
Hydrogen Bonding Interaction between Atmospheric Gaseous Amides and Methanol


Abstract
:1. Introduction
2. Results and Discussion
2.1. Geometric Analysis
2.2. Interaction Energy
2.3. OH-Stretching Vibrational Frequencies
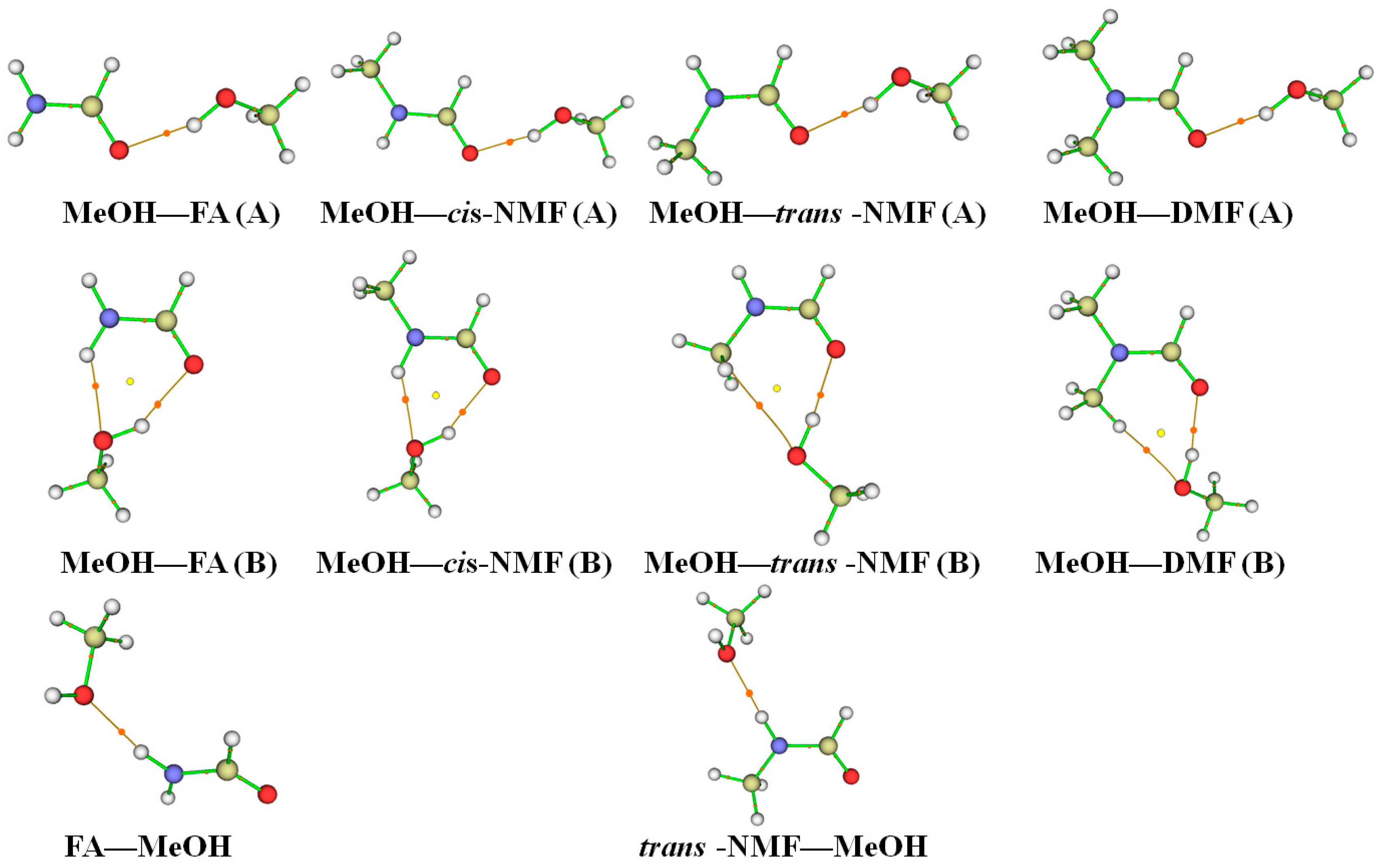
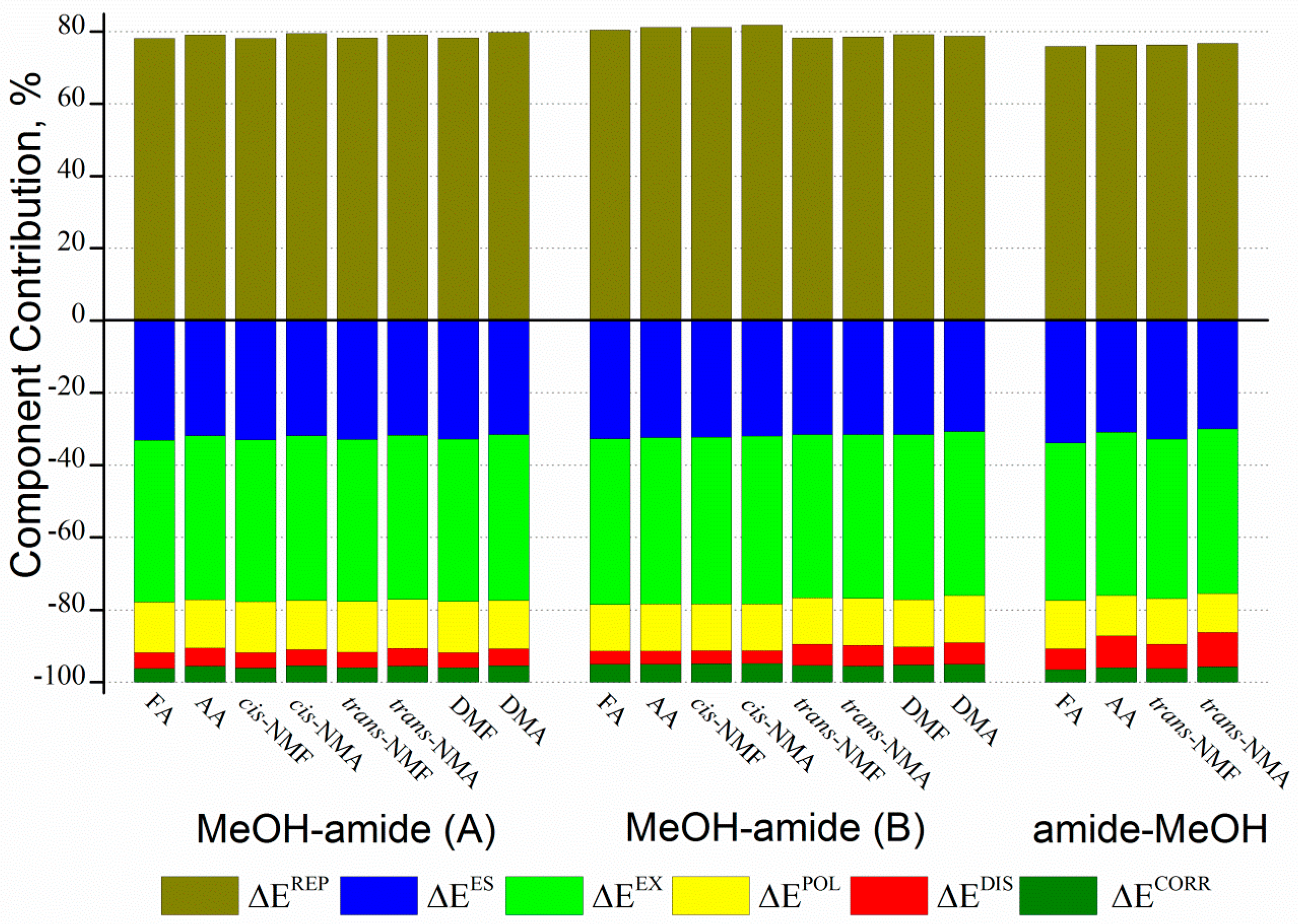
2.4. Topological and GKS-EDA Analysis
3. Computational Methods
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mikhonin, A.V.; Bykov, S.V.; Myshakina, N.S.; Asher, S.A. Peptide secondary structure folding reaction coordinate: Correlation between UV raman amide III frequency, ψ ramachandran angle, and hydrogen bonding. J. Phys. Chem. B 2006, 110, 1928–1943. [Google Scholar] [CrossRef] [PubMed]
- Manas, E.S.; Getahun, Z.; Wright, W.W.; DeGrado, W.F.; Vanderkooi, J.M. Infrared spectra of amide groups in α-helical proteins: Evidence for hydrogen bonding between helices and water. J. Am. Chem. Soc. 2000, 122, 9883–9890. [Google Scholar] [CrossRef]
- Walsh, S.T.R.; Cheng, R.P.; Wright, W.W.; Alonso, D.O.V.; Daggett, V.; Vanderkooi, J.M.; DeGrado, W.F. The hydration of amides in helices; a comprehensive picture from molecular dynamics, IR, and NMR. Protein Sci. 2003, 12, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Wexler, A.S.; Clegg, S.L. Atmospheric amines–Part I. A review. Atmos. Environ. 2011, 45, 524–546. [Google Scholar] [CrossRef]
- Zhu, L.; Schade, G.W.; Nielsen, C.J. Real-time monitoring of emissions from monoethanolamine-based industrial scale carbon capture facilities. Environ. Sci. Technol. 2013, 47, 14306–14314. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.J.; Herrmann, H.; Weller, C. Atmospheric chemistry and environmental impact of the use of amines in carbon capture and storage (ccs). Chem. Soc. Rev. 2012, 41, 6684–6704. [Google Scholar] [CrossRef] [PubMed]
- Bunkan, A.J.C.; Mikoviny, T.; Nielsen, C.J.; Wisthaler, A.; Zhu, L. Experimental and theoretical study of the OH-initiated photo-oxidation of formamide. J. Phys. Chem. A 2016, 120, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Leach, J.; Blanch, A.; Bianchi, A.C. Volatile organic compounds in an urban airborne environment adjacent to a municipal incinerator, waste collection centre and sewage treatment plant. Atmos. Environ. 1999, 33, 4309–4325. [Google Scholar] [CrossRef]
- Borduas, N.; da Silva, G.; Murphy, J.G.; Abbatt, J.P.D. Experimental and theoretical understanding of the gas phase oxidation of atmospheric amides with OH radicals: Kinetics, products, and mechanisms. J. Phys. Chem. A 2015, 119, 4298–4308. [Google Scholar] [CrossRef] [PubMed]
- Kulmala, M. How particles nucleate and grow. Science 2003, 302, 1000–1001. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, F.; Trostl, J.; Junninen, H.; Frege, C.; Henne, S.; Hoyle, C.R.; Molteni, U.; Herrmann, E.; Adamov, A.; Bukowiecki, N.; et al. New particle formation in the free troposphere: A question of chemistry and timing. Science 2016, 352, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Khalizov, A.; Wang, L.; Hu, M.; Xu, W. Nucleation and growth of nanoparticles in the atmosphere. Chem. Rev. 2012, 112, 1957–2011. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R. Getting to the critical nucleus of aerosol formation. Science 2010, 328, 1366–1367. [Google Scholar] [CrossRef] [PubMed]
