
Tumor-Derived Tissue Factor Aberrantly Activates Complement and Facilitates Lung Tumor Progression via Recruitment of Myeloid-Derived Suppressor Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
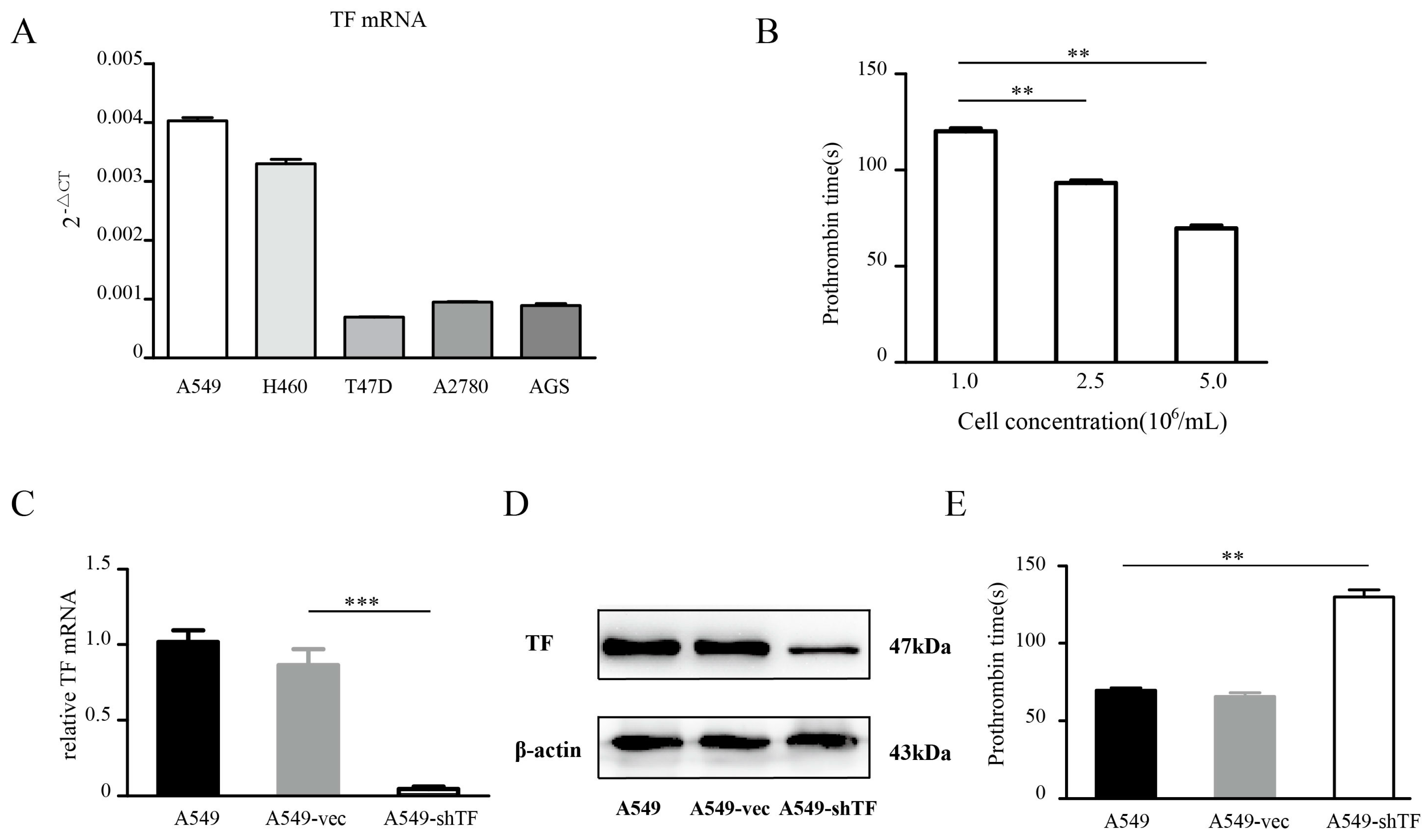
2.1. Tissue Factor Expression Knockdown Reduces the Procoagulant Activity of Lung Tumor Cells
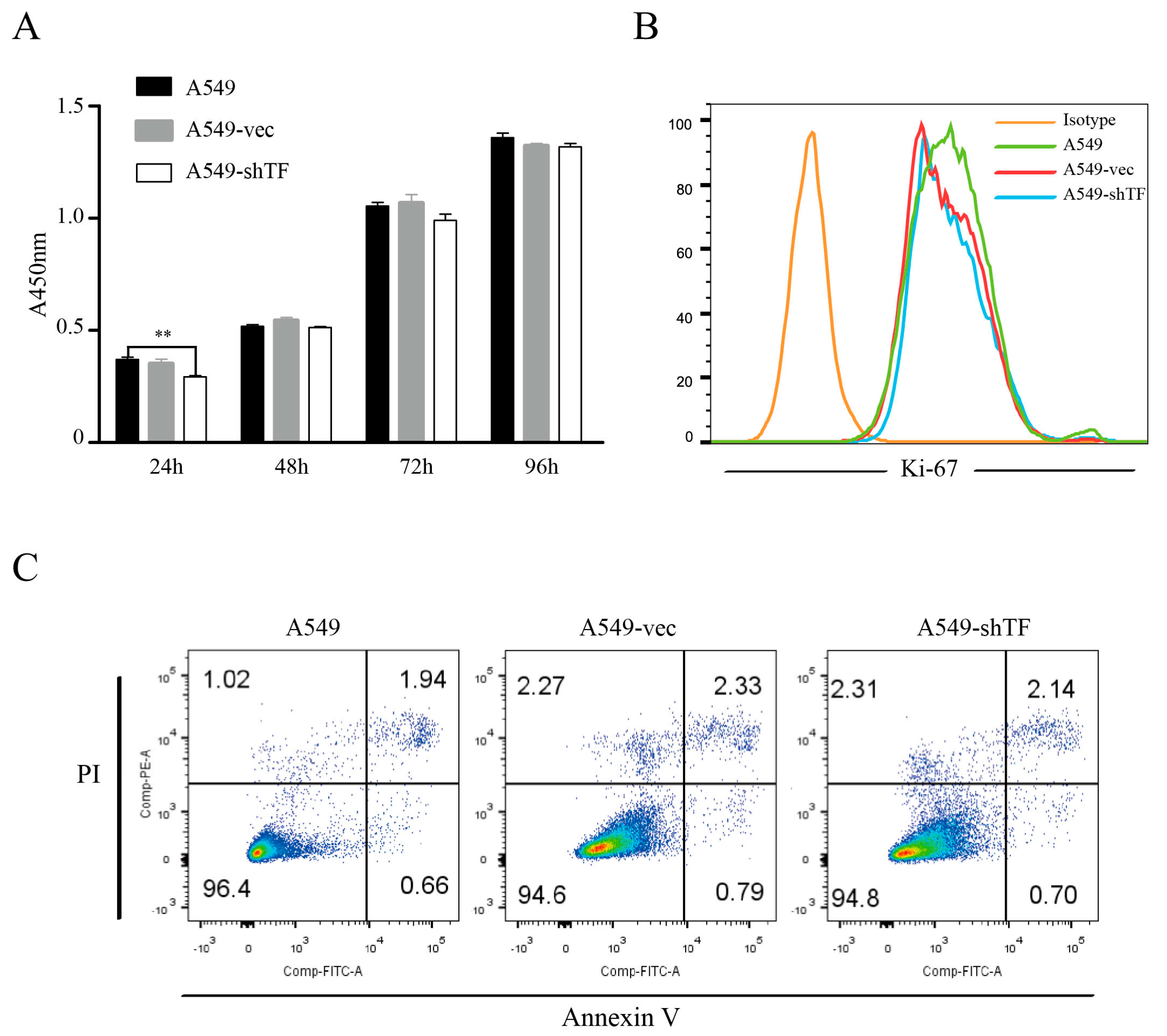
2.2. TF Knockdown Does Not Affect the Proliferation and Apoptosis of Lung Tumor Cells
2.3. TF-Facilitated Tumor Growth Is Associated with Local Coagulation Activation
2.4. Coagulation Induced by TF Activated Complement and Recruited MDSC
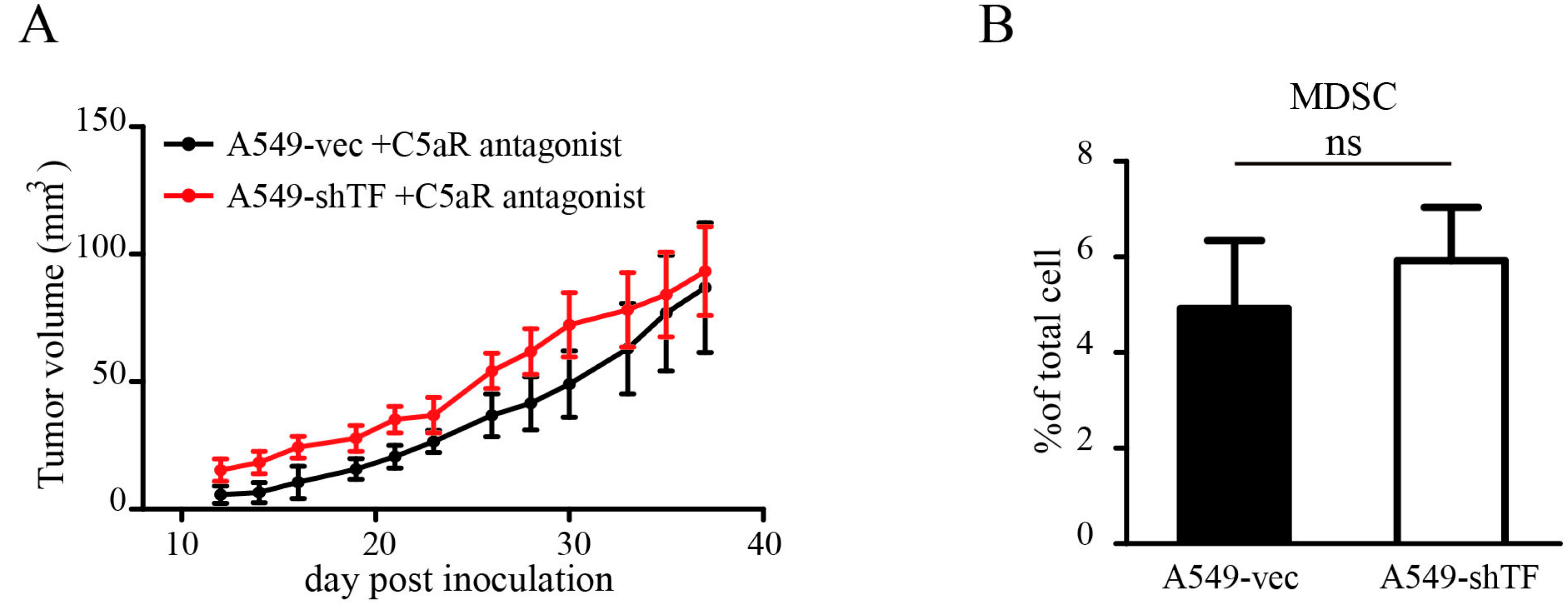
2.5. C5a Receptor Antagonism Blunted the Effect of TF by Inhibiting MDSC Recruitment
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. Transfection
4.3. RNA Isolation and Real-Time Polymerase Chain Reaction
4.4. Western Blot Analysis
4.5. Measurement of Prothrombin Time
4.6. In Vivo Xenograft Experiments
4.7. Immunohistochemical Analysis
4.8. Flow Cytometric Analysis
4.9. Cell Proliferation Assay
4.10. Statistics
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Falanga, A.; Russo, L. Epidemiology, risk and outcomes of venous thromboembolism in cancer. Hamostaseologie 2012, 32, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Cocco, E.; Hu, Z.; Richter, C.E.; Bellone, S.; Casagrande, F.; Bellone, M.; Todeschini, P.; Krikun, G.; Silasi, D.A.; Azodi, M.; et al. hI-con1, a factor VII-IgGFc chimeric protein targeting tissue factor for immunotherapy of uterine serous papillary carcinoma. Br. J. Cancer 2010, 103, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Zhang, Y.; Nayak, T.R.; Engle, J.W.; Wong, H.C.; Liu, B.; Barnhart, T.E.; Cai, W. Immuno-pet of tissue factor in pancreatic cancer. J. Nucl. Med. 2012, 53, 1748–1754. [Google Scholar] [CrossRef] [PubMed]
