
Post-Translational Modifications of the TAK1-TAB Complex


Abstract
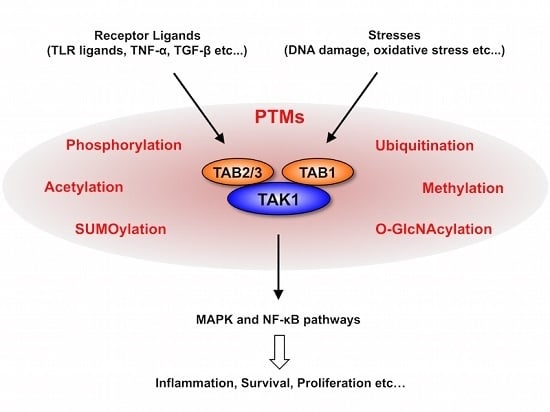
:
1. Introduction
2. TAK1 Signaling Pathway
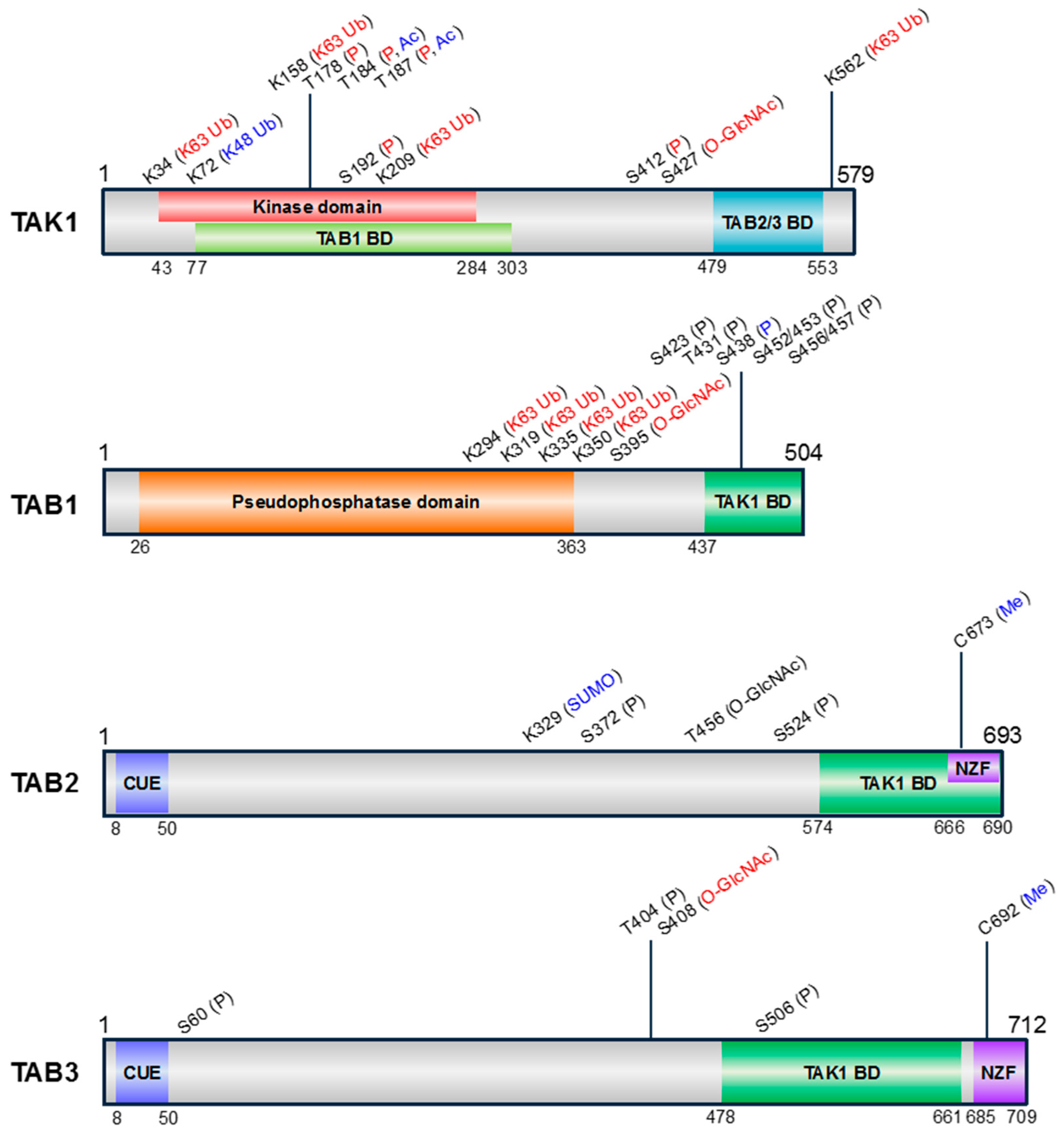
3. PTMs of TAK1 and TABs
3.1. Phosphorylation
3.2. Ubiquitination
3.3. SUMOylation
3.4. Acetylation
3.5. Methylation
3.6. O-GlcNAcylation
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Yamaguchi, K.; Shirakabe, K.; Shibuya, H.; Irie, K.; Oishi, I.; Ueno, N.; Taniguchi, T.; Nishida, E.; Matsumoto, K. Identification of a member of the MAPKKK family as a potential mediator of TGF-β signal transduction. Science 1995, 270, 2008–2011. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Aye Thu, C.; Liu, X.Y.; Xi, J.; Cheung, P.C. TAK1, more than just innate immunity. IUBMB Life 2012, 64, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Ajibade, A.A.; Wang, H.Y.; Wang, R.F. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 2013, 34, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in NF-κB signaling pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.C.; Campbell, D.G.; Nebreda, A.R.; Cohen, P. Feedback control of the protein kinase TAK1 by SAPK2a/p38α. EMBO J. 2003, 22, 5793–5805. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Stilmann, M.; Arslan, S.C.; Khanna, K.K.; Dittmar, G.; Scheidereit, C. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-κB activation. Mol. Cell 2010, 40, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.P.; Sun, L.; Chen, X.; Pineda, G.; Jiang, X.; Adhikari, A.; Zeng, W.; Chen, Z.J. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature 2009, 461, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.C.; Nebreda, A.R.; Cohen, P. TAB3, a new binding partner of the protein kinase TAK1. Biochem. J. 2004, 378, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Ishitani, T.; Takaesu, G.; Ninomiya-Tsuji, J.; Shibuya, H.; Gaynor, R.B.; Matsumoto, K. Role of the TAB2-related protein TAB3 in IL-1 and TNF signaling. EMBO J. 2003, 22, 6277–6288. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Sanjuan, I.; Bell, E.; Altmann, C.R.; Vonica, A.; Brivanlou, A.H. Gene profiling during neural induction in Xenopus laevis: Regulation of BMP signaling by post-transcriptional mechanisms and TAB3, a novel TAK1-binding protein. Development 2002, 129, 5529–5540. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, H.; Yamaguchi, K.; Shirakabe, K.; Tonegawa, A.; Gotoh, Y.; Ueno, N.; Irie, K.; Nishida, E.; Matsumoto, K. TAB1: An activator of the TAK1 MAPKKK in TGF-β signal transduction. Science 1996, 272, 1179–1182. [Google Scholar] [CrossRef] [PubMed]
- Takaesu, G.; Kishida, S.; Hiyama, A.; Yamaguchi, K.; Shibuya, H.; Irie, K.; Ninomiya-Tsuji, J.; Matsumoto, K. TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol. Cell 2000, 5, 649–658. [Google Scholar] [CrossRef]
- Ono, K.; Ohtomo, T.; Sato, S.; Sugamata, Y.; Suzuki, M.; Hisamoto, N.; Ninomiya-Tsuji, J.; Tsuchiya, M.; Matsumoto, K. An evolutionarily conserved motif in the TAB1 C-terminal region is necessary for interaction with and activation of TAK1 MAPKKK. J. Biol. Chem. 2001, 276, 24396–24400. [Google Scholar] [CrossRef] [PubMed]
- Besse, A.; Lamothe, B.; Campos, A.D.; Webster, W.K.; Maddineni, U.; Lin, S.C.; Wu, H.; Darnay, B.G. TAK1-dependent signaling requires functional interaction with TAB2/TAB3. J. Biol. Chem. 2007, 282, 3918–3928. [Google Scholar] [CrossRef] [PubMed]
- Holtmann, H.; Enninga, J.; Kalble, S.; Thiefes, A.; Dorrie, A.; Broemer, M.; Winzen, R.; Wilhelm, A.; Ninomiya-Tsuji, J.; Matsumoto, K.; et al. The MAPK kinase kinase TAK1 plays a central role in coupling the interleukin-1 receptor to both transcriptional and RNA-targeted mechanisms of gene regulation. J. Biol. Chem. 2001, 276, 3508–3516. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Nishi, A.; Sato, N.; Mizukami, J.; Miyoshi, H.; Sugita, T. TAK1–TAB1 fusion protein: A novel constitutively active mitogen-activated protein kinase kinase kinase that stimulates AP-1 and NF-κB signaling pathways. Biochem. Biophys. Res. Commun. 2002, 297, 1277–1281. [Google Scholar] [CrossRef]