- Temelso, B.; Morrell, T.E.; Shields, R.M.; Allodi, M.A.; Wood, E.K.; Kirschner, K.N.; Castonguay, T.C.; Archer, K.A.; Shields, G.C. Quantum mechanical study of sulfuric acid hydration: Atmospheric implications. J. Phys. Chem. A 2012, 116, 2209–2224. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Khalizov, A.F.; Zheng, J.; Xu, W.; Ma, Y.; Lal, V.; Zhang, R. Atmospheric nanoparticles formed from heterogeneous reactions of organics. Nat. Geosci. 2010, 3, 238–242. [Google Scholar] [CrossRef]
- Kulmala, M.; Kontkanen, J.; Junninen, H.; Lehtipalo, K.; Manninen, H.E.; Nieminen, T.; Petaja, T.; Sipila, M.; Schobesberger, S.; Rantala, P.; et al. Direct observations of atmospheric aerosol nucleation. Science 2013, 339, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Kulmala, M.; Petäjä, T.; Nieminen, T.; Sipilä, M.; Manninen, H.E.; Lehtipalo, K.; dal Maso, M.; Aalto, P.P.; Junninen, H.; Paasonen, P.; et al. Measurement of the nucleation of atmospheric aerosol particles. Nat. Protoc. 2012, 7, 1651–1667. [Google Scholar] [CrossRef] [PubMed]
- Nadykto, A.B.; Yu, F.Q.; Jakovleva, M.V.; Herb, J.; Xu, Y.S. Amines in the earth's atmosphere: A density functional theory study of the thermochemistry of pre-nucleation clusters. Entropy 2011, 13, 554–569. [Google Scholar] [CrossRef]
- Mellouki, A.; Wallington, T.J.; Chen, J. Atmospheric chemistry of oxygenated volatile organic compounds: Impacts on air quality and climate. Chem. Rev. 2015, 115, 3984–4014. [Google Scholar] [CrossRef] [PubMed]
- Legreid, G.; Lööv, J.B.; Staehelin, J.; Hueglin, C.; Hill, M.; Buchmann, B.; Prevot, A.S.H.; Reimann, S. Oxygenated volatile organic compounds (ovocs) at an urban background site in zürich (europe): Seasonal variation and source allocation. Atmos. Environ. 2007, 41, 8409–8423. [Google Scholar] [CrossRef]
- Rozenberg, M.; Loewenschuss, A.; Nielsen, C.J. Hydrogen bonding in the sulfuric acid–methanol–water system: A matrix isolation and computational study. J. Phys. Chem. A 2015, 119, 2271–2280. [Google Scholar] [CrossRef] [PubMed]
- Almeida, J.; Schobesberger, S.; Kuerten, A.; Ortega, I.K.; Kupiainen-Maatta, O.; Praplan, A.P.; Adamov, A.; Amorim, A.; Bianchi, F.; Breitenlechner, M.; et al. Molecular understanding of sulphuric acid-amine particle nucleation in the atmosphere. Nature 2013, 502, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Kurtén, T.; Loukonen, V.; Vehkamaki, H.; Kulmala, M. Amines are likely to enhance neutral and ion-induced sulfuric acid-water nucleation in the atmosphere more effectively than ammonia. Atmos. Chem. Phys. 2008, 8, 4095–4103. [Google Scholar] [CrossRef]
- Du, L.; Mackeprang, K.; Kjaergaard, H.G. Fundamental and overtone vibrational spectroscopy, enthalpy of hydrogen bond formation and equilibrium constant determination of the methanol-dimethylamine complex. Phys. Chem. Chem. Phys. 2013, 15, 10194–10206. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.R.; Contreras-Garcia, J.; Piquemal, J.-P.; Miller, B.J.; Kjaergaard, H.G. Are bond critical points really critical for hydrogen bonding? J. Chem. Theory Comput. 2013, 9, 3263–3266. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathi, R.; Subramanian, V.; Sathyamurthy, N. Hydrogen bonding without borders: An atoms-in-molecules perspective. J. Phys. Chem. A 2006, 110, 3349–3351. [Google Scholar] [CrossRef] [PubMed]