- Uno, K.; Homma, S.; Satoh, T.; Nakanishi, K.; Abe, D.; Matsumoto, K.; Oki, A.; Tsunoda, H.; Yamaguchi, I.; Nagasawa, T.; et al. Tissue factor expression as a possible determinant of thromboembolism in ovarian cancer. Br. J. Cancer 2007, 96, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Eisenreich, A.; Zakrzewicz, A.; Huber, K.; Thierbach, H.; Pepke, W.; Goldin-Lang, P.; Schultheiss, H.P.; Pries, A.; Rauch, U. Regulation of pro-angiogenic tissue factor expression in hypoxia-induced human lung cancer cells. Oncol. Rep. 2013, 30, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Goldin-Lang, P.; Tran, Q.V.; Fichtner, I.; Eisenreich, A.; Antoniak, S.; Schulze, K.; Coupland, S.E.; Poller, W.; Schultheiss, H.P.; Rauch, U. Tissue factor expression pattern in human non-small cell lung cancer tissues indicate increased blood thrombogenicity and tumor metastasis. Oncol. Rep. 2008, 20, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Unruh, D.; Sagin, F.; Adam, M.; van Dreden, P.; Woodhams, B.J.; Hart, K.; Lindsell, C.J.; Ahmad, S.A.; Bogdanov, V.Y. Levels of alternatively spliced tissue factor in the plasma of patients with pancreatic cancer may help predict aggressive tumor phenotype. Ann. Surg. Oncol. 2015, 22, 1206–1211. [Google Scholar] [CrossRef] [PubMed]
- Leppert, U.; Eisenreich, A. The role of tissue factor isoforms in cancer biology. Int. J. Cancer 2015, 137, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Gu, L.; Zhu, N.; Tang, H.; Alvarado, C.S.; Zhou, M. Tissue factor/FVIIa activates Bcl-2 and prevents doxorubicin-induced apoptosis in neuroblastoma cells. BMC Cancer 2008, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Aberg, M.; Johnell, M.; Wickstrom, M.; Siegbahn, A. Tissue factor/FVIIa prevents the extrinsic pathway of apoptosis by regulation of the tumor suppressor death-associated protein kinase 1 (DAPK1). Thromb. Res. 2011, 127, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jiang, P.; Capkova, K.; Xue, D.; Ye, L.; Sinha, S.C.; Mackman, N.; Janda, K.D.; Liu, C. Tissue factor-activated coagulation cascade in the tumor microenvironment is critical for tumor progression and an effective target for therapy. Cancer Res. 2011, 71, 6492–6502. [Google Scholar] [CrossRef] [PubMed]
