1181. [Google Scholar] [CrossRef] [PubMed]
- Sorbye, H.; Welin, S.; Langer, S.W.; Vestermark, L.W.; Holt, N.; Osterlund, P.; Dueland, S.; Hofsli, E.; Guren, M.G.; Ohrling, K.; et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): The NORDIC NEC study. Ann. Oncol. 2013, 24, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Rindi, G.; Klöppel, G.; Alhman, H.; Caplin, M.; Couvelard, A.; de Herder, W.W.; Erikssson, B.; Falchetti, A.; Falconi, M.; Komminoth, P.; et al. TNM staging of foregut (neuro) endocrine tumors: A consensus proposal including a grading system. Virchows Arch. 2006, 449, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.R.; Aaltonen, L.A. WHO Classification of Tumours of the Digestive System, 3rd ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2000. [Google Scholar]
- Klimstra, D.S.; Modlin, I.R.; Adsay, N.V.; Chetty, R.; Deshpande, V.; Gönen, M.; Jensen, R.T.; Kidd, M.; Kulke, M.H.; Lloyd, R.V.; et al. Pathology reporting of neuroendocrine tumors: Application of the delphic consensus process to the development of a minimum pathology data set. Am. J. Surg. Pathol. 2010, 34, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.H.; Gonen, M.; Hedvat, C.; Modlin, I.M.; Klimstra, D.S. Objective quantification of the Ki-67 proliferative index in neuroendocrine tumors of the gastroenteropancreatic system: A comparison of digital image analysis with manual methods. Am. J. Surg. Pathol. 2012, 36, 1761–1770. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Luong, T.V.; Watkins, J.; Toumpanakis, C.; Caplin, M.E.; Meyer, T. A comparison of Ki-67 and mitotic count as prognostic markers for metastatic pancreatic and midgut neuroendocrine neoplasms. Br. J. Cancer 2013, 108, 1838–1845. [Google Scholar] [CrossRef] [PubMed]
- McCall, C.M.; Shi, C.; Cornish, T.C.; Klimstra, D.S.; Tang, L.H.; Basturk, O.; Mun, L.J.; Ellison, T.A.; Wolfgang, C.L.; Choti, M.A.; et al. Grading of well-differentiated pancreatic neuroendocrine tumors is improved by the inclusion of both Ki-67 proliferative index and mitotic rate. Am. J. Surg. Pathol. 2013, 37, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Basturk, O.; Yang, Z.; Tang, L.H.; Hruban, R.H.; Adsay, V.; McCall, C.M.; Krasinskas, A.M.; Jang, K.T.; Frankel, W.L.; Balci, S.; et al. The high-grade (WHO G3) pancreatic neuroendocrine tumor category is morphologically and biologically heterogenous and includes both well differentiated and poorly differentiated neoplasms. Am. J. Surg. Pathol. 2015, 39, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Bastur, O.; Tang, L.; Hruban, R.H.; Adsay, V.; Yang, Z.; Krasinskas, A.M.; Vakiani, E.; La Rosa, S.; Jang, K.T.; Frankel, W.L.; et al. Poorly differentiated neuroendocrine carcinomas of the pancreas: A clinicopathologic analysis of 44 cases. Am. J. Surg. Pathol. 2014, 38, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Korse, C.M.; Taal, B.G.; van Velthuysen, M.L.; Visser, O. Incidence and survival of neuroendocrine tumours in the Netherlands according to histological grade: Experience of two decades of cancer registry. Eur. J. Cancer 2013, 49, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- Kunz, P.L.; Reidy-Lagunes, D.; Anthony, L.B.; Bertino, E.M.; Brendtro, K.; Chan, J.A.; Chen, H.; Jensen, R.T.; Kim, M.K.; Klimstra, D.S.; et al. Consensus guidelines for the management and treatment of neuroendocrine tumors. Pancreas 2013, 42, 557–577. [Google Scholar] [CrossRef] [PubMed]