- Su, P.F.; Jiang, Z.; Chen, Z.C.; Wu, W. Energy decomposition scheme based on the generalized kohn-sham scheme. J. Phys. Chem. A 2014, 118, 2531–2542. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Kurawaki, A.; Hamada, Y.; Shinya, K.; Ohno, K.; Tohara, A.; Sato, M. Conformational behavior of n-methylformamide in the gas, matrix, and solution states as revealed by IR and NMR spectroscopic measurements and by theoretical calculations. J. Mol. Struct. 2006, 791, 30–40. [Google Scholar] [CrossRef]
- Kang, Y.K. Ab initio MO and density functional studies on trans and cis conformers of n-methylacetamide. J. Mol. Struct. 2001, 546, 183–193. [Google Scholar] [CrossRef]
- Panuszko, A.; Gojlo, E.; Zielkiewicz, J.; Smiechowski, M.; Krakowiak, J.; Stangret, J. Hydration of simple amides. FTIR spectra of HDO and theoretical studies. J. Phys. Chem. B 2008, 112, 2483–2493. [Google Scholar] [CrossRef] [PubMed]
- Gilli, P.; Bertolasi, V.; Ferretti, V.; Gilli, G. Covalent nature of the strong homonuclear hydrogen bond: Study of the O-H···O system by crystal structure correlation methods. J. Am. Chem. Soc. 1994, 116, 909–915. [Google Scholar] [CrossRef]
- Alabugin, I.V.; Manoharan, M.; Peabody, S.; Weinhold, F. Electronic basis of improper hydrogen bonding: A subtle balance of hyperconjugation and rehybridization. J. Am. Chem. Soc. 2003, 125, 5973–5987. [Google Scholar] [CrossRef] [PubMed]
- Gilli, P.; Bertolasi, V.; Pretto, L.; Gilli, G. Outline of a transition-state hydrogen-bond theory. J. Mol. Struct. 2006, 790, 40–49. [Google Scholar] [CrossRef]
- Pakiari, A.H.; Eskandari, K. The chemical nature of very strong hydrogen bonds in some categories of compounds. J. Mol. Struct. 2006, 759, 51–60. [Google Scholar] [CrossRef]
- Xu, W.; Zhang, R. A theoretical study of hydrated molecular clusters of amines and dicarboxylic acids. J. Chem. Phys. 2013, 139, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Esseffar, M.; el Firdoussi, A.; Bouab, W.; Abboud, J.L.M.; Mo, O.; Yanez, M. Combined experimental and theoretical study on hydrogen-bonded complexes between cyclic ketones, lactones, and lactams with 3,4-dinitrophenol. J. Phys. Chem. A 2009, 113, 14711–14717. [Google Scholar] [CrossRef] [PubMed]
- El Firdoussi, A.; Esseffar, M.; Bouab, W.; Abboud, J.L.M.; Mo, O.; Yanez, M.; Ruasse, M.F. Density functional theory study of the hydrogen bond interaction between lactones, lactams, and methanol. J. Phys. Chem. A 2005, 109, 9141–9148. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Chang, J.; Du, L. Effect of hydrogen bonding on the spectroscopic properties of molecular complexes with aromatic rings as acceptors. Comput. Theor. Chem. 2016, 1084, 126–132. [Google Scholar] [CrossRef]
- Zhang, Q.; Du, L. Hydrogen bonding in the carboxylic acid–aldehyde complexes. Comput. Theor. Chem. 2016, 1078, 123–128. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, Q.; Du, L. Hydrogen bonding in cyclic complexes of carboxylic acid-sulfuric acid and their atmospheric implications. RSC Adv. 2016, 6, 71733–71743. [Google Scholar] [CrossRef]
- Tang, S.; Zhao, H.; Du, L. Hydrogen bonding in alcohol-ethylene oxide and alcohol-ethylene sulfide complexes. RSC Adv. 2016, 6, 91233–91242. [Google Scholar] [CrossRef]
- Du, L.; Kjaergaard, H.G. Fourier transform infrared spectroscopy and theoretical study of dimethylamine dimer in the gas phase. J. Phys. Chem. A 2011, 115, 12097–12104. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Tang, S.; Li, S.; Ding, L.; Du, L. Theoretical investigation of the hydrogen bond interactions of methanol and dimethylamine with hydrazone and its derivatives. Struct. Chem. 2016, 27, 1241–1253. [Google Scholar] [CrossRef]