- Gil-Bernabe, A.M.; Ferjancic, S.; Tlalka, M.; Zhao, L.; Allen, P.D.; Im, J.H.; Watson, K.; Hill, S.A.; Amirkhosravi, A.; Francis, J.L.; et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood 2012, 119, 3164–3175. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part I—Molecular mechanisms of activation and regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Heeger, P.S.; Kemper, C. Novel roles of complement in t effector cell regulation. Immunobiology 2012, 217, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, S.L.; Fantone, J.C.; Ward, P.A. Complement mediated inflammatory reactions. Pathobiol. Annu. 1981, 11, 127–154. [Google Scholar] [PubMed]
- Markiewski, M.M.; DeAngelis, R.A.; Benencia, F.; Ricklin-Lichtsteiner, S.K.; Koutoulaki, A.; Gerard, C.; Coukos, G.; Lambris, J.D. Modulation of the antitumor immune response by complement. Nat. Immunol. 2008, 9, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Ajona, D.; Rafail, S.; Lasarte, J.J.; Riezu-Boj, J.I.; Lambris, J.D.; Rouzaut, A.; Pajares, M.J.; Montuenga, L.M.; Pio, R. Anaphylatoxin C5a creates a favorable microenvironment for lung cancer progression. J. Immunol. 2012, 189, 4674–4683. [Google Scholar] [CrossRef] [PubMed]
- Huber-Lang, M.; Sarma, J.V.; Zetoune, F.S.; Rittirsch, D.; Neff, T.A.; McGuire, S.R.; Lambris, J.D.; Warner, R.L.; Flierl, M.A.; Hoesel, L.M.; et al. Generation of C5a in the absence of C3: A new complement activation pathway. Nat. Med. 2006, 12, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Amara, U.; Flierl, M.A.; Rittirsch, D.; Klos, A.; Chen, H.; Acker, B.; Bruckner, U.B.; Nilsson, B.; Gebhard, F.; Lambris, J.D.; et al. Molecular intercommunication between the complement and coagulation systems. J. Immunol. 2010, 185, 5628–5636. [Google Scholar] [CrossRef] [PubMed]
- Krisinger, M.J.; Goebeler, V.; Lu, Z.; Meixner, S.C.; Myles, T.; Pryzdial, E.L.; Conway, E.M. Thrombin generates previously unidentified C5 products that support the terminal complement activation pathway. Blood 2012, 120, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Bastarache, J.A.; Wang, L.; Geiser, T.; Wang, Z.; Albertine, K.H.; Matthay, M.A.; Ware, L.B. The