- Sorbye, H.; Strosberg, J.; Baudin, E.; Klimstra, D.S.; Yao, J.C. Gastroenteropancreatic high-grade neuroendocrine carcinoma. Cancer 2014, 120, 2814–2823. [Google Scholar] [CrossRef] [PubMed]
- Vélayoudom-Céphise, F.L.; Duvillard, P.; Foucan, L.; Hadoux, J.; Chougnet, C.N.; Leboulleux, S.; Malka, D.; Guigay, J.; Goere, D.; Debaere, T.; et al. Are G3 ENETS neuroendocrine neoplasms heterogeneous? Endocr. Relat. Cancer 2013, 20, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Coriat, R.; Walter, T.; Terris, B.; Couvelard, A.; Ruszniewski, P. Gastroenteropancreatic well-differentiated Grade 3 neuroendocrine tumors: Review and position statement. Oncologist 2016, 21, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Heetfeld, M.; Chougnet, C.N.; Olsen, I.H.; Rinke, A.; Borbath, I.; Crespo, G.; Barriuso, J.; Pavel, M.; O’Toole, D.; Walter, T.; et al. Characteristics and treatment of patients with G3 gastroenteropancreatic neuroendocrine neoplasms. Endocr. Relat. Cancer 2015, 22, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Fazio, N.; Milione, M. Heterogeneity of grade 3 gastroenteropancreatic neuroendocrine carcinomas: New insights and treatment implications. Cancer Treat. Rev. 2016, 50, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.H.; Basturk, O.; Sue, J.J.; Klimstra, D.S. A practical approach to the classification of WHO Grade 3 (G3) well-differentiated neuroendocrine tumor (WD-NET) and poorly differentiated neuroendocrine carcinoma (PD-NEC) of the pancreas. Am. J. Surg. Pathol. 2016, 40, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.H.; Untch, B.R.; Reidy, D.L.; O’Reilly, E.; Dhall, D.; Jih, L.; Basturk, O.; Allen, P.J.; Klimstra, D.S. Well-differentiated neuroendocrine tumors with a morphologically apparent high-grade component: A pathway distinct from poorly differentiated neuroendocrine carcinomas. Clin. Cancer Res. 2016, 22, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Milione, M.; Maisonneuve, P.; Spada, F.; Pellegrinelli, A.; Spaggiari, P.; Albarello, L.; Pisa, E.; Barberis, M.; Vanoli, A.; Buzzoni, R.; et al. The clinicopathologic heterogeneity of Grade 3 gastroenteropancreatic neuroendocrine neoplasms: Morphological differentiation and proliferation identify different prognostic categories. Neuroendocrinology 2017, 104, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- De Wilde, R.F.; Edil, B.H.; Hruban, R.H.; Maitra, A. Well-differentiated pancreatic neuroendocrine tumors: From genetics to therapy. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Corbo, V.; Dalai, I.; Scardoni, M.; Barbi, S.; Beghelli, S.; Bersani, S.; Albarello, L.; Doglioni, C.; Schott, C.; Capelli, P.; et al. MEN1 in pancreatic endocrine tumors: Analysis of gene and protein status in 169 sporadic neoplasms reveals alterations in the vast majority of cases. Endocr. Relat. Cancer 2010, 17, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef] [PubMed]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.L.; Westra, W.H.; et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; Liu, T.C.; Roncaioli, J.L.; Cao, D.; Zeh, H.J.; Zureikat, A.H.; Tsung, A.; Marsh, J.W.; Lee, K.K.; Hogg, M.E.; et al. Alterative lengthening of telomeres and loss of DAXX/ATRX expression predicts metastatic disease and poor survival in patients with pancreatic neuroendocrine tumors. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed]