- Howard, D.L.; Kjaergaard, H.G. Hydrogen bonding to divalent sulfur. Phys. Chem. Chem. Phys. 2008, 10, 4113–4118. [Google Scholar] [CrossRef] [PubMed]
- Dulce, M.; Faria, G.; Teixeira-Dias, J.J.C.; Fausto, R. Hydrogen bonding involving alpha-beta-unsaturated carboxylic esters and substituted phenols: An infrared spectroscopic study. J. Mol. Struct. 1991, 263, 87–94. [Google Scholar]
- D'Alva Torres, G.S.F.; Pouchan, C.; Teixeira-Dias, J.J.C.; Fausto, R. Hydrogen bonding between substituted phenols and CH3COOCH3 or CH2ClCOOCH3: An FTIR spectroscopic study. Spectrosc. Lett. 1993, 26, 913–922. [Google Scholar] [CrossRef]
- Putz, M.V. Markovian approach of the electron localization functions. Int. J. Quant. Chem. 2005, 105, 1–11. [Google Scholar] [CrossRef]
- Matito, E.; Putz, M.V. New link between conceptual density functional theory and electron delocalization. J. Phys. Chem. A 2011, 115, 12459–12462. [Google Scholar] [CrossRef] [PubMed]
- Hobza, P.; Havlas, Z. Blue-shifting hydrogen bonds. Chem. Rev. 2000, 100, 4253–4264. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Jemmis, E.D. Red-, blue-, or no-shift in hydrogen bonds: A unified explanation. J. Am. Chem. Soc. 2007, 129, 4620–4632. [Google Scholar] [CrossRef] [PubMed]
- Van der Veken, B.J.; Herrebout, W.A.; Szostak, R.; Shchepkin, D.N.; Havlas, Z.; Hobza, P. The nature of improper, blue-shifting hydrogen bonding verified experimentally. J. Am. Chem. Soc. 2001, 123, 12290–12293. [Google Scholar] [CrossRef] [PubMed]
- Delanoye, S.N.; Herrebout, W.A.; van der Veken, B.J. Blue shifting hydrogen bonding in the complexes of chlorofluoro haloforms with acetone-d6 and oxirane-d4. J. Am. Chem. Soc. 2002, 124, 11854–11855. [Google Scholar] [CrossRef] [PubMed]
- Blanco, S.; Pinacho, P.; López, J.C. Hydrogen-bond cooperativity in formamide2–water: A model for water-mediated interactions. Angew. Chem. Int. Ed. 2016, 55, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Mahadevi, A.S.; Sastry, G.N. Cooperativity in noncovalent interactions. Chem. Rev. 2016, 116, 2775–2825. [Google Scholar] [CrossRef] [PubMed]
- Koch, U.; Popelier, P. Characterization of CHO hydrogen bonds on the basis of the charge density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen bonding strength–measures based on geometric and topological parameters. J. Phys. Org. Chem. 2004, 17, 18–31. [Google Scholar] [CrossRef]
- Popelier, P. Characterization of a dihydrogen bond on the basis of the electron density. J. Phys. Chem. A 1998, 102, 1873–1878. [Google Scholar] [CrossRef]
- Su, P.; Li, H. Energy decomposition analysis of covalent bonds and intermolecular interactions. J. Chem. Phys. 2009, 131, 014102. [Google Scholar] [CrossRef] [PubMed]
- Vijay, D.; Sakurai, H.; Subramanian, V.; Sastry, G.N. Where to bind in buckybowls? The dilemma of a metal ion. Phys. Chem. Chem. Phys. 2012, 14, 3057–3065. [Google Scholar] [CrossRef] [PubMed]
- Senthilkumar, L.; Ghanty, T.K.; Ghosh, S.K. Electron density and energy decomposition analysis in hydrogen-bonded complexes of azabenzenes with water, acetamide, and thioacetamide. J. Phys. Chem. A 2005, 109, 7575–7582. [Google Scholar] [CrossRef] [PubMed]
- Thellamurege, N.; Hirao, H. Water complexes of cytochrome p450: Insights from energy decomposition analysis. Molecules 2013, 18, 6782. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01, Gaussian, Inc.: Wallingford, CT, USA, 2013.