- Brown, K.; Vial, S.C.; Dedi, N.; Long, J.M.; Dunster, N.J.; Cheetham, G.M. Structural basis for the interaction of TAK1 kinase with its activating protein TAB1. J. Mol. Biol. 2005, 354, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Ohtomo, T.; Ninomiya-Tsuji, J.; Tsuchiya, M. A dominant negative TAK1 inhibits cellular fibrotic responses induced by TGF-β. Biochem. Biophys. Res. Commun. 2003, 307, 332–337. [Google Scholar] [CrossRef]
- Sakurai, H.; Miyoshi, H.; Mizukami, J.; Sugita, T. Phosphorylation-dependent activation of TAK1 mitogen-activated protein kinase kinase kinase by TAB1. FEBS Lett. 2000, 474, 141–145. [Google Scholar] [CrossRef]
- Conner, S.H.; Kular, G.; Peggie, M.; Shepherd, S.; Schuttelkopf, A.W.; Cohen, P.; Van Aalten, D.M. TAK1-binding protein 1 is a pseudophosphatase. Biochem. J. 2006, 399, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Kanayama, A.; Seth, R.B.; Sun, L.; Ea, C.K.; Hong, M.; Shaito, A.; Chiu, Y.H.; Deng, L.; Chen, Z.J. TAB2 and TAB3 activate the NF-κB pathway through binding to polyubiquitin chains. Mol. Cell 2004, 15, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Shank, J.; Cusson, N.; Kelliher, M.A. The kinase activity of Rip1 is not required for tumor necrosis factor-α-induced IκB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2. J. Biol. Chem. 2004, 279, 33185–33191. [Google Scholar] [CrossRef] [PubMed]
- Kulathu, Y.; Akutsu, M.; Bremm, A.; Hofmann, K.; Komander, D. Two-sided ubiquitin binding explains specificity of the TAB2 NZF domain. Nat. Struct. Mol. Biol. 2009, 16, 1328–1330. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Yoshikawa, A.; Yamashita, M.; Yamagata, A.; Fukai, S. Structural basis for specific recognition of Lys 63-linked polyubiquitin chains by NZF domains of TAB2 and TAB3. EMBO J. 2009, 28, 3903–3909. [Google Scholar] [CrossRef] [PubMed]
- Symons, A.; Beinke, S.; Ley, S.C. Map kinase kinase kinases and innate immunity. Trends Immunol. 2006, 27, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef]
- Sun, L.; Deng, L.; Ea, C.K.; Xia, Z.P.; Chen, Z.J. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol. Cell 2004, 14, 289–301. [Google Scholar] [CrossRef]
- Shinohara, H.; Yasuda, T.; Aiba, Y.; Sanjo, H.; Hamadate, M.; Watarai, H.; Sakurai, H.; Kurosaki, T. PKC β regulates BCR-mediated IKK activation by facilitating the interaction between TAK1 and CARMA1. J. Exp. Med. 2005, 202, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.H.; Landstrom, M. The type I TGF-β receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-β. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Nagai, S.; Ninomiya-Tsuji, J.; Nishita, M.; Tamai, K.; Irie, K.; Ueno, N.; Nishida, E.; Shibuya, H.; Matsumoto, K. XIAP, a cellular member of the inhibitor of apoptosis protein family, links the receptors to TAB1–TAK1 in the BMP signaling pathway. EMBO J. 1999, 18, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Lin, S.C.; Huang, Y.; Kang, Y.J.; Rich, R.; Lo, Y.C.; Myszka, D.; Han, J.; Wu, H. XIAP induces NF-κB activation via the BIR1/TAB1 interaction and BIR1 dimerization. Mol. Cell 2007, 26, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.H.; Wong, E.T.; Shi, Y.; Niu, J.; Chen, Z.; Miyamoto, S.; Tergaonkar, V. ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-mediated IKK activation in response to genotoxic stress. Mol. Cell 2010, 40, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xia, F.; Hermance, N.; Mabb, A.; Simonson, S.; Morrissey, S.; Gandhi, P.; Munson, M.; Miyamoto, S.; Kelliher, M.A. A cytosolic ATM/NEMO/RIP1 complex recruits TAK1 to mediate the NF-κB and p38 mitogen-activated protein kinase (MAPK)/MAPK-activated protein 2 responses to DNA damage. Mol. Cell. Biol. 2011, 31, 2774–2786. [Google Scholar] [CrossRef] [PubMed]
- Ishitani, T.; Ninomiya-Tsuji, J.; Nagai, S.; Nishita, M.; Meneghini, M.; Barker, N.; Waterman, M.; Bowerman, B.; Clevers, H.; Shibuya, H.; et al. The TAK1-NLK-MAPK-related pathway antagonizes signalling between β-catenin and transcription factor TCF. Nature 1999, 399, 798–802. [Google Scholar] [PubMed]