- Raj, N.; Soumerai, T.; Valentino, E.; Hechtman, J.F.; Berger, M.F.; Reidy, D.L. Next-generation sequencing (NGS) in advanced well differentiated pancreatic neuroendocrine tumors (WD pNETs): A study using MSK-IMPACT. J. Clin. Oncol. 2016, 34 (Suppl. 4S). abstr 246. [Google Scholar]
- Dalgliesh, G.L.; Furge, K.; Greenman, C.; Chen, L.; Bignell, G.; Butler, A.; Davies, H.; Edkins, S.; Hardy, C.; Latimer, C.; et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Vakiani, E.; White, C.M.; Zhong, Y.; Saunders, T.; Morgan, R.; de Wilde, R.F.; Maitra, A.; Hicks, J.; Demarzo, A.M.; et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am. J. Surg. Pathol. 2012, 36, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Hijioka, S.; Hosoda, W.; Mizuno, N.; Hara, K.; Imaoka, H.; Bhatia, V.; Mekky, M.A.; Tajika, M.; Tanaka, T.; Ishihara, M.; et al. Does the WHO 2010 classification of pancreatic neuroendocrine neoplasms accurately characterize pancreatic neuroendocrine carcinomas? J. Gastroenterol. 2015, 50, 564–572. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Bergsland, E.K.; Roy, R.; Stephens, P.; Ross, J.S.; Bailey, M.; Olshen, A. Genomic profiling to distinguish poorly differentiated neuroendocrine carcinomas arising in different sites. J. Clin. Oncol. 2016, 34 (Suppl.). abstr 4020. [Google Scholar]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Miyamoto, H.; Fukuya, A.; Kitamura, S.; Okamoto, K.; Kimura, M.; Muguruma, N.; Ikemoto, T.; Shimada, M.; Yoneda, A.; et al. Neuroendocrine carcinoma of the pancreas with similar genetic alterations to invasive ductal adenocarcinoma. Clin. J. Gastroenterol. 2016, 9, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Orditura, M.; Petrillo, A.; Ventriglia, J.; Diana, A.; Laterza, M.M.; Fabozzi, A.; Savastano, B.; Franzese, E.; Conzo, G.; Santini, L.; et al. Pancreatic neuroendocrine tumors: Nosography, management and treatment. Int. J. Surg. 2016, 28, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Igarashi, H.; Jensen, R.T. Therapy of metastatic pancreatic neuroendocrine tumors (pNETs): Recent insights and advances. J. Gastroenterol. 2012, 47, 941–960. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.S.; McPhee, J.T.; McDade, T.P.; Zhou, Z.; Sullivan, M.E.; Whalen, G.F.; Tseng, J.F. Pancreatic neuroendocrine tumors: The impact of surgical resection on survival. Cancer 2009, 115, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Touzios, J.G.; Kiely, J.M.; Pitt, S.C.; Rilling, W.S.; Quebbeman, E.J.; Wilson, S.D.; Pitt, H.A. Neuroendocrine hepatic metastases: Does aggressive management improve survival? Ann. Surg. 2005, 241, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Fendrich, V.; Langer, P.; Celik, I.; Bartsch, D.K.; Zielke, A.; Ramaswamy, A.; Rothmund, M. An aggressive surgical approach leads to long-term survival in patients with pancreatic endocrine tumors. Ann. Surg. 2006, 244, 845–851. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, D.; Ruszniewski, P. Chemoembolization and other ablative therapies for liver metastases of gastrointestinal endocrine tumours. Best Pract. Res. Clin. Gastroenterol. 2005, 19, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Baudin, E.; Planchard, D.; Scoazec, J.Y.; Guigay, J.; Dromain, C.; Hadoux, J.; Debaere, T.; Elias, D.; Ducreux, M. Intervention in gastro-enteropancreatic neuroendocrine tumours. Best Pract. Res. Clin. Gastroenterol. 2012, 26, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Grozinsky-Glasberg, S.; Shimon, I.; Korbonits, M.; Grossman, A.B. Somatostatin analogues in the control of neuroendocrine tumours: Efficacy and mechanisms. Endocr. Relat. Cancer 2008, 15, 701–720. [Google Scholar] [CrossRef] [PubMed]