alveolar epithelium can initiate the extrinsic coagulation cascade through expression of tissue factor. Thorax 2007, 62, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Toomey, J.R.; Kratzer, K.E.; Lasky, N.M.; Broze, G.J. Effect of tissue factor deficiency on mouse and tumor development. Proc. Natl. Acad. Sci. USA 1997, 94, 6922–6926. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, H.H.; Schaffner, F.; Kerver, M.; Petersen, H.H.; Ahamed, J.; Felding-Habermann, B.; Takada, Y.; Mueller, B.M.; Ruf, W. Inhibition of tissue factor signaling suppresses tumor growth. Blood 2008, 111, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, A.K.; DeRuvo, N.; Chinswangwatanakul, V.; Tebbutt, S.; Williamson, R.C. Extrinsic-pathway activation in cancer with high factor VIIa and tissue factor. Lancet 1995, 346, 1004–1005. [Google Scholar] [CrossRef]
- Ueno, T.; Toi, M.; Koike, M.; Nakamura, S.; Tominaga, T. Tissue factor expression in breast cancer tissues: Its correlation with prognosis and plasma concentration. Br. J. Cancer 2000, 83, 164–170. [Google Scholar] [PubMed]
- Nitori, N.; Ino, Y.; Nakanishi, Y.; Yamada, T.; Honda, K.; Yanagihara, K.; Kosuge, T.; Kanai, Y.; Kitajima, M.; Hirohashi, S. Prognostic significance of tissue factor in pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2005, 11, 2531–2539. [Google Scholar] [CrossRef] [PubMed]
- Eisenreich, A.; Bolbrinker, J.; Leppert, U. Tissue factor: A conventional or alternative target in cancer therapy. Clin. Chem. 2016, 62, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Anand, M.; Brat, D.J. Oncogenic regulation of tissue factor and thrombosis in cancer. Thromb. Res. 2012, 129, 46–49. [Google Scholar] [CrossRef]
- Eisenreich, A.; Boltzen, U.; Malz, R.; Schultheiss, H.P.; Rauch, U. Overexpression of alternatively spliced tissue factor induces the pro-angiogenic properties of murine cardiomyocytic HL-1 cells. Circ. J. 2011, 75, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Delluc, A.; Rousseau, A.; Delluc, C.; Le Moigne, E.; Le Gal, G.; Mottier, D.; van Dreden, P.; Lacut, K. Venous