- Ishitani, T.; Kishida, S.; Hyodo-Miura, J.; Ueno, N.; Yasuda, J.; Waterman, M.; Shibuya, H.; Moon, R.T.; Ninomiya-Tsuji, J.; Matsumoto, K. The TAK1–NLK mitogen-activated protein kinase cascade functions in the Wnt-5a/Ca2+ pathway to antagonize Wnt/β-catenin signaling. Mol. Cell. Biol. 2003, 23, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Kanei-Ishii, C.; Ninomiya-Tsuji, J.; Tanikawa, J.; Nomura, T.; Ishitani, T.; Kishida, S.; Kokura, K.; Kurahashi, T.; Ichikawa-Iwata, E.; Kim, Y.; et al. Wnt-1 signal induces phosphorylation and degradation of c-Myb protein via TAK1, HIPK2, and NLK. Genes Dev. 2004, 18, 816–829. [Google Scholar] [CrossRef] [PubMed]
- Kurahashi, T.; Nomura, T.; Kanei-Ishii, C.; Shinkai, Y.; Ishii, S. The Wnt–NLK signaling pathway inhibits A-Myb activity by inhibiting the association with coactivator CBP and methylating histone H3. Mol. Biol. Cell 2005, 16, 4705–4713. [Google Scholar] [CrossRef] [PubMed]
- Smit, L.; Baas, A.; Kuipers, J.; Korswagen, H.; van de Wetering, M.; Clevers, H. Wnt activates the Tak1/Nemo-like kinase pathway. J. Biol. Chem. 2004, 279, 17232–17240. [Google Scholar] [CrossRef] [PubMed]
- Culver, C.; Sundqvist, A.; Mudie, S.; Melvin, A.; Xirodimas, D.; Rocha, S. Mechanism of hypoxia-induced NF-κB. Mol. Cell. Biol. 2010, 30, 4901–4921. [Google Scholar] [CrossRef] [PubMed]
- Blanco, S.; Santos, C.; Lazo, P.A. Vaccinia-related kinase 2 modulates the stress response to hypoxia mediated by TAK1. Mol. Cell. Biol. 2007, 27, 7273–7283. [Google Scholar] [CrossRef] [PubMed]
- Melvin, A.; Mudie, S.; Rocha, S. Further insights into the mechanism of hypoxia-induced NF-κB. Cell Cycle 2011, 10, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, W.C.; Omori, E.; Akira, S.; Matsumoto, K.; Ninomiya-Tsuji, J. Osmotic stress activates the TAK1–JNK pathway while blocking TAK1-mediated NF-κB activation: TAO2 regulates TAK1 pathways. J. Biol. Chem. 2006, 281, 28802–28810. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Shen, X.; Shen, F.; Zhong, W.; Wu, H.; Liu, S.; Lai, J. TAK1 activates AMPK-dependent cell death pathway in hydrogen peroxide-treated cardiomyocytes, inhibited by heat shock protein-70. Mol. Cell. Biochem. 2013, 377, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Martin, G.; Hoyer-Hansen, M.; Garcia-Garcia, C.; Fumarola, C.; Farkas, T.; Lopez-Rivas, A.; Jaattela, M. TAK1 activates AMPK-dependent cytoprotective autophagy in trail-treated epithelial cells. EMBO J. 2009, 28, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Jeong, S.; Jung, E.; Baik, K.H.; Chang, M.H.; Kim, S.A.; Shim, J.H.; Chun, E.; Lee, K.Y. AMP-activated protein kinase-α1 as an activating kinase of TGF-β-activated kinase 1 has a key role in inflammatory signals. Cell Death Dis. 2012, 3, e357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamidi, A.; von Bulow, V.; Hamidi, R.; Winssinger, N.; Barluenga, S.; Heldin, C.H.; Landstrom, M. Polyubiquitination of transforming growth factor β (TGF-β)-associated kinase 1 mediates nuclear factor-κB activation in response to different inflammatory stimuli. J. Biol. Chem. 2012, 287, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Fan, Y.; Cheng, J.; Cheng, D.; Zhao, Y.; Cao, B.; Ma, L.; An, L.; Jia, W.; Su, X.; et al. TAK1 ubiquitination regulates doxorubicin-induced NF-κB activation. Cell. Signal. 2013, 25, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Shi, Y.; Liu, S.; Mao, R.; An, L.; Zhao, Y.; Zhang, H.; Zhang, F.; Xu, G.; Qin, J.; et al. Lys48-linked TAK1 polyubiquitination at lysine-72 downregulates TNFα-induced NF-κB activation via mediating TAK1 degradation. Cell. Signal. 2012, 24, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Yu, Y.; Mao, R.; Zhang, H.; Yang, J. TAK1 Lys-158 but not Lys-209 is required for IL-1β-induced Lys63-linked TAK1 polyubiquitination and IKK/NF-κB activation. Cell. Signal. 2011, 23, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Yu, Y.; Shi, Y.; Sun, W.; Xie, M.; Ge, N.; Mao, R.; Chang, A.; Xu, G.; Schneider, M.D.; et al. Lysine 63-linked polyubiquitination of TAK1 at lysine 158 is required for tumor necrosis factor α- and interleukin-1β-induced IKK/NF-κB and JNK/AP-1 activation. J. Biol. Chem. 2010, 285, 5347–5360. [Google Scholar] [CrossRef] [PubMed]