- Viúdez, A.; de Jesus-Acosta, A.; Carvalho, F.L.; Vera, R.; Martín-Algarra, S.; Ramírez, N. Pancreatic neuroendocrine tumors: Challenges in an underestimated disease. Crit. Rev. Oncol. Hematol. 2016, 101, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Caplin, M.E.; Pavel, M.; Ćwikła, J.B.; Phan, A.T.; Raderer, M.; Sedláčková, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N. Engl. J. Med. 2014, 371, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Rinke, A.; Müller, H.H.; Schade-Brittinger, C.; Klose, K.J.; Barth, P.; Wied, M.; Mayer, C.; Aminossadati, B.; Pape, U.F.; Bläker, M.; et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID study group. J. Clin. Oncol. 2009, 27, 4656–4663. [Google Scholar] [CrossRef] [PubMed]
- Alexandraki, K.I.; Kaltsas, G. Gastroenteropancreatic neuroendocrine tumors: New insights in the diagnosis and therapy. Endocrine 2012, 41, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, E.I.; Teunissen, J.J.; Kam, B.L.; de Jong, M.; Krenning, E.P.; Kwekkeboom, D.J. Treatment of gastroenteropancreatic neuroendocrine tumors with peptide receptor radionuclide therapy. Neuroendocrinology 2013, 97, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Imhof, A.; Brunner, P.; Marincek, N.; Briel, M.; Schindler, C.; Rasch, H.; Mäcke, H.R.; Rochlitz, C.; Müller-Brand, J.; Walter, M.A. Response, survival, and long-term toxicity after therapy with the radiolabeled somatostatin analogue [90Y-DOTA]-TOC in metastasized neuroendocrine cancers. J. Clin. Oncol. 2011, 29, 2416–2423. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.R.; Wolin, E.M.; Chasen, B.; Kulke, M.H.; Bushnell, D.L.; Caplin, M.E.; Baum, R.P.; Kunz, P.L.; Hobday, T.J.; Hendifar, A.E.; et al. NETTER-1 phase III: Progression-free survival, radiographic response, and preliminary overall survival results in patients with midgut neuroendocrine tumors treated with 177-Lu-Dotatate. J. Clin. Oncol. 2016, 34 (Suppl. 4S). abstr 194. [Google Scholar]
- Moertel, C.G.; Lefkopoulo, M.; Lipsitz, S.; Hahn, R.G.; Klaassen, D. Streptozocin-doxorubicin, streptozocin-fluorouracil or chlorozotocin in the treatment of advanced islet-cell carcinoma. N. Engl. J. Med. 1992, 326, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.R.; Fine, R.L.; Choi, J.; Nasir, A.; Coppola, D.; Chen, D.T.; Helm, J.; Kvols, L. First-line chemotherapy with capecitabine and temozolomide in patients with metastatic pancreatic endocrine carcinomas. Cancer 2011, 117, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Okusaka, T.; Ueno, H.; Morizane, C.; Kondo, S.; Sakamoto, Y.; Takahashi, H.; Ohno, I.; Shimizu, S.; Mitsunaga, S.; Ikeda, M. Cytotoxic chemotherapy for pancreatic neuroendocrine tumors. J. Hepato-Biliary Pancreat. Sci. 2015, 22, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Gonzalez-Angulo, A.M. Targeting the mTOR signaling network for cancer therapy. J. Clin. Oncol. 2009, 27, 2278–2287. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; de Vries, E.G.; et al. Everolimus for advanced pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Fazio, N.; Singh, S.; Buzzoni, R.; Carnaghi, C.; Wolin, E.; Tomasek, J.; Raderer, M.; Lahner, H.; Voi, M.; et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): A randomised, placebo-controlled, phase 3 study. Lancet 2016, 387, 968–977. [Google Scholar] [CrossRef]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.; Kulke, M. Targeting the mTOR signaling pathway in neuroendocrine tumors. Curr. Treat. Opt. Oncol. 2014, 15, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Yan, H.; Frost, P.; Gera, J.; Lichtenstein, A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol. Cancer Ther. 2005, 4, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Salazar, R.; Verslype, C.; Baudin, E.; Libutti, S.K.; Yao, J.C.; Buzzoni, R.; Antonuzzo, L.; Hubner, R.; García-Carbonero, R.; Custodio, A.B.; et al. Phase II studies of BEZ235 in patients with advanced pancreatic neuroendocrine tumors (pNET). J. Clin. Oncol. 2015, 33 (Suppl.). abstr 4102. [Google Scholar]
- Zhang, J.; Francois, R.; Iyer, R.; Seshadri, M.; Zajac-Kaye, M.; Hochwald, S.N. Current understanding of the molecular biology of pancreatic neuroendocrine tumors. J. Natl. Cancer Inst. 2013, 105, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Raymond, E.; Dahan, L.; Raoul, J.L.; Bang, Y.J.; Borbath, I.; Lombard-Bohas, C.; Valle, J.; Metrakos, P.; Smith, D.; Vinik, A.; et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Phan, A. Overcoming antiangiogenic resistance. Clin. Cancer Res. 2011, 17, 5217–5219. [Google Scholar] [CrossRef] [PubMed]
- Hobday, T.J.; Qin, R.; Reidy-Lagunes, D.; Moore, M.J.; Strosberg, J.; Kaubisch, A.; Shah, M.; Kindler, H.L.; Lenz, H.J.; Chen, H.; et al. Multicenter phase II trial of temsirolimus and bevacizumab in pancreatic neuroendocrine tumors. J. Clin. Oncol. 2015, 33, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Kulke, M.H.; Niedzwiecki, D.; Foster, N.R.; Fruth, B.; Kunz, P.L.; Kennecke, H.F.; Wolin, E.M.; Venook, A.P. Randomized phase II study of everolimus (E) versus everolimus plus bevacizumab (E + B) in patients (pts) with locally advanced or metastatic pancreatic neuroendocrine tumors (pNET), CALGB 80701 (Alliance). J. Clin. Oncol. 2015, 33 (Suppl.). abstr 4005. [Google Scholar]
- Phan, A.T.; Halperin, D.M.; Chan, J.A.; Fogelman, D.R.; Hess, K.R.; Malinowski, P.; Regan, E.; Ng, C.S.; Yao, J.C.; Kulke, M.H. Pazopanib and depot octreotide in advanced, well-differentiated neuroendocrine tumours: A multicentre, single-group, phase 2 study. Lancet Oncol. 2015, 16, 695–703. [Google Scholar] [CrossRef]
- Kulke, M.H.; Stuart, K.; Enzinger, P.C.; Ryan, D.P.; Clark, J.W.; Muzikansky, A.; Vincitore, M.; Michelini, A.; Fuchs, C.S. Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J. Clin. Oncol. 2006, 24, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Papaxoinis, G.; Syrigos, K.; Saif, M.W. Novel therapeutic approaches and mechanisms in neuroendocrine tumors: The role of targeted agents. Discov. Med. 2016, 21, 391–402. [Google Scholar] [PubMed]