- Goerigk, L.; Grimme, S. A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions. Phys. Chem. Chem. Phys. 2011, 13, 6670–6688. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.S.; Du, L.; Kjaergaard, H.G. The effect of fluorine substitution in alcohol-amine complexes. Phys. Chem. Chem. Phys. 2014, 16, 22882–22891. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.S.; Du, L.; Kjaergaard, H.G. Positively charged phosphorus as a hydrogen bond acceptor. J. Phys. Chem. Lett. 2014, 5, 4225–4231. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies: Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Dunn, M.E.; Pokon, E.K.; Shields, G.C. Thermodynamics of forming water clusters at various temperatures and pressures by gaussian-2, gaussian-3, complete basis set-QB3, and complete basis set-APNO model chemistries: Implications for atmospheric chemistry. J. Am. Chem. Soc. 2004, 126, 2647–2653. [Google Scholar] [CrossRef] [PubMed]
- Feller, D. Application of systematic sequences of wave functions to the water dimer. J. Chem. Phys. 1992, 96, 6104–6114. [Google Scholar] [CrossRef]
- Bader, R.F. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]







{kind=link}
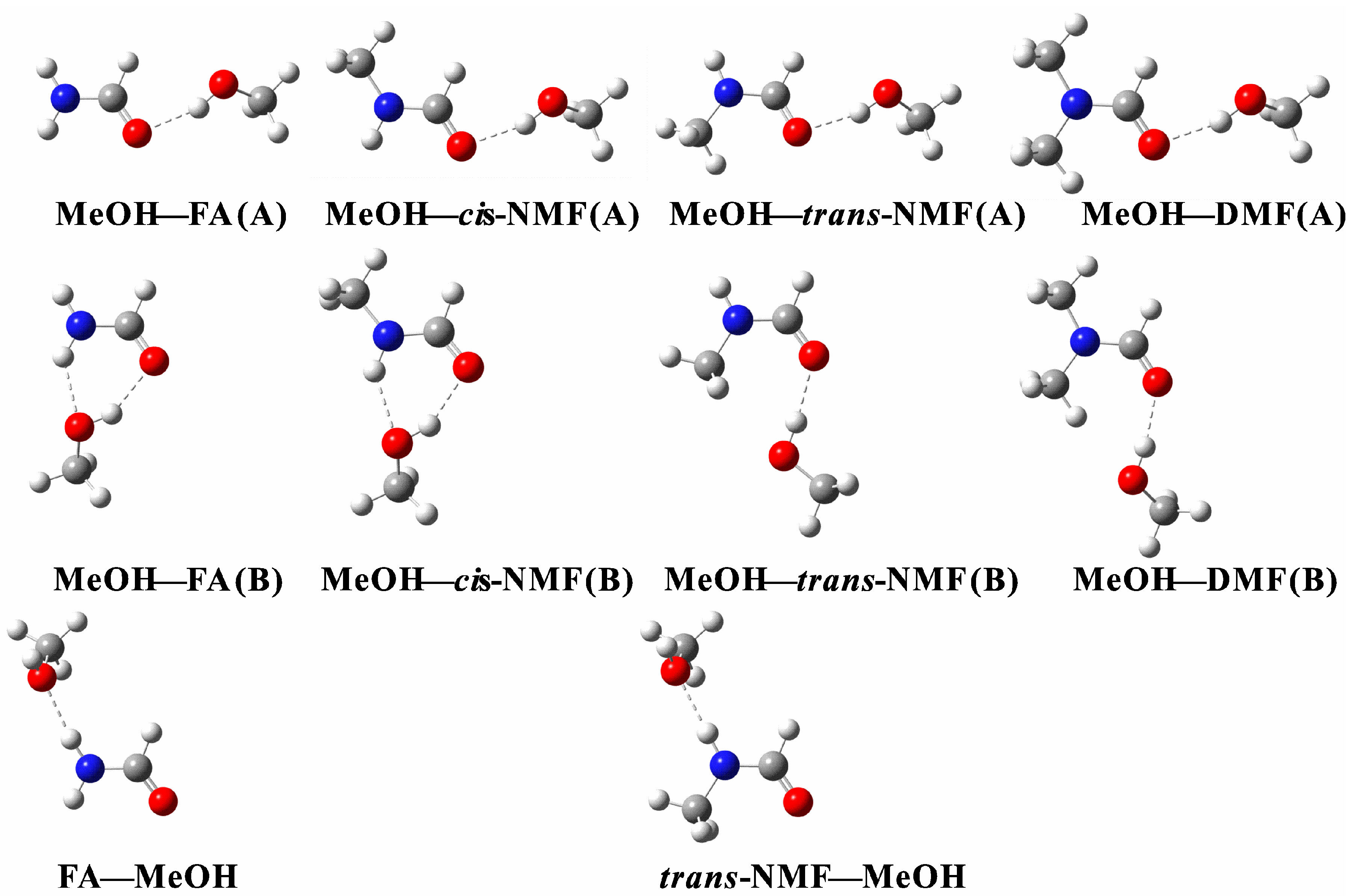{kind=link}
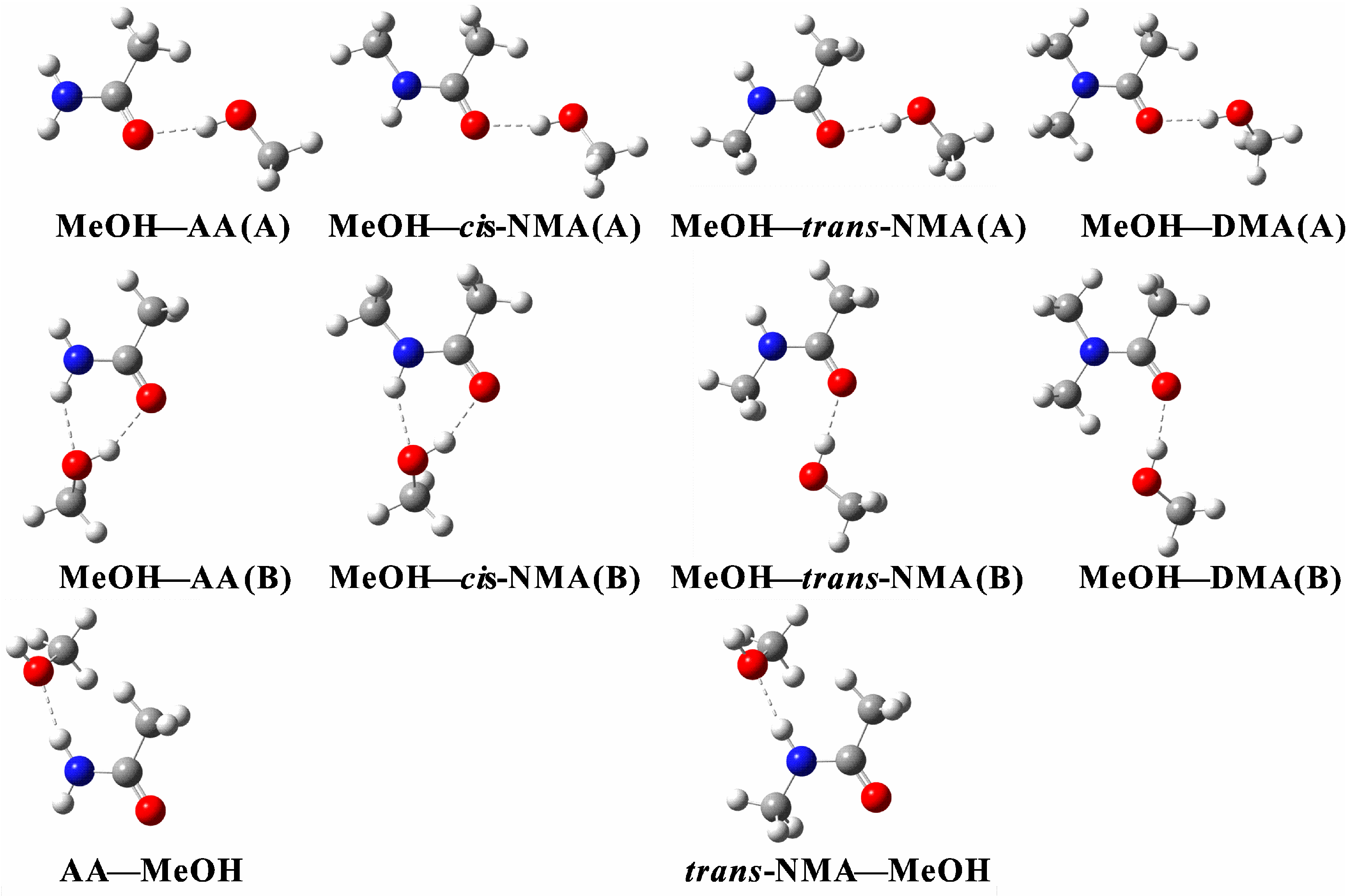{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Conformer | O–H···O | N–H···O | C=O | |||||
|---|---|---|---|---|---|---|---|---|---|
| Δr(OH) a | r(HB) b | θ(HB) c | d(O---O) d | Δr(NH) e | r(HB) b | θ(HB) c | Δr(C=O) f | ||
| MeOH–amide (A) | FA | 0.0102 | 1.8940 | 158.3 | 2.8193 | -- | -- | -- | 0.0080 |
| AA | 0.0126 | 1.8491 | 166.3 | 2.8043 | -- | -- | -- | 0.0087 | |
| cis-NMF | 0.0113 | 1.8735 | 160.4 | 2.8084 | -- | -- | -- | 0.0087 | |
| cis-NMA | 0.0134 | 1.8326 | 168.0 | 2.7929 | -- | -- | -- | 0.0088 | |
| trans-NMF | 0.0113 | 1.8715 | 161.5 | 2.8103 | -- | -- | -- | 0.0087 | |
| trans-NMA | 0.0135 | 1.8348 | 167.7 | 2.7945 | -- | -- | -- | 0.0091 | |
| DMF | 0.0119 | 1.8614 | 162.2 | 2.8035 | -- | -- | -- | 0.0091 | |
| DMA | 0.0136 | 1.8252 | 169.5 | 2.7891 | -- | -- | -- | 0.0091 | |
| MeOH–amide (B) | FA | 0.0137 | 1.8615 | 153.6 | 2.7992 | 0.0088 | 2.0569 | 139.5 | 0.0116 |
| AA | 0.0159 | 1.8615 | 153.6 | 2.7708 | 0.0089 | 2.0569 | 139.5 | 0.0126 | |
| cis-NMF | 0.0153 | 1.8789 | 152.1 | 2.7792 | 0.0085 | 2.0439 | 141.1 | 0.0125 | |