- Mao, R.; Fan, Y.; Mou, Y.; Zhang, H.; Fu, S.; Yang, J. TAK1 lysine 158 is required for TGF-β-induced TRAF6-mediated Smad-independent IKK/NF-κB and JNK/AP-1 activation. Cell. Signal. 2011, 23, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yan, J.; Mao, A.P.; Li, C.; Ran, Y.; Shu, H.B.; Wang, Y.Y. Tripartite motif 8 (TRIM8) modulates TNFα- and IL-1β-triggered NF-κB activation by targeting TAK1 for K63-linked polyubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 19341–19346. [Google Scholar] [CrossRef] [PubMed]
- Lamb, A.; Chen, J.; Blanke, S.R.; Chen, L.F. Helicobacter pylori activates NF-κB by inducing Ubc13-mediated ubiquitination of lysine 158 of TAK1. J. Cell. Biochem. 2013, 114, 2284–2292. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Ge, N.; Xie, M.; Sun, W.; Burlingame, S.; Pass, A.K.; Nuchtern, J.G.; Zhang, D.; Fu, S.; Schneider, M.D.; et al. Phosphorylation of Thr-178 and Thr-184 in the TAK1 T-loop is required for interleukin (IL)-1-mediated optimal NF-κB and AP-1 activation as well as IL-6 gene expression. J. Biol. Chem. 2008, 283, 24497–24505. [Google Scholar] [CrossRef]
- Paquette, N.; Conlon, J.; Sweet, C.; Rus, F.; Wilson, L.; Pereira, A.; Rosadini, C.V.; Goutagny, N.; Weber, A.N.; Lane, W.S.; et al. Serine/threonine acetylation of TGF-β-activated kinase (TAK1) by Yersinia Pestis YopJ inhibits innate immune signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 12710–12715. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Jin, J.; Chang, M.; Nakaya, M.; Hu, H.; Zou, Q.; Zhou, X.; Brittain, G.C.; Cheng, X.; Sun, S.C. TPL2 mediates autoimmune inflammation through activation of the TAK1 axis of IL-17 signaling. J. Exp. Med. 2014, 211, 1689–1702. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Li, Q.; Chen, R.; Zhang, J.; Ran, Y.; He, X.; Li, S.; Shu, H.B. The dual-specificity phosphatase DUSP14 negatively regulates tumor necrosis factor- and interleukin-1-induced nuclear factor-κB activation by dephosphorylating the protein kinase TAK1. J. Biol. Chem. 2013, 288, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Kajino, T.; Ren, H.; Iemura, S.; Natsume, T.; Stefansson, B.; Brautigan, D.L.; Matsumoto, K.; Ninomiya-Tsuji, J. Protein phosphatase 6 down-regulates TAK1 kinase activation in the IL-1 signaling pathway. J. Biol. Chem. 2006, 281, 39891–39896. [Google Scholar] [CrossRef] [PubMed]
- Hanada, M.; Ninomiya-Tsuji, J.; Komaki, K.; Ohnishi, M.; Katsura, K.; Kanamaru, R.; Matsumoto, K.; Tamura, S. Regulation of the TAK1 signaling pathway by protein phosphatase 2C. J. Biol. Chem. 2001, 276, 5753–5759. [Google Scholar] [CrossRef] [PubMed]
- Li, M.G.; Katsura, K.; Nomiyama, H.; Komaki, K.; Ninomiya-Tsuji, J.; Matsumoto, K.; Kobayashi, T.; Tamura, S. Regulation of the interleukin-1-induced signaling pathways by a novel member of the protein phosphatase 2C family (PP2Cepsilon). J. Biol. Chem. 2003, 278, 12013–12021. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Kwak, J.H.; Wang, L.; Choi, M.E. Protein phosphatase 2A is a negative regulator of transforming growth factor-β1-induced TAK1 activation in mesangial cells. J. Biol. Chem. 2008, 283, 10753–10763. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Busby, J.C.; Molkentin, J.D. Interaction between TAK1–TAB1–TAB2 and RCAN1–calcineurin defines a signalling nodal control point. Nat. Cell Biol. 2009, 11, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, K.; Matsumoto, K.; Ninomiya-Tsuji, J. TAK1 mitogen-activated protein kinase kinase kinase is activated by autophosphorylation within its activation loop. J. Biol. Chem. 2000, 275, 7359–7364. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, K.; Gohda, J.; Kanayama, A.; Miyamoto, Y.; Sakurai, H.; Yamamoto, M.; Akira, S.; Hayashi, H.; Su, B.; Inoue, J. Two mechanistically and temporally distinct NF-κB activation pathways in IL-1 signaling. Sci. Signal. 2009, 2, ra66. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, Y.; Yang, S.; Ren, F.; Xia, Y.; Chang, Z. Activation of TAK1 by Sef-S induces apoptosis in 293T cells. Cell. Signal. 2013, 25, 2039–2046. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Mizoguchi, T.; Take, I.; Kurihara, S.; Udagawa, N.; Takahashi, N. Prostaglandin E2 enhances osteoclastic differentiation of precursor cells through protein kinase A-dependent phosphorylation of TAK1. J. Biol. Chem. 2005, 280, 11395–11403. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, C.; Nie, L.; Gu, M.; Wu, A.; Han, X.; Wang, X.; Shao, J.; Xia, Z. Transforming growth factor (TGF)-β-activated kinase 1 (TAK1) activation requires phosphorylation of serine 412 by protein kinase a catalytic subunit α (PKACα) and X-linked protein kinase (PRKX). J. Biol. Chem. 2014, 289, 24226–24237. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Ouyang, C.; Lin, W.; Zhang, T.; Cao, X.; Xia, Z.; Wang, X. Phosphatase holoenzyme PP1/GADD34 negatively regulates TLR response by inhibiting TAK1 serine 412 phosphorylation. J. Immunol. 2014, 192, 2846–2856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Xu, Z.; Tao, T.; Liu, X.; Sun, X.; Ji, Y.; Han, L.; Qiu, H.; Zhu, G.; Shen, Y.; et al. Modification of TAK1 by O-linked N-acetylglucosamine facilitates TAK1 activation and promotes M1 macrophage polarization. Cell. Signal. 2016, 28, 1742–1752. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.T.; Hsu, P.H.; Hsu, W.C.; Chen, N.J.; Tseng, P.H. Polyubiquitination of transforming growth factor β-activated kinase 1 (TAK1) at lysine 562 residue regulates TLR4-mediated JNK and p38 MAPK activation. Sci. Rep. 2015, 5, 12300. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Xian, H.; Hu, J.; Tian, S.; Qin, Y.; Wang, R.F.; Cui, J. USP18 negatively regulates NF-κB signaling by targeting TAK1 and NEMO for deubiquitination through distinct mechanisms. Sci. Rep. 2015, 5, 12738. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, H.; Zhong, B.; Blonska, M.; Gorjestani, S.; Yan, M.; Tian, Q.; Zhang, D.E.; Lin, X.; Dong, C. USP18 inhibits NF-κB and NFAT activation during Th17 differentiation by deubiquitinating the TAK1-TAB1 complex. J. Exp. Med. 2013, 210, 1575–1590. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.H.; Yu, Y.; Mao, R.F.; Tan, X.J.; Xu, G.F.; Zhang, H.; Lu, X.B.; Fu, S.B.; Yang, J. USP4 targets TAK1 to downregulate TNFα-induced NF-κB activation. Cell Death Differ. 2011, 18, 1547–1560. [Google Scholar] [CrossRef] [PubMed]
- Reiley, W.W.; Jin, W.; Lee, A.J.; Wright, A.; Wu, X.; Tewalt, E.F.; Leonard, T.O.; Norbury, C.C.; Fitzpatrick, L.; Zhang, M.; et al. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase TAK1 and prevents abnormal T cell responses. J. Exp. Med. 2007, 204, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Zeng, M.; Sinha, I.; Polin, L.; Wei, W.Z.; Rathinam, C.; Flavell, R.; Massoumi, R.; Venuprasad, K. The E3 ligase itch and deubiquitinase Cyld act together to regulate TAK1 and inflammation. Nat. Immunol. 2011, 12, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Charlaftis, N.; Suddason, T.; Wu, X.; Anwar, S.; Karin, M.; Gallagher, E. The MEKK1 PHD ubiquitinates TAB1 to activate MAPKs in response to cytokines. EMBO J. 2014, 33, 2581–2596. [Google Scholar] [CrossRef] [PubMed]
- Pathak, S.; Borodkin, V.S.; Albarbarawi, O.; Campbell, D.G.; Ibrahim, A.; van Aalten, D.M. O-GlcNAcylation of TAB1 modulates TAK1-mediated cytokine release. EMBO J. 2012, 31, 1394–1404. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, H.; Campbell, D.G.; Burness, K.; Hastie, J.; Ronkina, N.; Shim, J.H.; Arthur, J.S.; Davis, R.J.; Gaestel, M.; Johnson, G.L.; et al. Roles for TAB1 in regulating the IL-1-dependent phosphorylation of the TAB3 regulatory subunit and activity of the TAK1 complex. Biochem. J. 2008, 409, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Li, J.P.; Chiu, L.L.; Lan, J.L.; Chen, D.Y.; Chuang, H.C.; Huang, C.Y.; Tan, T.H. Dual-specificity phosphatase 14 (DUSP14/MKP6) negatively regulates TCR signaling by inhibiting TAB1 activation. J. Immunol. 2014, 192, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Beuerlein, K.; Eckart, C.; Weiser, H.; Dickkopf, B.; Muller, H.; Sakurai, H.; Kracht, M. Identification and functional characterization of novel phosphorylation sites in TAK1-binding protein (TAB) 1. PLoS ONE 2011, 6, e29256. [Google Scholar] [CrossRef] [PubMed]