thromboembolism in patients with pancreatic cancer: Implications of circulating tissue factor. Blood Coagul. Fibrinolysis 2011, 22, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Zwicker, J.I.; Liebman, H.A.; Neuberg, D.; Lacroix, R.; Bauer, K.A.; Furie, B.C.; Furie, B. Tumor-derived tissue factor-bearing microparticles are associated with venous thromboembolic events in malignancy. Clin. Cancer Res. 2009, 15, 6830–6840. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Gui, Q.; Chen, W.; Wu, L.; Sun, W.; Zhang, N.; Xu, Q.; Wang, J.; Fu, X. Small interference RNA targeting tissue factor inhibits human lung adenocarcinoma growth in vitro and in vivo. J. Exp. Clin. Cancer Res. 2011, 30, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Hu, H.; Shi, W.; Ling, S.; Wang, T.; Wang, H. The expression and the functional roles of tissue factor and protease-activated receptor-2 on SW620 cells. Oncol. Rep. 2008, 20, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Ruf, W.; Dorfleutner, A.; Riewald, M. Specificity of coagulation factor signaling. J. Thromb. Haemost. 2003, 1, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Litwin, S.E.; Raya, T.E.; Anderson, P.G.; Daugherty, S.; Goldman, S. Abnormal cardiac function in the streptozotocin-diabetic rat. Changes in active and passive properties of the left ventricle. J. Clin. Investig. 1990, 86, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.L.; May, L.; Lhotak, V.; Shahrzad, S.; Shirasawa, S.; Weitz, J.I.; Coomber, B.L.; Mackman, N.; Rak, J.W. Oncogenic events regulate tissue factor expression in colorectal cancer cells: Implications for tumor progression and angiogenesis. Blood 2005, 105, 1734–1741. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Deng, Y.; Luther, T.; Muller, M.; Ziegler, R.; Waldherr, R.; Stern, D.M.; Nawroth, P.P. Tissue factor controls the balance of angiogenic and antiangiogenic properties of tumor cells in mice. J. Clin. Investig. 1994, 94, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Yousef, G.M.; Kopolovic, A.D.; Elliott, M.B.; Diamandis, E.P. Genomic overview of serine proteases. Biochem. Biophys. Res. Commun. 2003, 305, 28–36. [Google Scholar] [CrossRef]