| cis-NMA | 0.0171 | 1.8446 | 154.6 | 2.7600 | 0.0084 | 2.0448 | 143.8 | 0.0131 | |
| trans-NMF | 0.0110 | 1.8621 | 174.7 | 2.8312 | -- | -- | -- | 0.0079 | |
| trans-NMA | 0.0121 | 1.8393 | 176.6 | 2.8111 | -- | -- | -- | 0.0076 | |
| DMF | 0.0108 | 1.8473 | 170.4 | 2.8099 | -- | -- | -- | 0.0070 | |
| DMA | 0.0122 | 1.8357 | 171.7 | 2.8021 | -- | -- | -- | 0.0095 | |
| Amide–MeOH | FA | -- | -- | -- | -- | 0.0049 | 1.9530 | 169.5 | 0.0039 |
| AA | -- | -- | -- | -- | 0.0033 | 1.9908 | 164.1 | 0.0031 | |
| trans-NMF | -- | -- | -- | -- | 0.0057 | 1.9584 | 169.9 | 0.0043 | |
| trans-NMA | -- | -- | -- | -- | 0.0055 | 1.9921 | 164.4 | 0.0028 | |
| Type | Conformer | BE b | ZPVE | Δ | Δ |
|---|---|---|---|---|---|
| MeOH−amide (A) | FA | −23.8 | 6.0 | −23.0 | 8.9 |
| AA | −29.0 | 6.4 | −28.7 | 9.1 | |
| cis-NMF | −25.8 | 5.8 | −24.7 | 7.3 | |
| cis-NMA | −30.5 | 5.9 | −29.6 | 4.6 | |
| trans-NMF | −25.7 | 5.4 | −24.4 | 7.6 | |
| trans-NMA | −30.3 | 6.1 | −29.7 | 10.0 | |
| DMF | −26.7 | 5.5 | −25.4 | 6.3 | |
| DMA | −30.8 | 5.8 | −29.8 | 4.4 | |
| MeOH−amide (B) | FA | −35.6 | 8.1 | −36.3 | 2.6 |
| AA | −37.6 | 7.9 | −38.2 | 9.1 | |
| cis-NMF | −37.9 | 7.4 | −38.2 | 0.9 | |
| cis-NMA | −39.2 | 7.3 | −39.4 | 2.1 | |
| trans-NMF | −29.0 | 6.2 | −28.4 | 9.6 | |
| trans-NMA | −30.1 | 6.8 | −29.9 | 12.0 | |
| DMF | −27.9 | 5.7 | −26.6 | 5.3 | |
| DMA | −29.0 | 6.2 | −28.3 | 8.5 | |
| amide–MeOH | FA | −21.2 | 4.8 | −19.5 | 8.3 |
| AA | −21.5 | 5.3 | −20.2 | 13.1 | |
| trans-NMF | −22.0 | 3.7 | −19.7 | 9.8 | |
| trans-NMA | −21.9 | 4.8 | −20.3 | 17.0 |
| Type | Conformer | O–H | N–H | C=O | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Δ a | fD/fM b | Δ a | fD/fM b | Δ a | fD/fM b | |||||
| MeOH–amide (A) | FA | 3623 | 205 | 18.5 | -- | -- | -- | 1758 | 26 | 0.9 |
| AA | 3576 | 252 | 24.7 | -- | -- | -- | 1727 | 28 | 0.8 | |
| cis-NMF | 3601 | 227 | 20.1 | -- | -- | -- | 1747 | 24 | 0.9 | |
| cis-NMA | 3560 | 268 | 27.5 | -- | -- | -- | 1718 | 26 | 0.8 | |
| trans-NMF | 3602 | 226 | 23.0 | -- | -- | -- | 1737 | 28 | 0.8 | |
| trans-NMA | 3590 | 238 | 24.7 | -- | -- | -- | 1750 | 30 | 0.8 | |
| DMF | 3589 | 238 | 24.7 | -- | -- | -- | 1720 | 27 | 0.8 | |
| DMA | 3557 | 271 | 29.0 | -- | -- | -- | 1683 | 28 | 0.7 | |
| MeOH–amide (B) | FA | 3590 | 238 | 24.7 | 3684 (3452) | 28 (122) | 2.4 (3.8) | 1752 | 32 | 1.0 |
| AA | 3527 | 301 | 25.0 | 3695 (3456) | 28 (131) | 2.3 (1.9) | 1721 | 35 | 0.9 | |