- Theivanthiran, B.; Kathania, M.; Zeng, M.; Anguiano, E.; Basrur, V.; Vandergriff, T.; Pascual, V.; Wei, W.Z.; Massoumi, R.; Venuprasad, K. The E3 ubiquitin ligase itch inhibits p38α signaling and skin inflammation through the ubiquitylation of TAB1. Sci. Signal. 2015, 8, ra22. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jiang, J.; Lu, Y.; Shi, G.; Liu, R.; Cao, Y. TAB2, an important upstream adaptor of interleukin-1 signaling pathway, is subject to sumoylation. Mol. Cell. Biochem. 2014, 385, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.M.; Yang, Q.; Zhang, J.; Liu, S.M.; Zhang, Y.; Lin, H.; Huang, Z.F.; Wang, Y.Y.; Zhang, X.D.; Zhong, B.; et al. TRIM38 inhibits TNFα- and IL-1β-triggered NF-κB activation by mediating lysosome-dependent degradation of TAB2/3. Proc. Natl. Acad. Sci. USA 2014, 111, 1509–1514. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; He, Z.; Shao, Z.; Lu, H. TAB3 O-GlcNAcylation promotes metastasis of triple negative breast cancer. Oncotarget 2016, 7, 22807–22818. [Google Scholar] [PubMed]
- Zhang, L.; Ding, X.; Cui, J.; Xu, H.; Chen, J.; Gong, Y.N.; Hu, L.; Zhou, Y.; Ge, J.; Lu, Q.; et al. Cysteine methylation disrupts ubiquitin-chain sensing in NF-κB activation. Nature 2012, 481, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Muhlen, S.; Oates, C.V.; Pearson, J.S.; Hartland, E.L. Identification of a distinct substrate-binding domain in the bacterial cysteine methyltransferase effectors NleE and OspZ. J. Biol. Chem. 2016, 291, 20149–20162. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhang, Y.; Zhong, B.; Wang, Y.Y.; Diao, F.C.; Wang, R.P.; Zhang, M.; Chen, D.Y.; Zhai, Z.H.; Shu, H.B. RBCK1 negatively regulates tumor necrosis factor- and interleukin-1-triggered NF-κB activation by targeting TAB2/3 for degradation. J. Biol. Chem. 2007, 282, 16776–16782. [Google Scholar] [CrossRef] [PubMed]
- Scholz, R.; Sidler, C.L.; Thali, R.F.; Winssinger, N.; Cheung, P.C.; Neumann, D. Autoactivation of transforming growth factor β-activated kinase 1 is a sequential bimolecular process. J. Biol. Chem. 2010, 285, 25753–25766. [Google Scholar] [CrossRef] [PubMed]
- Singhirunnusorn, P.; Suzuki, S.; Kawasaki, N.; Saiki, I.; Sakurai, H. Critical roles of threonine 187 phosphorylation in cellular stress-induced rapid and transient activation of transforming growth factor-β-activated kinase 1 (TAK1) in a signaling complex containing TAK1-binding protein TAB1 and TAB2. J. Biol. Chem. 2005, 280, 7359–7368. [Google Scholar] [CrossRef] [PubMed]
- Prickett, T.D.; Ninomiya-Tsuji, J.; Broglie, P.; Muratore-Schroeder, T.L.; Shabanowitz, J.; Hunt, D.F.; Brautigan, D.L. TAB4 stimulates TAK1–TAB1 phosphorylation and binds polyubiquitin to direct signaling to NF-κB. J. Biol. Chem. 2008, 283, 19245–19254. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, F.; Dikic, I. Atypical ubiquitin chains: New molecular signals. EMBO Rep. 2008, 9, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Rieser, E.; Cordier, S.M.; Walczak, H. Linear ubiquitination: A newly discovered regulator of cell signalling. Trends Biochem. Sci. 2013, 38, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Deng, W.; Bi, E.; Mao, K.; Ji, Y.; Lin, G.; Wu, X.; Tao, Z.; Li, Z.; Cai, X.; et al. TRIM30 α negatively regulates TLR-mediated NF-κB activation by targeting TAB2 and TAB3 for degradation. Nat. Immunol. 2008, 9, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Huang, F.; Xiao, H.; Sun, B.; Yang, R. TRIM22 inhibits the TRAF6-stimulated NF-κB pathway by targeting TAB2 for degradation. Virol. Sin. 2013, 28, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Mu, R.; Chang, Y.; Wang, Y.B.; Wu, M.; Tu, H.Q.; Zhang, Y.C.; Guo, S.S.; Qin, X.H.; Li, T.; et al. RNF4 negatively regulates NF-κB signaling by down-regulating TAB2. FEBS Lett. 2015, 589, 2850–2858. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Kerscher, O.; Felberbaum, R.; Hochstrasser, M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 2006, 22, 159–180. [Google Scholar] [CrossRef] [PubMed]