- Haugvik, S.P.; Janson, E.T.; Österlund, P.; Langer, S.W.; Falk, R.S.; Labori, K.J.; Vestermark, L.W.; Grønbæk, H.; Gladhaug, I.P.; Sorbye, H. Surgical treatment as a principle for patients with high-grade pancreatic neuroendocrine carcinoma: A nordic multicenter comparative study. Ann. Surg. Oncol. 2016, 23, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Fazio, N.; Spada, F.; Giovannini, M. Chemotherapy in gastroenteropancreatic (GEP) neuroendocrine carcinomas (NEC): A critical view. Cancer Treat. Rev. 2013, 39, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; di Maio, M.; Chiodini, P.; Rudd, R.M.; Okamoto, H.; Skarlos, D.V.; Früh, M.; Qian, W.; Tamura, T.; Samantas, E.; et al. Carboplatin- or cisplatin-based chemotherapy in first-line treatment of small-cell lung cancer: The COCIS meta-analysis of individual patient data. J. Clin. Oncol. 2012, 30, 1692–1698. [Google Scholar] [CrossRef] [PubMed]
- Hanna, N.; Bunn, P.A.; Langer, C.; Einhorn, L.; Guthrie, T.; Beck, T.; Ansari, R.; Ellis, P.; Byrne, M.; Morrison, M.; et al. Randomized phase III trial comparing irinotecan/cisplatin with etoposide/cisplatin in patients with previously untreated extensive-stage disease small-cell lung cancer. J. Clin. Oncol. 2006, 24, 2038–2043. [Google Scholar] [CrossRef] [PubMed]
- Brennan, S.M.; Gregory, D.L.; Stillie, A.; Herschtal, A.; Mac Manus, M.; Ball, D.L. Should extrapulmonary small cell cancer be managed like small cell lung cancer? Cancer 2010, 116, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Cicin, I.; Karagol, H.; Uzunoglu, S.; Uygun, K.; Usta, U.; Kocak, Z.; Caloglu, M.; Saynak, M.; Tokatli, F.; Uzal, C. Extrapulmonary small-cell carcinoma compared with small-cell lung carcinoma: A retrospective single-center study. Cancer 2007, 110, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Terashima, T.; Morizane, C.; Hiraoka, N.; Tsuda, H.; Tamura, T.; Shimada, Y.; Kaneko, S.; Kushima, R.; Ueno, H.; Kondo, S.; et al. Comparison of chemotherapeutic treatment outcomes of advanced extrapulmonary neuroendocrine carcinomas and advanced small-cell lung carcinoma. Neuroendocrinology 2012, 96, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Morizane, C.; Machida, N.; Honma, Y.; Okusaka, T.; Boku, N.; Kato, K.; Mizusawa, J.; Katayama, H.; Hiraoka, N.; Taniguchi, H.; et al. Randomized phase III study of etoposide plus cisplatin versus irinotecan plus cisplatin in advanced neuroendocrine carcinoma of the digestive system: A Japan clinical oncology group study (JCOG1213). J. Clin. Oncol. 2015, 33 (Suppl.). abstr TPS4143. [Google Scholar]
- Welin, S.; Sorbye, H.; Sebjornsen, S.; Knappskog, S.; Busch, C.; Oberg, K. Clinical effect of temozolomide-based chemotherapy in poorly differentiated endocrine carcinoma after progression on first-line chemotherapy. Cancer 2011, 117, 4617–4622. [Google Scholar] [CrossRef] [PubMed]
- Hentic, O.; Hammel, P.; Couvelard, A.; Rebours, V.; Zappa, M.; Palazzo, M.; Maire, F.; Goujon, G.; Gillet, A.; Lévy, P.; et al. FOLFIRI regimen: An effective second-line chemotherapy after failure of etoposide-platinum combination in patients with neuroendocrine carcinomas grade 3. Endocr. Relat. Cancer 2012, 19, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; López-Martin, J.A.; Bendell, J.; Ott, P.A.; Taylor, M.; Eder, J.P.; Jäger, D.; Pietanza, M.C.; Le, D.T.; de Braud, F.; et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): A multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 883–895. [Google Scholar] [CrossRef]
- Ott, P.A.; Fernandez, M.E.E.; Hiret, S.; Kim, D.W.; Moss, R.A.; Winser, T.; Yuan, S.; Cheng, J.D.; Piperdi, B.; Mehnert, J.M. Pembrolizumab (MK-3475) in patients (pts) with extensive-stage small cell lung cancer (SCLC): Preliminary safety and efficacy results from KEYNOTE-028. J. Clin. Oncol. 2015, 33 (Suppl.). abstr 7502. [Google Scholar]