| cis-NMF | 3539 | 288 | 20.7 | 3442 | 149 | 4.9 | 1745 | 27 | 1.0 | |
| cis-NMA | 3507 | 320 | 29.2 | 3452 | 160 | 0.9 | 1714 | 29 | 1.0 | |
| trans-NMF | 3610 | 217 | 21.0 | -- | -- | -- | 1742 | 22 | 0.8 | |
| trans-NMA | 3590 | 238 | 24.7 | -- | -- | -- | 1712 | 23 | 0.7 | |
| DMF | 3681 | 147 | 21.9 | -- | -- | -- | 1731 | 16 | 0.8 | |
| DMA | 3584 | 244 | 22.8 | -- | -- | -- | 1681 | 30 | 0.8 | |
| Amide–MeOH | FA | -- | -- | -- | 3568 (3487) | 53 (87) | 3.0 (10.2) | 1773 | 11 | 0.9 |
| AA | -- | -- | -- | 3675 (3521) | 48 (66) | 3.1 (7.0) | 1746 | 10 | 0.9 | |
| trans-NMF | -- | -- | -- | 3591 | 35 | 16.6 | 1752 | 12 | 0.9 | |
| trans-NMA | -- | -- | -- | 3557 | 95 | 11.4 | 1727 | 7 | 0.9 | |
| Type | Conformer | O–H···O (C–H···O) | N–H···O (C–H···O) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Δq(H) a | ρ(BCP) b | ∇2ρ(BCP) c | r d | Δq(H) a | ρ(BCP) b | ∇2ρ(BCP) c | r d | ||
| MeOH–amide (A) | FA | 0.036 | 0.0230 | 0.1255 | -- | -- | -- | -- | -- |
| AA | 0.046 | 0.0243 | 0.1419 | 1.9492 | (0.073) | (0.0070) | (0.0329) | (0.9857) | |
| cis-NMF | 0.040 | 0.0238 | 0.1318 | -- | -- | -- | -- | -- | |
| cis-NMA | 0.049 | 0.0248 | 0.1478 | 1.8697 | (0.045) | (0.0081) | (0.0359) | (1.0279) | |
| trans-NMF | 0.039 | 0.0239 | 0.1322 | -- | -- | -- | -- | -- | |
| trans-NMA | 0.050 | 0.0249 | 0.1462 | 2.0597 | (0.034) | (0.0063) | (0.0300) | (0.7400) | |
| DMF | 0.043 | 0.0232 | 0.1433 | -- | -- | -- | -- | -- | |
| DMA | 0.050 | 0.0252 | 0.1513 | 1.8371 | (0.031) | (0.0083) | (0.0356) | (0.9545) | |
| MeOH–amide (B) | FA | 0.044 | 0.0226 | 0.1254 | 1.6165 | 0.060 | 0.0178 | 0.0939 | 1.5660 |
| AA | 0.050 | 0.0244 | 0.1411 | 1.6115 | 0.059 | 0.0178 | 0.0922 | 1.5223 | |
| cis-NMF | 0.048 | 0.0238 | 0.1346 | 1.6220 | 0.060 | 0.0184 | 0.0932 | 1.5306 | |
| cis-NMA | 0.056 | 0.0251 | 0.1476 | 1.5958 | 0.053 | 0.0183 | 0.0939 | 1.4802 | |
| trans-NMF | 0.039 | 0.0239 | 0.1322 | -- | -- | -- | -- | -- | |
| trans-NMA | 0.050 | 0.0249 | 0.1462 | 2.0597 | (0.034) | (0.0063) | (0.0300) | (0.7400) | |
| DMF | 0.042 | 0.0244 | 0.1369 | -- | -- | -- | -- | -- | |
| DMA | 0.050 | 0.0252 | 0.1513 | 1.8371 | (0.031) | (0.0083) | (0.0356) | (0.9545) | |
| Amide–MeOH | FA | -- | -- | -- | -- | 0.061 | 0.0204 | 0.1052 | -- |
| AA | (0.016) | (0.0063) | (0.0225) | (0.8941) | 0.055 | 0.0199 | 0.1013 | 1.8910 | |