- Drazic, A.; Myklebust, L.M.; Ree, R.; Arnesen, T. The world of protein acetylation. Biochim. Biophys. Acta 2016, 1864, 1372–1401. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R.; Peak-Chew, S.Y.; McMahon, H.T. Acetylation of MEK2 and IκB kinase (IKK) activation loop residues by YopJ inhibits signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 18574–18579. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Keitany, G.; Li, Y.; Wang, Y.; Ball, H.L.; Goldsmith, E.J.; Orth, K. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 2006, 312, 1211–1214. [Google Scholar] [CrossRef] [PubMed]
- Biggar, K.K.; Li, S.S. Non-histone protein methylation as a regulator of cellular signalling and function. Nat. Rev. Mol. Cell Biol. 2015, 16, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Hardiville, S.; Hart, G.W. Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metab. 2014, 20, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, J.F.; Gong, C.X.; Monroe, M.E.; Aldrich, J.T.; Clauss, T.R.; Purvine, S.O.; Wang, Z.; Camp, D.G.; Shabanowitz, J.; Stanley, P.; et al. Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc. Natl. Acad. Sci. USA 2012, 109, 7280–7285. [Google Scholar] [CrossRef] [PubMed]
- Trinidad, J.C.; Barkan, D.T.; Gulledge, B.F.; Thalhammer, A.; Sali, A.; Schoepfer, R.; Burlingame, A.L. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol. Cell. Proteom. 2012, 11, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Nagel, A.K.; Schilling, M.; Comte-Walters, S.; Berkaw, M.N.; Ball, L.E. Identification of O-linked N-acetylglucosamine (O-GlcNAc)-modified osteoblast proteins by electron transfer dissociation tandem mass spectrometry reveals proteins critical for bone formation. Mol. Cell. Proteom. 2013, 12, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, Y.; Shibuya, H.; Takeda, N.; Ninomiya-Tsuji, J.; Yasui, T.; Miyado, K.; Sekimoto, T.; Ueno, N.; Matsumoto, K.; Yamada, G. Targeted disruption of the TAB1 gene causes embryonic lethality and defects in cardiovascular and lung morphogenesis. Mech. Dev. 2002, 119, 239–249. [Google Scholar] [CrossRef]
- Sanjo, H.; Takeda, K.; Tsujimura, T.; Ninomiya-Tsuji, J.; Matsumoto, K.; Akira, S. TAB2 is essential for prevention of apoptosis in fetal liver but not for interleukin-1 signaling. Mol. Cell. Biol. 2003, 23, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Jadrich, J.L.; O’Connor, M.B.; Coucouvanis, E. The TGF-β activated kinase TAK1 regulates vascular development in vivo. Development 2006, 133, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol. Sci. 2012, 33, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.Y.; Tesch, G.H.; Ozols, E.; Xie, M.; Schneider, M.D.; Nikolic-Paterson, D.J. TGF-β1-activated kinase-1 regulates inflammation and fibrosis in the obstructed kidney. Am. J. Physiol. Ren. Physiol. 2011, 300, 1410–1421. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.G.; Wang, Y.; Hao, W.; Wan, Y.Y. An essential role for TAK1 in the contact hypersensitivity response. Cell. Mol. Immunol. 2011, 8, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Neubert, M.; Ridder, D.A.; Bargiotas, P.; Akira, S.; Schwaninger, M. Acute inhibition of TAK1 protects against neuronal death in cerebral ischemia. Cell Death Differ. 2011, 18, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Wade, E.M.; Daniel, P.B.; Jenkins, Z.A.; McInerney-Leo, A.; Leo, P.; Morgan, T.; Addor, M.C.; Ades, L.C.; Bertola, D.; Bohring, A.; et al. Mutations in MAP3K7 that alter the activity of the TAK1 signaling complex cause frontometaphyseal dysplasia. Am. J. Hum. Genet. 2016, 99, 392–406. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Receptor-Mediated Signaling | Stress Response |
|---|---|
| IL-1α, IL-1β | DNA damage |
| TLR ligands | Oxidative stress |