- Veldman, A.; Hoffman, M.; Ehrenforth, S. New insights into the coagulation system and implications for new therapeutic options with recombinant factor VIIa. Curr. Med. Chem. 2003, 10, 797–811. [Google Scholar] [CrossRef] [PubMed]
- Ghebrehiwet, B.; Silverberg, M.; Kaplan, A.P. Activation of the classical pathway of complement by hageman factor fragment. J. Exp. Med. 1981, 153, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Hakulinen, J.; Meri, S. Complement-mediated killing of microtumors in vitro. Am. J. Pathol. 1998, 153, 845–855. [Google Scholar] [CrossRef]
- Sayegh, E.T.; Bloch, O.; Parsa, A.T. Complement anaphylatoxins as immune regulators in cancer. Cancer Med. 2014, 3, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.; Long, H.; He, L.; Han, X.; Lin, K.; Liang, Z.; Zhuo, W.; Xie, R.; Zhu, B. Interleukin-17 produced by tumor microenvironment promotes self-renewal of CD133+ cancer stem-like cells in ovarian cancer. Oncogene 2015, 34, 165–176. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, X.; Zha, H.; Yang, F.; Guo, B.; Zhu, B. Tumor-Derived Tissue Factor Aberrantly Activates Complement and Facilitates Lung Tumor Progression via Recruitment of Myeloid-Derived Suppressor Cells. Int. J. Mol. Sci. 2017, 18, 22. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18010022
Han X, Zha H, Yang F, Guo B, Zhu B. Tumor-Derived Tissue Factor Aberrantly Activates Complement and Facilitates Lung Tumor Progression via Recruitment of Myeloid-Derived Suppressor Cells. International Journal of Molecular Sciences. 2017; 18(1):22. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18010022
Chicago/Turabian StyleHan, Xiao, Haoran Zha, Fei Yang, Bo Guo, and Bo Zhu. 2017. "Tumor-Derived Tissue Factor Aberrantly Activates Complement and Facilitates Lung Tumor Progression via Recruitment of Myeloid-Derived Suppressor Cells" International Journal of Molecular Sciences 18, no. 1: 22. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18010022