| trans-NMF | -- | -- | -- | -- | 0.066 | 0.0204 | 0.1036 | -- | |
| trans-NMA | (0.016) | (0.0059) | (0.0210) | (0.7357) | 0.052 | 0.0198 | 0.1032 | 1.9016 | |
| Type | Conformer | ΔEES | ΔEEX | ΔEREP | ΔEPOL | ΔEDISP | ΔECORR | ΔEINT |
|---|---|---|---|---|---|---|---|---|
| MeOH–amide (A) | FA | −45.3 | −61.1 | 106.8 | −19.2 | −5.9 | −5.3 | −30.1 |
| AA | −54.4 | −77.3 | 134.8 | −22.9 | −8.3 | −7.7 | −35.8 | |
| cis-NMF | −48.1 | −65.1 | 113.8 | −20.6 | −6.1 | −5.8 | −31.9 | |
| cis-NMA | −57.3 | −81.8 | 142.8 | −24.5 | −7.9 | −8.2 | −36.8 | |
| trans-NMF | −47.7 | −64.8 | 113.4 | −20.4 | −6.1 | −5.9 | −31.5 | |
| trans-NMA | −56.1 | −79.8 | 139.4 | −24.0 | −8.6 | −7.9 | −36.9 | |
| DMF | −49.4 | −67.2 | 117.6 | −21.4 | −6.3 | −6.0 | −32.7 | |
| DMA | −58.1 | −84.3 | 147.0 | −24.8 | −8.4 | −8.5 | −37.1 | |
| MeOH–amide (B) | FA | −74.9 | −104.3 | 183.9 | −29.7 | −8.3 | −11.3 | −44.5 |
| AA | −80.3 | −113.8 | 201.1 | −32.5 | −8.7 | −12.5 | −46.6 | |
| cis-NMF | −79.2 | −112.5 | 198.6 | −31.8 | −8.8 | −12.5 | −46.3 | |
| cis-NMA | −83.8 | −121.4 | 214.5 | −34.0 | −9.3 | −13.6 | −47.5 | |
| trans-NMF | −51.6 | −73.7 | 127.8 | −21.0 | −9.5 | −7.6 | −35.5 | |
| trans-NMA | −54.4 | −77.7 | 135.0 | −22.5 | −9.7 | −7.7 | −37.1 | |
| DMF | −53.1 | −76.5 | 133.0 | −22.0 | −8.2 | −8.1 | −34.8 | |
| DMA | −53.1 | −78.2 | 136.0 | −22.6 | −10.2 | −8.7 | −36.8 | |
| Amide–MeOH | FA | −36.3 | −46.5 | 81.1 | −14.3 | −6.1 | −3.8 | −26.0 |
| AA | −34.8 | −50.7 | 85.9 | −12.6 | −9.9 | −4.5 | −26.6 | |
| trans-NMF | −35.6 | −47.6 | 82.6 | −13.8 | −7.1 | −4.2 | −25.6 | |
| trans-NMA | −34.3 | −51.9 | 87.7 | −12.3 | −10.8 | −4.9 | −26.4 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, H.; Tang, S.; Xu, X.; Du, L. Hydrogen Bonding Interaction between Atmospheric Gaseous Amides and Methanol. Int. J. Mol. Sci. 2017, 18, 4. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18010004
Zhao H, Tang S, Xu X, Du L. Hydrogen Bonding Interaction between Atmospheric Gaseous Amides and Methanol. International Journal of Molecular Sciences. 2017; 18(1):4. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18010004
Chicago/Turabian StyleZhao, Hailiang, Shanshan Tang, Xiang Xu, and Lin Du. 2017. "Hydrogen Bonding Interaction between Atmospheric Gaseous Amides and Methanol" International Journal of Molecular Sciences 18, no. 1: 4. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18010004