| TNF-α | Osmotic stress |
| TGF-β | Hypoxia |
| BMP | – |
| T-cell antigens | – |
| B-cell antigens | – |
| Wnt | – |
| Protein | Site | PTM | Reaction | Catalyzed By | Effect on TAK1 Activity | Inducing Stimuli | Reference |
|---|---|---|---|---|---|---|---|
| TAK1 | K34 | K63 Ub | ubiquitination | TRAF6 | + | TGF-β, TNF-α, LPS, IL-1β | [31,49] |
| K72 | K48 Ub | ubiquitination | ITCH | – | TNF-α, Doxorubicin | [50,51] | |
| K158 | K63 Ub | ubiquitination | TRAF2, TRAF6, TRIM8 | + | TNF-α, IL-1β, TGF-β | [52,53,54,55] | |
| ? | + | Doxorubicin | [50] | ||||
| TRAF6, Ubc13 | + | H. pylori infection (CagA) | [56] | ||||
| deubiquitination | USP4 | – | Doxorubicin | [50] | |||
| T178 | P | autophoshporylation | + | IL-1β, TAB1 co-expression, etc. | [57] | ||
| T184 | P | autophosphorylation | + | TAB1 co-expression, etc. | [19] | ||
| Ac | Acetylation | YopJ | – | bacterial infection | [58] | ||
| T187 | P | autophosphorylation | + | TAB1 co-expression, etc. | [57] | ||
| phosphorylation | TPL-2 | + | IL-17 | [59] | |||
| dephosphorylation | DUSP14 (MKP6) | – | TNF-α, IL-1β | [60] | |||
| PP6, PP2Cβ-1, PP2Cε | – | IL-1β | [61,62,63] | ||||
| PP2A, calcineurin | – | TGF-β | [64,65] | ||||
| Ac | Acetylation | YopJ | – | bacterial infection | [58] | ||
| S192 | P | autophosphorylation | ? | + | IL-1 *, TAB1 co-expression, etc. | [66] | |
| K209 | K63 Ub | ubiquitination | TRAF6, Ubc13 | + | IL-1 * | [67] | |
| ? | + | Sef-S expression | [68] | ||||
| S412 | P | phosphorylation | PKA | + | TNF-α, RANKL | [69] | |
| PKACα, PRKX | + | IL-1β, LPS, TNF-α | [70] | ||||
| dephosphorylation | PP1 | – | TLR ligands | [71] | |||
| S427 | O-GlcNAc | O-GlcNAcylation | OGT | + | IL-1α, osmotic stress | [72] | |
| K562 | K63 Ub | ubiquitination | TRAF6 | + | LPS | [73] | |
| deubiquitination | USP18 | − | LPS, TNF-α, TCR ligands | [74,75] | |||
| deubiquitination | USP4, CYLD | – | TNF-α | [76,77,78] | |||
| TAB1 | K294/319/335/350 | K63 Ub | ubiquitination | MEKK1 (PHD), UBE2N | + | TGF-β | [79] |
| S395 | O-GlcNAc | O-GlcNAcylation | OGT | + | IL-1α, osmotic stress | [80] | |
| S423/T431 | P | phosphorylation | p38α | ? | LPS, IL-1α, TNF-α, H2O2, UV-C, anisomycin, sorbitol | [5] | |
| S438 | P | phosphorylation | ERK, JNK | ? | LPS, IL-1α, IL-1β, TNF-α, H2O2, UV-C, anisomycin, sorbitol | [5,81] | |
| dephosphorylation | DUSP14 (MKP6) | – | TCR ligands | [82] | |||
| S452/453/456/457 | P | phosphorylation | TAK1, p38 | ? | IL-1α, anisomycin, sorbitol | [83] | |
| dephosphorylation | calcineurin | ? | ? | [65] | |||
| K48 Ub | ubiquitination | ITCH | – | TNF-α | [84] | ||
| TAB2 | K329 | SUMO | SUMOylation | PIAS3 | – | ? | [85] |
| S372/S524 | P | phosphorylation | ? | ? | IL-1β | [63] | |
| T456 | O-GlcNAc | O-GlcNAcylation | OGT | ? | ? | [80] | |
| C673 | Me | Methylation | NleE | – | bacterial infection | [86,87] | |
| K48 Ub | ubiquitination | RBCK1 | – | IL-1β, TNF-α | [78] | ||
| TAB3 | S60/T404 | P | phosphorylation | p38α | ? | IL-1α, IL-1β | [81] |
| S408 | O-GlcNAc | O-GlcNAcylation | OGT | + | IL-1β | [88] | |
| S506 | P | phosphorylation | p38α, MAPKAP-K2/K3 | ? | IL-1α, IL-1β | [81] | |
| C692 | Me | Methylation | NleE, OspZ | – | bacterial infection | [89,90] | |
| K48 Ub | ubiquitination | RBCK1 | – | IL-1β, TNF-α | [91] |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirata, Y.; Takahashi, M.; Morishita, T.; Noguchi, T.; Matsuzawa, A. Post-Translational Modifications of the TAK1-TAB Complex. Int. J. Mol. Sci. 2017, 18, 205. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18010205
Hirata Y, Takahashi M, Morishita T, Noguchi T, Matsuzawa A. Post-Translational Modifications of the TAK1-TAB Complex. International Journal of Molecular Sciences. 2017; 18(1):205. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18010205
Chicago/Turabian StyleHirata, Yusuke, Miki Takahashi, Tohru Morishita, Takuya Noguchi, and Atsushi Matsuzawa. 2017. "Post-Translational Modifications of the TAK1-TAB Complex" International Journal of Molecular Sciences 18, no. 1: 205. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18010205