- Rudin, C.M.; Pietanza, M.C.; Bauer, T.M.; Spigel, D.R.; Ready, N.; Morgensztern, D.; Glisson, B.S.; Byers, L.A.; Johnson, M.L.; Burris, H.A.; et al. Safety and efficacy of single-agent rovalpituzumab tesirine (SC16LD6.5), a delta-like protein 3 (DLL3)-targeted antibody-drug conjugate (ADC) in recurrent or refractory small cell lung cancer (SCLC). J. Clin. Oncol. 2016, 34 (Suppl.). abstr LBA8505. [Google Scholar]
- Pietanza, M.C.; Spira, A.I.; Jotte, R.M.; Gadgeel, S.M.; Mita, A.C.; Hart, L.H.; Gluck, W.L.; Chiang, A.C.; Liu, S.V.; Kapoun, A.M.; et al. Final results of phase Ib of tarextumab (TRXT, OMP-59R5, anti-Notch2/3) in combination with etoposide and platinum (EP) in patients (pts) with untreated extensive-stage small-cell lung cancer (ED-SCLC). J. Clin. Oncol. 2015, 33 (Suppl.). abstr 7508. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification | Mitotic Count (per 10 hpf) | Ki-67 Index (%) |
|---|---|---|
| NET G1 | <2 | <3 |
| NET G2 | 2–20 | 3–20 |
| NEC | >20 | >20 |
| Gene | Jiao et al. [26] | Raj et al. [33] |
|---|---|---|
| MEN1 | 44% | 61% |
| ATRX | 18% | 25% |
| DAXX | 25% | 41% |
| PTEN | 7% | 11% |
| TSC1/TSC2 | 9% | 18% |
| PIK3CA | 1% | NA |
| ARID1A | NA | 14% |
| SETD2 | NA | 21% |
| Gene | Yachida et al. [35] | Bergsland et al. [38] |
|---|---|---|
| TP53 | 57% | 18% |
| RB1 | 71% | 10% |
| CDKN2A | 0% | 21% |
| CDKN2B | NA | 16% |
| KRAS | 29% | 7% |
| MEN1 | NA | 33% |
| DAXX | NA | 20% |
| Agent | Mechanism | Phase | Status |
|---|---|---|---|
| Romidepsin | HDAC inhibitor | II | Terminated |
| Motesanib + Octreotide | Multi-tyrosine kinase inhibitor | II | Completed |
| Ganitumab | Human anti–insulin-like growth factor receptor type I monoclonal antibody | II | Ongoing |
| MK-2206 | AKT-inhibitor | II | Completed |
| Cabozantinib | Multi-tyrosine kinase inhibitor | II | Ongoing |
| X-82 + everolimus | VEGFR tyrosine Kinase Inhibitor | I/II | Ongoing |
| Endostatin + temozolomide/dacarbazine-based chemotherapy | The 20 kDa C-terminal fragment of collagen XVIII | II | Ongoing |
| Famitinib | Multi-tyrosine kinase inhibitor | II | Ongoing |
| Fosbretabulin | Microtubule destabilizing agent | II | Ongoing |
| Carfilzomib | Proteasome inhibitor | II | Ongoing |
| Ribociclib | CDK 4/6 inhibitor | II | Ongoing |
| Sulfatinib | Tyrosine kinase inhibitor of VEGFR 1, 2 and 3 and FGFR 1 | III | Ongoing |
| Ibrutinib | Bruton’s tyrosine kinase inhibitor | II | Ongoing |
| Palbociclib | CDK 4/6 inhibitor | II | Ongoing |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohmoto, A.; Rokutan, H.; Yachida, S. Pancreatic Neuroendocrine Neoplasms: Basic Biology, Current Treatment Strategies and Prospects for the Future. Int. J. Mol. Sci. 2017, 18, 143. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18010143
Ohmoto A, Rokutan H, Yachida S. Pancreatic Neuroendocrine Neoplasms: Basic Biology, Current Treatment Strategies and Prospects for the Future. International Journal of Molecular Sciences. 2017; 18(1):143. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18010143
Chicago/Turabian StyleOhmoto, Akihiro, Hirofumi Rokutan, and Shinichi Yachida. 2017. "Pancreatic Neuroendocrine Neoplasms: Basic Biology, Current Treatment Strategies and Prospects for the Future" International Journal of Molecular Sciences 18, no. 1: 143. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18010143